
Abstract
Chemical substances shall not migrate from food contact materials (FCM) at levels that are potentially harmful for the consumers. Each of the current analytical methods applied to verify the migration of substances from FCM covers only one or few substances. There is a very limited number of publications on the development of analytical methods allowing the simultaneous determination of several classes of FCM substances, and almost none of them reported methods entirely dedicated to the ones in the positive list of Commission Regulation (EU) No. 10/2011 for plastic FCMs. Therefore, a simple, sensitive and reliable multi-analyte method was developed for the analysis of FCM substances in food simulants. It employs an optimised liquid-liquid extraction with dichloromethane as extraction solvent in the presence of 10% m/v NaCl, followed by quantitative analysis with gas chromatography coupled to mass spectrometry (GC-MS). A combination of total ion chromatograms (TICs) and extracted ion chromatograms (EICs) was used. The optimisation and validation of the method have been carried out according to current international guidelines. Adequate sensitivity was demonstrated in the selected concentration ranges for most of the analytes, with limits of quantification (LOQs) at least three times lower than the legislative limit, when existing. The results showed that the method is sufficiently accurate for the majority of substances, with recoveries between 70 and 115% and relative standard deviations (RSDs) smaller than 20% at three concentration levels. The method was applied to the analysis of some FCM multilayers. The method allows, for the first time, the simultaneous quantification of 84 FCM substances in two of the official food simulants (A and C) at levels of a few ng g−1.

Graphical abstract
Similar content being viewed by others

Introduction
Human exposure to chemicals from food contact materials (FCM) occurs mainly as a result of migration from materials into foodstuffs. The extent of this migration is one of the key factors for the human health risk which a packaging material could represent [1].
The European Union (EU) has set up with Regulation (EU) No. 10/2011 a positive list of substances that are allowed to be used in plastic FCM [2]. These substances may have specific migration limits (SMLs) or other restrictions for their application. Another class of frequently found but not regulated substances in FCM is the so-called non-intentionally added substances (NIAS). These are by-products from production processes or they originate from the degradation of materials and could migrate into food.
Official food simulants that mimic the use and properties of real food are also described in the Regulation, and must be used by laboratories when performing migration experiments. The European Union Reference Laboratory for Food Contact Materials (EURL-FCM) maintains and updates a repository of approved FCM additives. From almost 900 regulated substances, less than 600 are commercially available from trustworthy suppliers [2]. Official Control Laboratories (OCLs) all over the EU perform the monitoring of substances used in plastic FCM. Few CEN methods are in place, and therefore, OCLs apply in-house validated methods which focus mainly on one or a few substances.
The availability of validated methods for the simultaneous analysis of large groups of regulated substances listed in the Regulation (EU) No. 10/2011 would improve considerably the efficiency of compliance testing in the plastic FCM field. However, the challenges for the development of such methods are considerable. The Regulation includes not only individual organic substances but also many mixtures, natural products, resins, monomers, oxides, silicates and more. Therefore, the development of multi-analyte methods has to take into account a multitude of different chemical structures and physical-chemical properties. Additionally, such methods cannot be only focused on their instrumental separation and quantification steps, but have to include also a robust sample preparation step that can be applied to the official food simulants. Those may vary from ethanolic solutions to vegetable oils, acetic acid solutions and poly(2,6-diphenyl-p-phenylene oxide), a simulant for dry foods. The methods need also to be very versatile in their sensitivity, as existing SMLs can range from 10 μg kg−1 to 30 mg kg−1 levels. In some cases, the legislation refers to a maximum mass fraction of substance(s) in the FCM that has to be controlled, typically via extraction followed by measurement techniques. In such cases, sample preparation techniques are even more important due to the complexity of the matrices to be investigated.
Only a limited number of papers related with the analysis of multiple analytes in the FCM field are available in the literature, most of them on materials not regulated at EU level (e.g. paper and board). Representative examples are presented in Table 1.
Most of the target analytes covered by the methods of Table 1 are not regulated in the EU and, therefore, have no legal limits (LLs) in place. The majority of the reported analytical methods aim to detect the presence of analytes in extracts/migration solutions. It also appears that the majority of the target substances were selected either based on a shared chemical nature or on their function as substance in the FCM items. Complex sample preparation steps are often employed, depending on the nature of the article and target substances.
