
Abstract
Ischemia–reperfusion (I/R) injury is common during surgery and often results in organ dysfunction. The mechanisms of I/R injury are complex, diverse, and not well understood. RNA methylation is a novel epigenetic modification that is involved in the regulation of various biological processes, such as immunity, response to DNA damage, tumorigenesis, metastasis, stem cell renewal, fat differentiation, circadian rhythms, cell development and differentiation, and cell division. Research on RNA modifications, specifically N6-methyladenosine (m6A), have confirmed that they are involved in the regulation of organ I/R injury. In this review, we summarized current understanding of the regulatory roles and significance of m6A RNA methylation in I/R injury in different organs.
Similar content being viewed by others
Facts
m6A modifications have revealed relatively complete enzymatic systems, and the related regulatory mechanisms are becoming increasingly well understood.
m6A mRNA modifications have been found in myocardial, brain, and renal ischemia–reperfusion (I/R) in in vivo and in vitro studies.
m6A RNA modifications have been identified as an important mechanism in I/R-induced organ injury, where inhibition of m6A methylation protects organs from I/R injury.
Most of the known functions of m6A have been inferred from the phenotypic consequences of manipulating m6A “writers” and “erasers” during I/R.
Questions
Does m6A modification play a regulatory role in ischemia or in reperfusion stage?
What are the individual mRNA m6A modification sites during I/R?
Can m6A mRNA methylation optimize ischemic cardiac preconditioning or post-conditioning in I/R therapy?
Introduction
Ischemia, occurs following restricted blood supply to tissues, is common in patients undergoing surgeries. Re-establishment (reperfusion) of blood flow is mandatory to salvage the ischemic tissues1,2,3,4. In most cases, post-ischemic reperfusion can restore the normal functions of tissues and organs and repair damaged structures, but in some cases, reperfusion itself may lead to further tissue damage/dysfunction and eventually cause organ failure. This phenomenon is known as ischemia–reperfusion (I/R) injury. Despite incredible advancements of techniques in reducing tissue ischemia such as thrombolytic therapy, percutaneous coronary angioplasty and cardiopulmonary bypass, post-operative morbidity and mortality owing to I/R injury remain high1,2,3,4. Extensive studies have focused on investigating the underline mechanisms of I/R injury. Different mechanisms have been suggested, such as burst of reactive oxygen species (ROS) during reperfusion5,6,7, elevation for inflammatory response, mitochondrial dysfunction, and calcium overload. However, the full picture of the pathophysiology of I/R injury is far from complete and further research is warranted.
Epigenetic modifications, including histone modification and DNA methylation, have been demonstrated to play key roles in I/R injury8,9. Chromatin structure depends on electrostatic interactions between positive charges on histones and negative charges on DNA. Histones acetylation on lysine residues can neutralize the positive charge, thereby disrupting the stability of histone and DNA interactions, and subsequently changes the condensed chromatin into an open, loosely assembled chromatin structure that allows for the mobilization of gene transcription regulators10. DNA methylation is catalyzed by DNA methyltransferase, which adds methyl groups to DNA nucleotides, leading to chromatin condensation and gene expression changes11. Similarly, RNA nucleotides can also have covalent modifications that regulate gene expression by affecting RNA stability and translation. RNA modifications are types of post-transcriptional regulation, and over 150 types of RNA modifications have been identified. They are widely distributed in various types of RNA, including messenger RNA (mRNA), transfer RNA (tRNA), ribosomal RNA (rRNA), small non-coding RNA, and long non-coding RNA (lncRNA). RNA methylation accounts for over 60% of all RNA modifications12,13. Methylation modifications have been identified on all four ribonucleotides (A, U, C, and G) including N6-methyladenosine (m6A), 5-methylcytosine (m5C)14, 3-methyluracil (m3U)15, N7-methylguanosine (m7G)16, and so on. Among them, m6A methylation is the most common type of RNA methylation in mRNA17. m6A methylation primarily occurs at the sixth site of adenine in RRACH RNA sequences, which can regulate the splicing, transportation, localization, translation, and degradation of target RNAs18. m6A is widely present in various eukaryotes, including yeast19, plants20, Drosophila21, and mammals18, and it is also the most common modification in lncRNA in higher organisms22,23,24. However, specific molecular functions of m6A are still not well understood.
In recent years, studies on m6A RNA modification have confirmed that it dynamically and reversibly regulates the development and progression of I/R injury in different organs. In this review, we summarized the known modifications, regulations, and significances of m6A RNA methylation in I/R injury in different organs.
m6A RNA methylation machinery
m6A modifications on mRNA was first reported by Desrosiers et al.18 in the 1970s, but studies on RNA modification have lagged behind studies on DNA modifications, and precise functions of m6A modifications were largely unclear until recently. The core question that remains unresolved regarding RNA modifications is whether such modifications regulate gene expression and whether they are dynamic and reversible.
The functions of m6A methylation are determined by “writers”, “erasers”, and “readers”25 (Fig. 1). “Writers”, also known as methyltransferases, are proteins that induce specific RNA methylation. Methyltransferase-like protein 3 (METTL3), methyltransferase-like protein 14 (METTL14), Wilm’s tumor 1 associated protein (WTAP), KIAA1429, METTL3, METTL14, and WTAP are methyltransferases that catalyze m6A modifications26. METTL3 and its homolog METTL14 are localized in splicing factor-rich nuclear subcellular compartments known as nuclear speckles, suggesting that m6A modifications may be associated with RNA splicing. WTAP interacts with METTL3–METTL14 dimers and colocalizes with nuclear speckles, which affects the efficiency of methylation and mRNA splicing27. KIAA1429 is a candidate subunit of the methyltransferase complex that has been shown to be necessary for methylation28.

a m6A modification occurs primarily at sixth site of adenine in RRACH sequences (R indicates A or G; H indicates A, U, or C). Dynamic and reversible mRNA m6A modification: the m6A methyltransferase complex METTL3/METTL14/WTAP catalyzes the transformation of A to m6A, and the demethylases FTO and ALKBH5 catalyze its demethylation. The YTH family protein YTHDC1 binds to m6A modifications in the nucleus, and YTHDF2/1 binds to cytoplasmic m6A. MicroRNAs regulate m6A production by regulating METTL3 activity.
In contrast to “writers”, “erasers” are proteins that remove specific RNA methylation, also known as demethylases, which include fat mass and obesity-associated (FTO) and ALKBH5. FTO is a member of the ALKB family and was the first demethylase discovered. It has been shown to affect the RNA-binding ability of the splicing factor SRSF2, thus regulates pre-mRNA splicing29,30,31,32. In vitro studies incubating wild-type and mutant FTO proteins with methylated substrates showed that FTO proteins exert demethylating activity on m6A in single-stranded RNA33. ALKBH5 is another member of the ALKB family that found to have demethylation activity34. ALKBH5 colocalizes with nuclear speckles in an RNase A-sensitive manner and can directly catalyze the demethylation of m6A-methylated adenosine, which is in contrast to the oxidation reaction catalyzed by FTO35. Demethylation by ALKBH5 also affects the efficiency of nascent mRNA synthesis and splicing36.
m6A is recognized by “readers”. “Readers” are a large class of proteins or domains, which can specifically identify different types of RNA methylation and link RNA methylation modification to specific biological function37. At present, known m6A-binding proteins are YT521-B homology (YTH) domain proteins, including YT521-B homology domain family 1 (YTHDF1), YTHDF2, YTHDF3, YT521-B homology domain containing 1 (YTHDC1), YTHDC2, and the heterogeneous nuclear ribonucleoprotein (HNRNP) family proteins HNRNPA2B1 and HNRNPC38. m6A mRNA modifications function in primarily two ways: modulation of the structure of the methylated transcript to prevent or induce protein-RNA interactions and direct recognition by m6A-binding proteins, which induces subsequent reactions25. A class of proteins containing YTH functional domains has been shown to bind to m6A39. Among these, YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2 have been confirmed to be m6A-binding proteins39. YTHDF1 primarily affects the gene translations by m6A modification, whereas YTHDF2 primarily affects their degradation, and YTHDC1 affects splicing. On the other hand, HNRNPC is an abundant nuclear RNA-binding protein that is involved in pre-mRNA processing40. Study has shown that HNRNPC regulates the abundance and alternative splicing of target transcripts via binding to m6A and RNA41. Dynamic and reversible mRNA m6A modification is shown in Fig. 2.

m6A also exists in the intronic regions of pre-mRNA precursors, suggesting that methylation modifications may regulate the alternative splicing of mRNA precursors to form mature mRNAs. m6A also affects the nuclear export, translation, and degradation of mRNA; mRNA degradation is mediated by YTHDF2, mRNA translation is mediated by YTHDF1, and alternative splicing is mediated by YTHDC1; other biological functions are yet to be elucidated. The potential regulatory functions of m6A in mRNA processing and metabolism, including splicing, nuclear export, localization, translation, and degradation (stability), occur after the transcription of precursor pre-mRNA, and m6A is an important reversible chemical modification of mRNA that may serve as a novel cis-regulatory element for these processes.
Epigenetic regulations in ischemia–reperfusion injury
I/R injury occurs in various tissues and organs, including the heart42, liver43,44,45, kidneys46, skin47, lungs48,49,50, muscle51,52, eye53,54, brain55,56, blood vessels57, and mesentery58,59. The pathophysiology of I/R injury in these organs is generally similar60,61. I/R injury is primarily characterized by neutrophil infiltration and the burst production of ROS. This excess production of ROS results in oxidative stress in tissues, which leads to cell death and eventually organ dysfunction. The reperfusion injury salvage kinase (RISK)62,63,64,65, survival activating factor enhancement (SAFE)66,67,68, and cyclic guanosine monophosphate (cGMP)-protein kinase G (cGMP-dependent kinase) signaling pathways69,70 are the major pathways involved in the protective effects of various interventions in different organs against I/R injury (Fig. 3). Recently, epigenetic regulation has been suggested to play important roles in I/R injury.

The RISK pathway is activated by opioid peptides, bradykinins, adenosine, receptor tyrosine kinase G protein-coupled receptors, erythropoietin, cytokines, insulin, or insulin-like growth factor-1. The cGMP/PKG pathway is activated by external stimuli (ANP and BNP) via the activation of natriuretic peptide receptors A and B. The SAFE pathway is activated by IL-6, IL-11, leukemia inhibitory factor (LIF), or TNF-α via the glycoprotein 130 receptor or TNF-α type 2 receptor. Among these pathways, the potential m6A methylation targets have been labeled with pink circle.