The scope of this study was to develop a method dedicated to the simultaneous quantification of a large group of substances that can be present in plastic FCM. This method should be simple and accurate, as well as being applicable to the analysis of certain official (liquid) food simulants from EU regulation. The need of limited sample preparation was also one of the desired characteristics for the method, together with the possibility of quantification at the legislated migration limits. Its applicability to some real plastic FCM films has been tested.
Considerations for the development of a dedicated multi-analyte method
The positive list of the Regulation (EU) No. 10/2011 contains more than 900 additives, belonging to different chemical classes/nature and different physical-chemical properties. These substances can range from inorganic to organic, from polar to apolar, from volatile to non-volatile substances, from low molecular masses to masses higher than 1000 Da [2].
In order to select the group of target analytes for the development of the multi-analyte method, a strategy had to be developed. It has been taken into account for the present study the chemical structure of substances potentially migrating from plastic FCMs, the availability of well characterised analytical standards and a preference for the instrumental approaches intended to be used. A schematic illustration of the process applied to select the final group of analytes to be addressed by the method is presented in Fig. 1.

Selection path for the final group of analytes to be targeted by the multi-analyte method
The first step consisted of removing from the initial number of regulated substances the ones that are not available in the EURL repository for plastic FCM additives. A second step eliminated the substances considered as “inorganic” (oxides, hydroxides, silicates, etc.). Gas chromatography (GC) has been selected due to its versatility and availability in most control laboratories as the analytical separation technique to be used for this method. Therefore, all the non-volatile substances were removed (sugars, waxes, resins, monomers and co-monomers).
The selection of the analytical column is a crucial step for the development of any GC-based method. Here, a 5% phenyl methyl siloxane column has been chosen. This type of stationary phase was not only adequate for the analysis of substances with the chemical nature of most of the selected substances, but allows also a good efficiency and performance at the high oven temperatures expected for the chromatographic programme [15]. Additionally, it was important to select a column offering some resistance towards water as the method should be applied to extracts from aqueous/ethanolic simulants, which may contain traces of water. Therefore, the possibility of an ultra-inert (UI) feature was considered during the column selection. Taking the suitability for using a common 5% phenyl methyl siloxane GC column into account, acids, alcohols and amines were excluded as analytes. Finally, substances which showed a poor solubility during the initial testing in the selected solvents have been excluded. The resulting group covered aldehydes, ketones, esters and some aromatic substances, in total 81 regulated substances as presented in Table 2 (“Chemicals” section). In addition, some NIAS of interest were included as analytical targets, elevating the final number of substances to 84.
The analytical method was developed for use with official food simulants A and C (10% and 20% v/v aqueous ethanol, respectively). These food simulants tend to be relatively simple matrices, requiring limited sample preparation (extraction/clean-up) steps. However, a “change of solvent” step has to be included for GC methods, as water-containing samples should not be analysed directly. This solvent change is commonly achieved through a simple liquid-liquid extraction (LLE) with a proper organic solvent. The challenge here was to identify an organic solvent that acts as an efficient extraction solvent and can also solubilise the broad range of target substances. The solvents have also to be compatible with GC-MS, i.e. they should not add any analytical interference or shorten the lifetime of the analytical column.
Materials and methods
Chemicals
Ethanol (EtOH; CAS: 64-17-5), n-hexane (Hex; CAS: 110-54-3), isooctane (Iso; CAS: 540-84-1), tert-butyl methyl ether (MTBE; CAS: 1634-04-4) and dichloromethane (DCM; 75-09-2) were Chromasolv grade purity and obtained from Sigma-Aldrich (Steinheim, Germany). Ultrapure water (18.2 MΩ), for the preparation of the official food simulants (A and C, 10% and 20% v/v aqueous ethanol, respectively) and solutions, was obtained from a Milli-Q system (Millipore, Bedford, USA). Sodium chloride (NaCl; ≥ 99.5%) was supplied from Fluka (Steinheim, Germany). PTFE 17 mm, 0.2 μm membrane filters were supplied from CPS Analitica (Milan, Italy).