Two major types of epigenetic regulation, histone acetylation and DNA methylation, have been implicated in the pathogenesis of I/R injury. During I/R-induced lung injury, the histone acetylation inhibitor trichostatin A (TSA) has been shown to suppress lung inflammation by inhibiting apoptosis and the phosphorylation of ERK, JNK, and p3871. Histone acetylation can also promote the acetylation and release of HMGB1 from hepatocyte nuclei72, inhibit MPO activity in lung tissue73, and promote the production of antioxidant enzymes, such as FoxO3a and SOD, in cardiac cells74, suggesting that histone acetylation can effectively regulate inflammatory infiltration and the oxidative stress response in I/R injury in different tissues and organs. Recent studies on DNA methylation in I/R injury have focused on the kidneys75,76, heart77,78,79, and brain80, which showed that inhibition of DNA methylation can protect organs from different kinds of I/R injury.
Recently, studies have reported that m6A is closely associated with oxidative stress. Li et al.81 demonstrated that p21 can induce oxidative stress and regulate cellular senescence through m6A and m5C RNA modifications. They showed that oxidative stress altered m6A levels, thereby affecting mRNA translation82. Recently, Xiang Zhong et al.83 reported that knockout of the liver circadian clock gene Bmal1 in mice resulted in abnormal liver lipid metabolism, accompanied with increase of mRNA m6A levels and a loss of mRNA m6A circadian rhythm. Moreover, in Bmal1 deletion mice, m6A-seq showed elevated m6A methylation of PPARα, a key transcription factor that regulates lipid and lipoprotein metabolism in the liver, suggesting that Bmal1 may regulate hepatic lipid metabolism through m6A RNA methylation of PPARα83. Inhibition of m6A RNA methylation increased the stability and longevity of PPARα mRNA through YTHDF2 by regulating PPARα transcription and translation, reducing lipid accumulation in hepatocytes in vitro83. These studies suggest that oxidative stress is involved in m6A methylation and that m6A may also affect mRNA translation by inducing oxidative stress, indicating that m6A and oxidative stress regulate each other. However, whether this interaction exists in I/R injury remains as a question.
Interestingly, m6A has been reported in recovery of translational efficiency following hypoxic stress, affecting cell survival84. Myocardial infarction-associated transcript (MIAT), a hypoxia-responsive lncRNA, has been identified as a target gene of ALKBH1-related m6A mRNA modification. Wu L et al. found that m6A mRNA, but not 5-methylcytosine DNA methylation, in leukocyte was reduced in atherosclerosis patients with increased carotid plaque size. They further found that LDL was an independent risk factor in reducing the level of m6A and the progression of plaque formation85. Mechanistically, they showed that ox-LDL-induced m6A demethylation facilitated HIF1α binding to the ALKBH1-demethylated MIAT promoter and activating MIAT transcription85. These studies indicate mRNA modifications by m6A may take part in hypoxic-related ischemia or I/R disease.
m6A RNA methylation regulates ischemia/reperfusion injury in the heart
Current therapies for ischemic heart disease and adverse post-ischemic cardiac remodeling have limited efficacy. Although the roles of various transcription factors and transcription coactivators have been evaluated, studies on the post-transcriptional regulation of mRNAs that regulate the key proteins and cardiac function are still at the initial stage. Most recently, using whole-genome m6A sequencing analysis, Dom et al.86 revealed the presence of m6A and its dynamic changes in cardiomyocyte hypertrophy. In this study, they found that METTL3 is a key RNA-modifying protein that catalyzes m6A methylation of specific mRNA subgroups, which lead to cardiomyocyte hypertrophy. They further showed that enhancement of METTL3 is sufficient to induce cardiomyocyte hypertrophy in the absence of additional stimulation in vitro. Inhibition of METTL3 had no effect on cardiomyocyte hypertrophy under normal serum condition but effectively blocked the development of cardiomyocyte hypertrophy when additional serum was provided86. Increase in m6A results in the adaptive growth of cardiomyocytes, whereas decrease in m6A induces eccentricity and adverse cardiomyocyte geometry. Therefore, alterations in METTL3 level affects m6A metabolism and induces spontaneous cardiomyocyte remodeling86. This indicates that METTL3-mediated m6A modification is sufficient to regulate genes that responsible for cardiac remodeling and function. Thus, METTL3 may be a potential therapeutic target for pathological cardiac remodeling.
Cardiomyocyte death occurs during myocardial I/R and has a critical role in myocardial I/R injury. m6A RNA methylation has been shown to closely to the pathogenesis of myocardial I/R injury. Cardiac METTL3 protein level was increased accompanied with decreased myocardial cell viability in mice subjected to myocardial I/R87. In line with these findings, in neonatal cardiomyocytes subjected to hypoxia/reoxygenation, upregulation of METTL3 decreased autophagic flux and promoted the cell apoptosis, whereas knockout of METTL3 enhanced the cell viability87. These findings highlight the important role of METTL3, or m6A modifications in general, in myocardial I/R injury. Indeed, in the same model in cardiomyocytes subjected to hypoxia/reoxygenation, overexpression of RNA demethylase ALKBH5 reversed METTL3-induced cell injury87, confirming the role of m6A modifications in myocardial I/R injury.