All the analytical standards were obtained either from Sigma-Aldrich (Steinheim, Germany) or from TCI Chemicals (Tokyo, Japan). All the relevant information regarding the target substances are presented in Table 2, including FCM numbers, CAS numbers, molecular masses, purity (as stated by the supplier) and MS data regarding the selected abundant ions (m/z) for extraction ion chromatograms (EIC) [2]. In addition, three NIAS were included, i.e. di-n-butyl maleate, diisobutyl phthalate (DiBP) and diethyl phosphite. Tributyl aconitate, a by-product of acetyl tributyl citrate, was identified but it has not been quantified (please see “Experimental issues in the quantification of some FCM substances” section). The DiBP was included because of EFSA’s reassessment of phthalates in FCM [16], while diethyl phosphite originates from the degradation of FCM No. 293. The method can also qualitatively assess the degradation of FCM No. 138 (tri-n-butyl acetyl citrate) into tributyl aconitate.
Preparation of standard solutions
Stock solutions containing 10 mg mL−1 of each analyte and internal standards (IS) were prepared using ethanol as solvent and were stored at − 18 °C. Appropriate working solution mixtures were prepared by diluting the stock standard solutions with ethanol and sonicated (59 kHz) at 25 °C for 15 min. Stock solutions were stored at − 18 °C, while the working solutions were stored at 4 °C. Fresh working standards were produced every week. Both stock and working standard solutions were prepared in amber vials in order to prevent any light-induced degradation or isomeric conversion of the substances. As contamination with phthalates is very common during sample preparation procedures [3, 4, 17, 18], only glassware properly cleaned and rinsed was used. Briefly, all glassware was rinsed twice with acetone and hexane and stored in a desiccator over aluminium oxide [17, 18].
Analysis of real FCM samples
Fifteen plastic FCM samples coming from a range of EU plastic producers have been tested as to check the applicability of the method to real samples. Samples were stored in wrapped aluminium foils at room temperature (20 ± 5 °C). The test conditions of the migration experiments were based on the intended use of the material according to Regulation (EU) No. 10/2011 [2]. All the samples were cut into square pieces (approximately 1 dm2; 10 × 10 cm) prior to the test. Data regarding the type of material, the intended use, the type of food simulant, the type of migration experiment, the amount of food simulant and the contact time and temperature conditions are presented in Table 3.
Liquid-liquid extraction
The used LLE method was based on the sample preparation procedure known as QuEChERS (quick, easy, cheap, effective, rugged and safe) [19] and on previous work carried at the EURL-FCM, with slight modifications [20]. In the present study, a specimen of 5 mL of the food simulant (A or C), containing 400 mg of NaCl, the 2 selected IS and 2 mL of DCM were added to a tube. The tube was vigorously vortexed for 1 min and centrifuged using an Eppendorf 5810 R refrigerated centrifuge, set at 20 °C and 2500 rpm (1280g) for 5 min. The procedure was repeated for a second time by adding 1 mL of DCM. The DCM extracts were removed (bottom solvent layers), filtered with PTFE 0.22 μm filters and transferred to another glass tube, where they have been concentrated to 150 μL by a gentle stream of nitrogen at 25 °C, and the volume fixed to 300 μL by adding DCM.
GC-MS analysis
The method was designed to rely mainly on the separation power of the chromatographic step rather than taking advantage of using an MS detector. That means chromatographic resolution was a crucial factor to be considered. Therefore, a 60-m-column has been selected (HP-5MS UI 5%, 60 m × 250 μm, 0.25 μm, Agilent Technologies, USA) to allow for a proper separation of the total number of substances. Such a long analytical column results normally in a longer analysis time and potentially interfering peaks of the last eluting substances. Although the former effect was observed for the method studied here, the latter was not. The use of the selected column allowed the simultaneous analysis of such a large number of compounds in a single run with good resolution for the majority of substances.
Chromatographic analyses were performed in a GC equipped with a single quadrupole mass detector. The chromatographic column was supplied by Agilent Technologies Inc. (USA). All the GC-MS parameters are presented in Table 4.