FTO, an m6A demethylase that regulates transcriptomic m6A modifications in mRNA88, is associated with metabolic disorders such as diabetes and obesity, diseases in which the heart is vulnerable to I/R injury. In mammals, cardiac FTO expression is decreased in the failing hearts of human, pig, and mouse, as well as in mouse cardiomyocytes under hypoxia89. In cardiomyocytes under hypoxia or mice with heart failure, overexpression of FTO attenuates ischemia-induced cardiac remodeling, increases cardiac contractile protein expression, and improves cardiac contractility89, suggesting that FTO would be a therapeutic target for heart failure. Moreover, studies indicated that the cardioprotective mechanism of FTO is mediated by the selective demethylation of transcripts related to cardiac contraction under ischemia, which increases mRNA stability and protein expression90.
In addition to its role in I/R injury, m6A modifications have been shown to have a role in anesthetic post-conditioning cardioprotection. In H2O2-induced senescent H9c2 cells, hypoxia/reoxygenation was found to increase the level of m6A methylation globally, where post-conditioning with dexmedetomidine, a widely used anesthetic, reduce m6A methylation and attenuate cell death91. They further showed that dexmedetomidine post-conditioning increases lncRNA H19 by upregulating ALKBH5 and reduces hypoxia/reoxygenation-induced cell death91. However, whether the hypoxia/reoxygenation-induced global m6A changes apply to genes other than H19 and whether they play roles in dexmedetomidine post-conditioning cellular protective effects are unclear. Thus, exploring the precise mechanism of m6A methylation may facilitate the optimization of ischemic cardiac preconditioning or post-conditioning in protecting the heart against I/R injury.
m6A RNA methylation regulates ischemia/reperfusion injury in the brain
The fate of neurons after brain I/R injury is determined by a series of complex biochemical and molecular events, including excitotoxicity92, ion imbalance93, oxidative stress94, endoplasmic reticulum stress95, apoptosis96, and inflammation97. These processes can facilitate rapid changes in the ischemic-sensitive transcriptome. In addition, post-ischemic pathophysiological changes can be fine-tuned by the regulation of post-transcriptional RNA levels98 via non-coding RNAs, RNA-binding proteins, and epigenetic post-transcriptional modifications99,100. The “eraser” of m6A methylation, FTO, is highly enriched in the brain, where its deletion results in impaired dopaminergic neurotransmission and congenital microcephaly101. In mammals, m6A modifications are the most abundant in the brain and serve to regulate synaptic plasticity, axonal growth, learning and memory, and stress responses102. Using the Arraystar mouse epitranscriptome chip, Chokkalla et al.103 first demonstrated that m6A modifications were increased in 122 mRNAs and 17 lncRNAs and decreased in 15 mRNAs and 3 lncRNAs after 12 hours of transient ischemia and reperfusion in mice. GO/pathway analysis revealed that these mRNAs with altered m6A modifications were enriched in biological processes such as inflammation, apoptosis, and transcriptional regulation. Most recently, Diao et al.104 showed that, in primary hippocampal neurons, hypoxia/reoxygenation-induced activation of PTEN along with increased cell pyroptosis, which reversed by hypothermia. Interestingly, global m6A-methylated RNA, and PTEN methylation in specific, were increased in neurons subjected to hypoxia/reoxygenation, whereas these changes were decreased after hypothermia treatment. Similarly, inhibition of PTEN transcription, the PTEN RNA decay rate was reduced to baseline level after hypothermia treatment104, suggesting that hypothermia may confer protective effects through m6A modification of PTEN. Together, these results provide evidence of the involvement of m6A modifications in I/R injury in the brain.
m6A RNA methylation regulates ischemia/reperfusion injury in the kidney
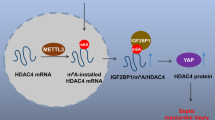
During renal I/R injury, excess ROS production results in renal tubular necrosis and renal dysfunction105,106. Recent studies have shown that FTO mediates m6A mRNA demethylation in the 3′-UTR of peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) in renal cells, which increases PGC-1α mRNA stability and protein expression. This increased PGC-1α expression enhanced mitochondrial activity and induced oxidative stress in HEK 293 T renal cells, suggesting that the regulation of m6A methylation is closely associated with oxidative stress, the major adverse event of renal I/R injury, in renal cells107. During renal I/R injury, acute kidney injury often progresses to chronic kidney injury, and interstitial fibrosis has an important role in this process108. Yes-associated protein 1 (YAP1) is involved in kidney regeneration and fibrosis through its proliferation-promoting and pro-fibrosis functions. Interstitial fibrosis and abnormal tubular differentiation are associated with a continuous increase in the activation of YAP1. YAP1 mRNA methylation levels were reduced in HK-2 cells with METTL14 knockdown and in the kidney from METTL14 knockout mice. Loss of METTL14 function resulted in decreased YAP1 methylation levels and increased YAP1 protein translation109. Inhibition of the YAP1-TEAD pathway by peptide 17 eliminated the protective effect of METTL14 on renal I/R in vivo and in vitro, indicating that the role of METTL14 in renal I/R is dependent on the activation of YAP1 and the YAP1-TEAD pathway by RNA methylation109. This suggests that the regulation of m6A RNA methylation is involved in the development and progression of renal I/R injury.