Method performance
The proposed method was evaluated in terms of linearity, precision and trueness, limits of detection (LODs) and quantification (LOQs) according to method performance validation guidelines [21, 22]. The linearity was assessed by analysing standard solution mixtures at six concentration levels for each of the target analytes. The calibration curve was constructed with the ratio of the analyte peak area to the IS peak area. Two IS were used, namely 3-(4-isopropylphenyl)-2-methylpropionaldehyde and diphenyl phthalate. The former was employed for the quantification of the substances eluting up to its retention time (tr = 32.17 min), hence the most volatile ones. Diphenyl phthalate was used as IS for all the remaining substances (tr = 47.93 min). The linearity was evaluated by calculating the linear regression coefficient (R2). LODs and LOQs were evaluated from the chromatographic signal-to-noise ratio S/N. Mean value and standard deviation of the S/N were obtained from 5 chromatograms of blanks and the lowest calibration level for each substance, respectively. The LOD was estimated as analyte concentration providing an S/N of 3, while the LOQ was calculated as 3 times the LOD [21, 22].
Trueness and precision were assessed in food simulants A and C [2]. The respective food simulant was fortified at three concentration levels for all the selected analytes, based on their linear range, along with the IS. For short-term repeatability, six replicates of the fortified samples were analysed during the day, while for intermediate precision, six replicates of the aforementioned samples were analysed on three consecutive days. The trueness assessment of the analytical method was based on the calculation of the relative recovery as amount found in the fortified sample divided by the known amount added and expressed as percentage. The three tested concentrations for the short-term repeatability and intermediate precision have been selected either based on the SML [2] or on their LOQ [21, 22]. All results concerning trueness and precision are presented as Electronic Supplementary Material (ESM, Tables S1 to S4).
Results and discussion
Optimisation of the extraction from food simulant solutions
The main challenge was the selection of an organic solvent that could extract simultaneously and with good efficiency all the selected target FCM substances from the tested food simulants A and C.
Different organic solvents, namely hexane, isooctane, MTBE and DCM, were tested regarding the extraction efficiency for substances in simulant A, which is considered to be the “worst case” [2]. This efficiency was evaluated by comparing the amounts of each target analyte extracted from fortified food simulants (containing 250 ng mL−1) with the results obtained using their analytical standard solutions at the same concentration level. The results are presented in Table 5.
It can be concluded that DCM with the presence of 10% w/v NaCl is the most suitable extraction solvent as it provided the best overall efficiency for a larger number of substances. Some of them could only be extracted with DCM, like ethylene glycol monomethacrylate (FCM No. 374), caprolactone (FCM No. 342), caprolactam (FCM No. 212) or dapsone (FCM No. 153). An extraction with isooctane presented subpar efficiency for a considerable number of substances. The addition of 10% m/v NaCl to DCM increased the extraction efficiency for many of the substances [20]. This effect was of particular importance for substances like caprolactam (FCM No. 212) and some parabens such as methyl paraben (FCM No. 189) and ethyl paraben (FCM No. 287). A notable example is hexadecyltrimethylammonium bromide (FCM No. 104), which showed low recoveries with MTBE and isooctane (< 48 %), an acceptable recovery with hexane (75 %) and up to 95% with DCM plus 10% NaCl. The effect of the salt addition during the LLE seems to be more significant with polar substances (such as phenol, FCM No. 241). However, a slight decrease in the DCM extraction efficiency was observed for substances with a lower polarity, where aprotic solvents are more efficient. Also, other substances, such as vinyltrimethoxysilane (FCM No. 453) or triethylphosphite (FCM No. 293), showed higher recoveries when using hexane and isooctane instead of DCM. In the end, a compromise had to be found and therefore DCM with the addition of 10% m/v NaCl has been selected because of the best overall results.
This extraction study was not performed under optimised precision and accuracy conditions because the objective at this stage was to screen and compare extraction efficiencies and not to validate the method. For instance, a complete baseline resolution was not obtained for certain substances and solvents, which has influenced the peak area calculation. Therefore, some of the reported extraction efficiencies are well above 100% in Table 5.
Method validation
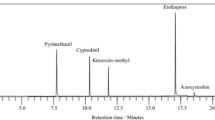
In Fig. 2 are presented examples of the total ion chromatograms (TICs) of solutions resulting from the extraction of fortified food simulants with DCM + 10% NaCl.

GC-MS total ion chromatograms of a fortified and extracted food simulant A at 2nd concentration level. b Fortified and extracted food simulant C at 2nd concentration level; IS 1: 3-(4-isopropylphenyl)-2-methylpropionaldehyde (0.5 μg mL−1); IS 2: diphenyl phthalate (0.5 μg mL−1)
The method was validated in-house and its LODs, LOQs and upper linear limits in food simulants A and C are presented in Table 6.