Summary and outlook
In summary, research on m6A modifications has revealed key regulatory enzymes and proteins involved in the process, including methyltransferases, demethylases, and binding proteins. The related regulatory mechanisms, such as microRNAs, are becoming better understood as well. RNA modifications with m6A play important regulatory roles in RNA processing and I/R injury (Table 1). Through the combined action of “writers” and “erasers,” the m6A levels on RNA can be dynamically regulated, and fine regulation of RNA processing can be achieved through the recruitment of various binding proteins. However, most of the currently known functions of m6A have been inferred from the phenotypic consequences of manipulating m6A “writers” and “erasers.” The identification of individual mRNA m6A sites is still challenging owing to the limitations of current experimental and sequencing technologies. m6A modification site mismatches have been achieved at single-base accuracy by modifying DNA polymerases to detect m6A transcriptome modification levels110. Techniques for the direct detection of RNA modifications on transcripts using nanopore sequencing111 have become popular, and these technologies are essential for elucidating the dynamic regulatory mechanisms of m6A modification and their molecular functions. The RNA modification mechanisms regulating gene expression during I/R injury will certainly become clearer with additional research and the development of more advanced technologies.
References
Kalogeris, T., Baines, C. P., Krenz, M. & Korthuis, R. J. Ischemia/reperfusion. Compr. Physiol. 7, 113–170 (2016).
Abela, C. B. & Homer-Vanniasinkham, S. Clinical implications of ischaemia-reperfusion injury. Pathophysiology 9, 229–240 (2003).
Bronicki, R. A. & Hall, M. Cardiopulmonary bypass-induced inflammatory response: pathophysiology and treatment. Pediatr. Crit. Care Med. 17, S272–S278 (2016).
Kosieradzki, M. & Rowinski, W. Ischemia/reperfusion injury in kidney transplantation: mechanisms and prevention. Transplant. Proc. 40, 3279–3288 (2008).
Chouchani, E. T. et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 (2014).
Braunersreuther, V. & Jaquet, V. Reactive oxygen species in myocardial reperfusion injury: from physiopathology to therapeutic approaches. Curr. Pharm. Biotechnol. 13, 97–114 (2012).
Kuznetsov, A. V. et al. The role of mitochondria in the mechanisms of cardiac ischemia-reperfusion injury. Antioxidants (Basel) 8, 454 (2019).
Tang, J. & Zhuang, S. Histone acetylation and DNA methylation in ischemia/reperfusion injury. Clin. Sci. (Lond.) 133, 597–609 (2019).
Tang, J. & Zhuang, S. Epigenetics in acute kidney injury. Curr. Opin. Nephrol. Hypertens. 24, 351–358 (2015).
Tessarz, P. & Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 15, 703–708 (2014).
Ficz, G. New insights into mechanisms that regulate DNA methylation patterning. J. Exp. Biol. 218, 14–20 (2015).
Jonkhout, N. et al. The RNA modification landscape in human disease. RNA 23, 1754–1769 (2017).
Batista, P. J. The RNA Modification N(6)-methyladenosine and Its Implications in Human Disease. Genomics Proteom. Bioinf. 15, 154–163 (2017).
Liu, R. J., Long, T., Li, J., Li, H. & Wang, E. D. Structural basis for substrate binding and catalytic mechanism of a human RNA:m5C methyltransferase NSun6. Nucleic Acids Res. 45, 6684–6697 (2017).
Glasner, H., Riml, C., Micura, R. & Breuker, K. Label-free, direct localization and relative quantitation of the RNA nucleobase methylations m6A, m5C, m3U, and m5U by top-down mass spectrometry. Nucleic Acids Res. 45, 8014–8025 (2017).
Malbec, L. et al. Dynamic methylome of internal mRNA N(7)-methylguanosine and its regulatory role in translation. Cell Res. 29, 927–941 (2019).
Tuck, M. T. The formation of internal 6-methyladenine residues in eucaryotic messenger RNA. Int J. Biochem. 24, 379–386 (1992).
Desrosiers, R., Friderici, K. & Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 71, 3971–3975 (1974).
Yadav, P. K. & Rajasekharan, R. The m(6)A methyltransferase Ime4 and mitochondrial functions in yeast. Curr. Genet. 64, 353–357 (2018).
Yue, H., Nie, X., Yan, Z. & Weining, S. N6-methyladenosine regulatory machinery in plants: composition, function and evolution. Plant Biotechnol. J. 17, 1194–1208 (2019).
Lence, T. et al. m(6)A modulates neuronal functions and sex determination in Drosophila. Nature 540, 242–247 (2016).
Coker, H., Wei, G. & Brockdorff, N. m6A modification of non-coding RNA and the control of mammalian gene expression. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 310–318 (2019).
Erson-Bensan, A. E. & Begik, O. m6A Modification and Implications for microRNAs. MicroRNA 6, 97–101 (2017).
Ma, S. et al. The interplay between m6A RNA methylation and noncoding RNA in cancer. J. Hematol. Oncol. 12, 121 (2019).
Fu, Y., Dominissini, D., Rechavi, G. & He, C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat. Rev. Genet 15, 293–306 (2014).
Scholler, E. et al. Interactions, localization, and phosphorylation of the m(6)A generating METTL3-METTL14-WTAP complex. RNA 24, 499–512 (2018).
Liu, J. et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95 (2014).
Schwartz, S. et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 8, 284–296 (2014).
Zhao, X. et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res 24, 1403–1419 (2014).
Fu, Y. et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat. Commun. 4, 1798 (2013).
Dina, C. et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet. 39, 724–726 (2007).
Boissel, S. et al. Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am. J. Hum. Genet. 85, 106–111 (2009).
Jia, G. et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 (2011).
Ensfelder, T. T. et al. ALKBH5-induced demethylation of mono- and dimethylated adenosine. Chem. Commun. (Camb.) 54, 8591–8593 (2018).
Zheng, G. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013).
Zhang, S. et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 31, 591–606 e596 (2017).
Zaccara, S., Ries, R. J. & Jaffrey, S. R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 20, 608–624 (2019).
Berlivet, S., Scutenaire, J., Deragon, J. M. & Bousquet-Antonelli, C. Readers of the m(6)A epitranscriptomic code. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 329–342 (2019).
Liao, S., Sun, H. & Xu, C. YTH domain: a family of N(6)-methyladenosine (m(6)A) readers. Genomics Proteom. Bioinf. 16, 99–107 (2018).
Cienikova, Z., Damberger, F. F., Hall, J., Allain, F. H. & Maris, C. Structural and mechanistic insights into poly(uridine) tract recognition by the hnRNP C RNA recognition motif. J. Am. Chem. Soc. 136, 14536–14544 (2014).
Liu, N. et al. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564 (2015).
Vilalva, K. H. et al. Use of methylene blue to treat hypovolemic shock followed by ischemia-reperfusion injury in the postoperative orthotopic liver transplant patient: a case report. Exp. Clin. Transplant. 16, 511–514 (2018).
Chies, A. B. et al. Rivastigmine prevents injury induced by ischemia and reperfusion in rat liver. Acta Cir. Bras. 33, 775–784 (2018).
Nakazato, P. C. G. et al. Liver ischemia and reperfusion injury. Pathophysiology and new horizons in preconditioning and therapy. Acta Cir. Bras. 33, 723–735 (2018).
Yao, W. et al. Intravenous anesthetic protects hepatocyte from reactive oxygen species-induced cellular apoptosis during liver transplantation in vivo. Oxid. Med. Cell Longev. 2018, 4780615 (2018).
Wu, H. et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Invest. 117, 2847–2859 (2007).
Koutsogiannidis, C. P. & Johnson, E. O. Pharmacological neuroprotection in cardiac surgery: effectiveness of pharmacologic-preconditioning with erythromycin. Curr. Vasc. Pharm. 16, 329–335 (2018).
de Perrot, M., Liu, M., Waddell, T. K. & Keshavjee, S. Ischemia-reperfusion-induced lung injury. Am. J. Respir. Crit. Care Med. 167, 490–511 (2003).
Eppinger, M. J., Deeb, G. M., Bolling, S. F. & Ward, P. A. Mediators of ischemia-reperfusion injury of rat lung. Am. J. Pathol. 150, 1773–1784 (1997).
Yao, W. et al. Neutrophil elastase inhibitors suppress oxidative stress in lung during liver transplantation. Oxid. Med. Cell Longev. 2019, 7323986 (2019).
Weiser, M. R. et al. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J. Exp. Med. 183, 2343–2348 (1996).
Menger, M. D., Pelikan, S., Steiner, D. & Messmer, K. Microvascular ischemia-reperfusion injury in striated muscle: significance of “reflow paradox”. Am. J. Physiol. 263, H1901–H1906 (1992).
Junk, A. K. et al. Erythropoietin administration protects retinal neurons from acute ischemia-reperfusion injury. Proc. Natl Acad. Sci. USA 99, 10659–10664 (2002).
Oharazawa, H. et al. Protection of the retina by rapid diffusion of hydrogen: administration of hydrogen-loaded eye drops in retinal ischemia-reperfusion injury. Invest. Ophthalmol. Vis. Sci. 51, 487–492 (2010).
Oliver, C. N. et al. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc. Natl Acad. Sci. USA 87, 5144–5147 (1990).
Santos, M. R. et al. The protective effect of cilostazol on isolated rabbit femoral arteries under conditions of ischemia and reperfusion: the role of the nitric oxide pathway. Clinics 67, 171–178 (2012).
Ciscato, J. G. Jr. et al. Vascular relaxation of canine visceral arteries after ischemia by means of supraceliac aortic cross-clamping followed by reperfusion. Scand. J. Trauma Resusc. Emerg. Med. 18, 41 (2010).
Bertoni, S., Ballabeni, V., Barocelli, E. & Tognolini, M. Mesenteric ischemia-reperfusion: an overview of preclinical drug strategies. Drug Disco. Today 23, 1416–1425 (2018).
Zhao, W. et al. Resveratrol suppresses gut-derived NLRP3 inflammasome partly through stabilizing mast cells in a rat model. Mediators Inflamm. 2018, 6158671 (2018).
Eltzschig, H. K. & Eckle, T. Ischemia and reperfusion–from mechanism to translation. Nat. Med. 17, 1391–1401 (2011).
Yao, W., Tai, L. W., Liu, Y., Hei, Z. & Li, H. Oxidative stress and inflammation interaction in ischemia reperfusion injury: role of programmed cell death. Oxid. Med. Cell Longev. 2019, 6780816 (2019).
Hausenloy, D. J. & Yellon, D. M. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the reperfusion injury salvage kinase (RISK)-pathway. Cardiovasc. Res. 61, 448–460 (2004).