For all the studied substances, the linear regression coefficients (R2) were higher than 0.99, indicating good linearity of the calibration curves. Regarding sensitivity, the followed guidelines [21, 22] and EU legislation requests that the LOQs should be at least 3 times lower than the SML of the substance. This was achieved for most of the substances in both food simulants A and C (Table 6). This requirement was even fulfilled for many of the substances with low SMLs (50.0 μg kg−1), like FCMs No. 175, 385, 187, 342, 197, 206, 186, 185, 788, 420, 434, 337, 437, 463, 487, 152, 438 and 492. The only two substances that could not be quantified at this SML were FCM No. 453 and 142, two silane-type substances. Also, FCM No. 220 (glycidyl methacrylate), with an SML of 20.0 ng g−1 and a method’s LOQ of 12.5 ng g−1 is challenging to be reliably quantified at its SML. For substance FCM No. 199, the required non-detection limit of 10 ng g−1 could not be reached. This was also the case for the individual substances forming FCM No. 405 for which the sum of divinylbenzenes and ethylvinylbenzenes should be non-detectable at a level of 10 ng g−1. For the substances without an SML, the existence of a proper analytical method as sensitive as possible is compulsory for their quantification in official food simulants A and C. The current method achieved low LOQs for all these substances. Overall, the LOQs for almost all substances are sufficient for their quantification at trace levels in official food simulants A and C [2, 21, 22].
The trueness and precision characteristics of the method were established with fortified food simulants A and C. All the results for the measurements in food simulant A are given in Tables S1 and S2 (see ESM), and the ones in food simulant C in Tables S3 and S4 (see ESM), respectively.
The results demonstrate the good precision of the method, with RSDs for the repeatability and intermediate precision below 15% for the determination of the majority of the substances. Some exceptions were observed for analytes belonging to the acrylates, namely FCM No. 206, 218, 355, 371 and 463. For these substances, RSDs were as high as 19.4% at some of the studied concentration levels. Recoveries were for the majority of substances between 70 and 115%.
Analysis of real FCM samples
In order to evaluate the applicability of the method, 15 commercial FCM polymeric films were investigated. Migration tests were performed using different types of films, migration test conditions and food simulants according to their intended use (see Table 3 for migration test conditions). Results for the identified and quantified FCM substances are presented in Table 7.
Eight out of the 14 commercial films, i.e. S5, S20, S25, S29, S31, S47, S49 and S62, did not release any of the 84 substances under the defined test conditions. The remaining six films released substances either below LOQ or up to 2.7 mg kg−1 food. The latter parameter has been calculated by taking into account the surface area which was in contact with the food simulant (A or C) in the migration test and a standard surface-to-food mass ratio of 6 dm2 kg−1 food that is prescribed for FCM films according to the Reg. (EU) No. 10/2011 [2]. The analysis of real FCM samples with food simulants A or C showed that the migrated amounts of the regulated substances of all tested materials were compliant with the requirements in the corresponding Regulation [2].
In addition to regulated substances, several NIAS were identified. Whether their migration is compliant with the Regulation depends on their quantification and risk assessment as reported by the producer.
Experimental issues in the quantification of some FCM substances
During the method development, several observations with respect to the analysis of some of the substances have been made and are presented below.
During the quantification of triethylphosphite (FCM No. 293) in food simulants A and C, the presence of a second chromatographic peak was observed at a different retention time and with m/z of 82.0 Da, whereas this peak was not observed using a standard solution in organic solvents. This second peak was tentatively identified by using MS libraries as diethyl phosphite. This substance is not included in the positive list of the Reg. (EU) No. 10/2011; therefore, it is considered as a NIAS.
The presence of diethyl phosphite in aqueous food simulants could be a result of a hydrolysis, generating smaller alkyl phosphites or phosphorous acid [23, 24]. Another study using 17O-NMR investigated the hydrolysis of alkyl phosphites, including triethylphosphite, and observed the appearance of the di-alkyl product (Fig, 3). Since the Reg. (EU) No. 10/2011 specifies the use of aqueous food simulants, there is a priori a high probability that hydrolysis reactions would occur during the migration test. This could be especially the case during migration at high temperatures which could accelerate the hydrolysis rate [25, 26]. Therefore, the analysis of FCM No. 293 may become relatively difficult in aqueous simulants.