Rossello, X. & Yellon, D. M. The RISK pathway and beyond. Basic Res. Cardiol. 113, 2 (2018).
Ghaboura, N. et al. Diabetes mellitus abrogates erythropoietin-induced cardioprotection against ischemic-reperfusion injury by alteration of the RISK/GSK-3beta signaling. Basic Res. Cardiol. 106, 147–162 (2011).
Hausenloy, D. J. & Yellon, D. M. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev. 12, 217–234 (2007).
Hadebe, N., Cour, M. & Lecour, S. The SAFE pathway for cardioprotection: is this a promising target? Basic Res. Cardiol. 113, 9 (2018).
Lecour, S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: does it go beyond the RISK pathway? J. Mol. Cell Cardiol. 47, 32–40 (2009).
Lecour, S. & James, R. W. When are pro-inflammatory cytokines SAFE in heart failure? Eur. Heart J. 32, 680–685 (2011).
Inserte, J. & Garcia-Dorado, D. The cGMP/PKG pathway as a common mediator of cardioprotection: translatability and mechanism. Br. J. Pharm. 172, 1996–2009 (2015).
Cohen, M. V., Yang, X. M., Liu, Y., Solenkova, N. V. & Downey, J. M. Cardioprotective PKG-independent NO signaling at reperfusion. Am. J. Physiol. Heart Circ. Physiol. 299, H2028–H2036 (2010).
Hsu, H. H. et al. Protection against reperfusion lung injury via aborgating multiple signaling cascades by trichostatin A. Int. Immunopharmacol. 25, 267–275 (2015).
Evankovich, J. et al. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J. Biol. Chem. 285, 39888–39897 (2010).
Kim, K. et al. Effect of valproic acid on acute lung injury in a rodent model of intestinal ischemia reperfusion. Resuscitation 83, 243–248 (2012).
Guo, Y. et al. Trichostatin A attenuates oxidative stress-mediated myocardial injury through the FoxO3a signaling pathway. Int. J. Mol. Med 40, 999–1008 (2017).
Huang, N., Tan, L., Xue, Z., Cang, J. & Wang, H. Reduction of DNA hydroxymethylation in the mouse kidney insulted by ischemia reperfusion. Biochem. Biophys. Res. Commun. 422, 697–702 (2012).
Tampe, B. et al. Low-dose hydralazine prevents fibrosis in a murine model of acute kidney injury-to-chronic kidney disease progression. Kidney Int. 91, 157–176 (2017).
Li, J. et al. Genome-wide analysis of DNA methylation and acute coronary syndrome. Circ. Res. 120, 1754–1767 (2017).
Li, D. et al. Genome-wide DNA methylome alterations in acute coronary syndrome. Int J. Mol. Med. 41, 220–232 (2018).
Ke, J. et al. Role of DNA methylation in perinatal nicotine-induced development of heart ischemia-sensitive phenotype in rat offspring. Oncotarget 8, 76865–76880 (2017).
Endres, M. et al. DNA methyltransferase contributes to delayed ischemic brain injury. J. Neurosci. 20, 3175–3181 (2000).
Li, Q. et al. NSUN2-mediated m5C methylation and METTL3/METTL14-mediated m6A methylation cooperatively enhance p21 translation. J. Cell Biochem. 118, 2587–2598 (2017).
Anders, M. et al. Dynamic m(6)A methylation facilitates mRNA triaging to stress granules. Life Sci. Alliance 1, e201800113 (2018).
Zhong, X. et al. Circadian clock regulation of hepatic lpid metabolism by modulation of m(6)A mRNA methylation. Cell Rep. 25, 1816–1828 e1814 (2018).
Fry, N. J., Law, B. A., Ilkayeva, O. R., Holley, C. L. & Mansfield, K. D. N(6)-methyladenosine is required for the hypoxic stabilization of specific mRNAs. RNA 23, 1444–1455 (2017).
Wu, L. et al. Association of N(6)-methyladenine DNA with plaque progression in atherosclerosis via myocardial infarction-associated transcripts. Cell Death Dis. 10, 909 (2019).
Dorn, L. E. et al. The N(6)-methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation 139, 533–545 (2019).
Song, H. et al. METTL3 and ALKBH5 oppositely regulate m(6)A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy 15, 1419–1437 (2019).
Shen, F. et al. Decreased N(6)-methyladenosine in peripheral blood RNA from diabetic patients is associated with FTO expression rather than ALKBH5. J. Clin. Endocrinol. Metab. 100, E148–E154 (2015).
Mathiyalagan, P. et al. FTO-dependent N(6)-methyladenosine regulates cardiac function during remodeling and repair. Circulation 139, 518–532 (2019).
Wang, X. et al. FTO is required for myogenesis by positively regulating mTOR-PGC-1alpha pathway-mediated mitochondria biogenesis. Cell Death Dis. 8, e2702 (2017).
Xuan, Z. et al. Dexmedetomidine postconditioning alleviates hypoxia/reoxygenation injury in senescent myocardial cells by regulating lncRNA H19 and m6A modification. Oxid. Med. Cell Longev. 2020, 9250512 (2020).
Saxena, R., Weintraub, N. L. & Tang, Y. Optimizing cardiac ischemic preconditioning and postconditioning via epitranscriptional regulation. Med. Hypotheses 135, 109451 (2020).