Potential hydrolysis of triethylphosphite (FCM No. 293) to diethylphosphite (NIAS)
Consequently, also diethyl phosphite has been included in the present multi-analyte method for being able to quantify FCM No. 293 indirectly. No hydrolysis of diethyl phosphite in fortified food simulants A and C was observed. The LOQ for the determination of diethyl phosphite was 25.0 ng mL−1 and accurate results were obtained in both food simulants A and C at the lowest concentration level studied. It is worth to note that according to the Reg. (EU) No. 10/2011, the verification of compliance of FCM products regarding FCM No. 293 is pending due to the unavailability of a proper analytical method. This may be related to the potential hydrolysis during the migration test in aqueous food simulants.
The hydrolysis of substance FCM No. 138, acetyl tributyl citrate, may also occur during the migration test in aqueous food simulants. This substance could be hydrolysed to tributyl aconitate. This was confirmed by studying a commercial standard and comparing its mass spectrum with those in MS libraries. The reaction product is also not listed in the Reg. (EU) No. 10/2011 and should be considered as a NIAS (Fig. 4). Although the hydrolysis rate seemed to be much lower than for FCM No. 293, it may have still affected the quantification.

Hydrolysis of acetyl tributyl citrate (FCM No. 138) to tributyl aconitate (NIAS)
Conclusions
The multi-analyte method described here should support the efficient compliance control of FCM products regarding more than a few substances. The achieved method performance characteristics demonstrate that 84 substances in food simulants A and C can be analysed simultaneously. This number of analytes represents about 9% of the total number of authorised substances listed in the Regulation (EU) No. 10/2011.
References
Fatima Pocas M, Hogg T. Exposure assessment of chemicals from packaging materials in foods: a review. Trends Food Sci Technol. 2007;18:219–30.
Commission Regulation (EU) No. 10/2011 of 14 January 2011 on plastic materials and article intended to come into contact with food. Off J Eur Comm. L 12:1.
Xun Z, Huang J, Li X-Y, Lin S, He S, Guo X, et al. Simultaneous determination of seven acrylates in food contact paper products by GC/MS and modified QuEChERS. Anal Methods. 2016;8:3953–8.
Guo X, Xian Y, Luo H, Luo D, Chen Y, Lu Y, et al. Quantitative determinations of seven fluorescent whitening agents in polystyrene and polyvinyl chloride plastics by ultrahigh performance liquid chromatography–tandem mass spectrometry. Anal Methods. 2013;5:6086–93.
Wu Z, Xu Y, Li M, Guo X, Xian Y, Dong H. Simultaneous determination of fluorescent whitening agents (FWAs) and photoinitiators (PIs) in food packaging coated paper products by the UPLC-MS/MS method using ESI positive and negative switching modes. Anal Methods. 2016;8:1052–9.
Alberto Lopes JA, Tsochatzis ED, Emons H, Hoekstra E. Development and validation of an HPLC method with fluorescence detection for the determination of fluorescent whitening agents migrating from plastic beverage cups. Food Addit Contam. 2018;35:1438–46.
Cesen M, Lambropoulou D, Laimou-Geraniou, Blaznik U, Heath D, Heath E. Determination of bisphenols and related compounds in honey and their migration from selected food contact materials. J Agric Food Chem. 2016;64:8866–75.
Onghena M, van Hoeck E, Vervliet P, Scippo ML, Simon C, van Loco J, et al. Development and application of a non-targeted extraction method for the analysis of migrating compounds from plastic baby bottles by GC-MS. Food Addit Contam. 2014;31:2090–102.
de los Santos M, Batlle R, Salafranca J, Nerin C. Subcritical water and dynamic sonication-assisted solvent extraction of fluorescent whitening agents and azo dyes in paper samples. J Chromatogr A. 2005;1064:135–41.
Jiang D, Chen L, Fu W, Qiu H. Simultaneous determination of 11 fluorescent whitening agents in food-contact paper and board by ion-pairing high-performance liquid chromatography with fluorescence detection. J Sep Sci. 2015;38:605–11.
Jung T, Simat TJ. Multi-analyte methods for the detection of photoinitiators and amine synergists in food contact materials and foodstuffs – part II: UHPLC-MS/MS analysis of materials and dry foods. Food Addit Contam. 2014;31:743–66.
Lin Q-B, Li B, Song H, Li X-M. Determination of 7 antioxidants, 8 ultraviolet absorbents, and 2 fire retardants in plastic food package by ultrasonic extraction and ultra-performance liquid chromatography. J Liq Chromatogr Relat Technol. 2011;34:730–43.
Aznar M, Alfaro P, Nerin C, Kabir A, Furton KG. Fabric phase sorptive extraction: an innovative sample preparation approach applied to the analysis of specific migration from food packaging. Anal Chim Acta. 2016;936:97–107.
Vavrous A, Ševčík V, Dvořáková M, Čabala R, Moulisová A, Vrbik K. Easy and inexpensive method for multiclass analysis of 41 food contact related contaminants in fatty food by liquid chromatography−tandem mass spectrometry. J Agric Food Chem. 2019;67:10968–76.
Barry EF, Grob RL. Columns for gas chromatography: performance and selection. 1st ed. Hoboken: Wiley; 2007. p. 94–186.
European Food Safety Authority. Update of the risk assessment of di-butylphthalate (DBP), butyl-benzyl-phthalate (BBP), bis(2-ethylhexyl)phthalate (DEHP), di-isononylphthalate (DINP)and di-isodecylphthalate (DIDP) for use in food contact materials. EFSA J. 2019;17:5838. https://doi.org/10.2903/j.efsa.2019.5838.
European Commission. 2013. Standard operating procedure for the determination of phthalates in wine and spirits (in-house validated by the EC-JRC-IRMM April 2013). https://ec.europa.eu/jrc/sites/jrcsh/files/SOP/phthalates/by/L_L/partitioning/GCMS_final.pdf Accessed 15 March 2019.
Tsochatzis E, Gika E, Tzimou-Tsitouridou R. Analytical methodologies for the assessment of phthalate exposure in humans. Crit Rev Anal Chem. 2017;47:279–97.
Anastassiades M, Lehotay SJ, Stajnbaher D, Shenck FJ. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int. 2006;86:412–31.
Tsochatzis E, Mieth A, Simoneau C. Report of an inter-laboratory comparison from the European Union Reference Laboratory for Food Contact Materials ILC02 2015 – specific migration from a multilayer in food simulant A. JRC100837, Publications Office of the European Union, Luxembourg. 2016. (ISBN: 978-92-79-57669-0)
Bratinova S, Raffael B, Simoneau C. Guidelines for performance criteria and validation procedures of analytical methods used in controls of food contact materials, European Commission, European Commission, Luxembourg, 2009. (ISBN: 978-92-79-14483-7)
Thompson M, Ellison SLR, Wood R. Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC Technical Report). Pure Appl Chem. 2002;74:835–55.
Bart CJ. Polymer additive analytics: industrial practice and case studies. Firenze: Firenze University Press; 2006. p. 150–90.
Westheimer FH, Huang S, Covitz F. Rates and mechanisms of hydrolysis of esters of phosphorous acid. J Am Chem Soc. 1988;110:181–5.
Tsochatzis ED, Alberto Lopes JA, Dehouck P, Robouch P, Hoekstra E. Proficiency test on the determination of polyethylene and polybutylene terephthalate cyclic oligomers in a food simulant. Food Packag Shelf Life. 2020;23:100441.
Alberto Lopes JA, Tsochatzis ED, Robouch P, Hoekstra E. Influence of pre-heating of food contact polypropylene cups on its physical structure and on the migration of additives. Food Packag Shelf Life. 2019;20:100305.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Disclaimer
Certain commercial equipment, instruments and materials are identified in this paper/report to specify adequately the experimental procedure. In no case does such identification imply recommendation or endorsement by the European Commission, nor does it imply that the material or equipment is necessarily the best available for the purpose.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 214 kb).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tsochatzis, E.D., Alberto Lopes, J., Hoekstra, E. et al. Development and validation of a multi-analyte GC-MS method for the determination of 84 substances from plastic food contact materials. Anal Bioanal Chem 412, 5419–5434 (2020). https://doi.org/10.1007/s00216-020-02758-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-020-02758-7