Leng, T., Shi, Y., Xiong, Z. G. & Sun, D. Proton-sensitive cation channels and ion exchangers in ischemic brain injury: new therapeutic targets for stroke? Prog. Neurobiol. 115, 189–209 (2014).
Galkin, A. Brain ischemia/reperfusion injury and mitochondrial complex I damage. Biochemistry 84, 1411–1423 (2019).
Xin, Q. et al. Endoplasmic reticulum stress in cerebral ischemia. Neurochem. Int. 68, 18–27 (2014).
Liu, J. et al. Neuronal apoptosis in cerebral ischemia/reperfusion area following electrical stimulation of fastigial nucleus. Neural Regen. Res 9, 727–734 (2014).
Yang, Q., Huang, Q., Hu, Z. & Tang, X. Potential neuroprotective treatment of stroke: targeting excitotoxicity, oxidative stress, and inflammation. Front. Neurosci. 13, 1036 (2019).
DeGracia, D. J. Disease of mRNA regulation: relevance for ischemic brain injury. Transl. Stroke Res. 9, 251–257 (2018).
Chen, Y. & Zhou, J. LncRNAs: macromolecules with big roles in neurobiology and neurological diseases. Metab. Brain Dis. 32, 281–291 (2017).
Yang, J., Chen, M., Cao, R. Y., Li, Q. & Zhu, F. The role of circular RNAs in cerebral ischemic diseases: ischemic stroke and cerebral ischemia/reperfusion injury. Adv. Exp. Med. Biol. 1087, 309–325 (2018).
Hess, M. E. et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat. Neurosci. 16, 1042–1048 (2013).
Widagdo, J. & Anggono, V. The m6A-epitranscriptomic signature in neurobiology: from neurodevelopment to brain plasticity. J. Neurochem. 147, 137–152 (2018).
Chokkalla, A. K. et al. Transient focal ischemia significantly alters the m(6)A epitranscriptomic tagging of RNAs in the brain. Stroke 50, 2912–2921 (2019).
Diao, M. Y. et al. Hypothermia protects neurons against ischemia/reperfusion-induced pyroptosis via m6A-mediated activation of PTEN and the PI3K/Akt/GSK-3beta signaling pathway. Brain Res. Bull. 159, 25–31 (2020).
Malek, M. & Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Ren. Inj. Prev. 4, 20–27 (2015).
Chen, C. et al. Crosstalk between connexin32 and mitochondrial apoptotic signaling pathway plays a pivotal role in renal ischemia reperfusion-induced acute kidney injury. Antioxid. Redox Signal. 30, 1521–1538 (2018).
Zhuang, C. et al. N6-methyladenosine demethylase FTO suppresses clear cell renal cell carcinoma through a novel FTO-PGC-1alpha signalling axis. J. Cell Mol. Med. 23, 2163–2173 (2019).
Fiorentino, M., Grandaliano, G., Gesualdo, L. & Castellano, G. Acute kidney injury to chronic kidney disease transition. Contrib. Nephrol. 193, 45–54 (2018).
Xu, Y. et al. The N6-methyladenosine mRNA methylase METTL14 promotes renal ischemic reperfusion injury via suppressing YAP1. J. Cell Biochem. 121, 524–533 (2020).
Aschenbrenner, J. et al. Engineering of a DNA polymerase for direct m(6) A sequencing. Angew. Chem. Int. Ed. Engl. 57, 417–421 (2018).
Garalde, D. R. et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 15, 201–206 (2018).
Francis, A. & Baynosa, R. Ischaemia-reperfusion injury and hyperbaric oxygen pathways: a review of cellular mechanisms. Diving Hyperb. Med. 47, 110–117 (2017).
Pak, S. et al. Platelet adhesion in the sinusoid caused hepatic injury by neutrophils after hepatic ischemia reperfusion. Platelets 21, 282–288 (2010).
Kalogeris, T., Baines, C. P., Krenz, M. & Korthuis, R. J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 298, 229–317 (2012).
Acknowledgements
The study was supported in part by grants from Natural Science Foundation of China (no. 81974081 for Weifeng Yao).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by S. Lavandero
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yao, W., Han, X., Ge, M. et al. N6-methyladenosine (m6A) methylation in ischemia–reperfusion injury. Cell Death Dis 11, 478 (2020). https://doi.org/10.1038/s41419-020-2686-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-2686-7
This article is cited by
-
Demethylase FTO-Mediated m6A Modification of lncRNA MEG3 Activates Neuronal Pyroptosis via NLRP3 Signaling in Cerebral Ischemic Stroke
Molecular Neurobiology (2024)
-
Key m6A regulators mediated methylation modification pattern and immune infiltration characterization in hepatic ischemia-reperfusion injury
BMC Medical Genomics (2023)
-
Methyltransferase-like 3 aggravates endoplasmic reticulum stress in preeclampsia by targeting TMBIM6 in YTHDF2-dependent manner
Molecular Medicine (2023)
-
Identification of m6A methylation-related genes in cerebral ischaemia‒reperfusion of Breviscapus therapy based on bioinformatics methods
BMC Medical Genomics (2023)
-
MiR-144-5p/CCL12 Signaling Axis Modulates Ischemic Preconditioning-Mediated Cardio-protection by Reducing Cell Viability, Enhancing Cell Apoptosis, Fibrosis, and Pyroptosis
Applied Biochemistry and Biotechnology (2023)
