
Abstract
2-keto-3-L-arabinonate dehydratase (L-KdpD) and 2-keto-3-D-xylonate dehydratase (D-KdpD) are the third enzymes in the Weimberg pathway catalyzing the dehydration of respective 2-keto-3-deoxy sugar acids (KDP) to α-ketoglutaric semialdehyde (KGSA). The Weimberg pathway has been explored recently with respect to the synthesis of chemicals from L-arabinose and D-xylose. However, only limited work has been done toward characterizing these two enzymes. In this work, several new L-KdpDs and D-KdpDs were cloned and heterologously expressed in Escherichia coli. Following kinetic characterizations and kinetic stability studies, the L-KdpD from Cupriavidus necator (CnL-KdpD) and D-KdpD from Pseudomonas putida (PpD-KdpD) appeared to be the most promising variants from each enzyme class. Magnesium had no effect on CnL-KdpD, whereas increased activity and stability were observed for PpD-KdpD in the presence of Mg2+. Furthermore, CnL-KdpD was not inhibited in the presence of L-arabinose and L-arabinonate, whereas PpD-KdpD was inhibited with D-xylonate (I50 of 75 mM), but not with D-xylose. Both enzymes were shown to be highly active in the one-step conversions of L-KDP and D-KDP. CnL-KdpD converted > 95% of 500 mM L-KDP to KGSA in the first 2 h while PpD-KdpD converted > 90% of 500 mM D-KDP after 4 h. Both enzymes in combination were able to convert 83% of a racemic mixture of D,L-KDP (500 mM) after 4 h, with both enzymes being specific toward the respective stereoisomer.
Key points • L-KdpDs and D-KdpDs are specific toward L- and D-KDP, respectively. • Mg2+ affected activity and stabilities of D-KdpDs, but not of L-KdpDs. • CnL-KdpD and PpD-KdpD converted 0.5 M of each KDP isomer reaching 95 and 90% yield. • Both enzymes in combination converted 0.5 M racemic D,L-KDP reaching 83% yield. |
Similar content being viewed by others


Introduction
Petrochemicals have been utilized by humans for almost a century in producing chemicals and other building blocks that are used in everyday life. The utilization of fossil resources, which are the primary source of petrochemicals, has long been a concern as regards sustainability and climate change. The use of plant-derived raw materials has been proposed in order to alleviate our dependency on petrochemicals. In the first generation, starchy materials were utilized, mainly for bioethanol fermentation and lactic acid production (Naik et al. 2010; Mohr and Raman 2015). This approach, while being more environmentally friendly, is still under debate, in particular in relation to competition with food sources (Naik et al. 2010; Mohr and Raman 2015; Rulli et al. 2016). The second-generation bio-production of fuel and chemicals has been developed in order to avoid the potential of competition. In this approach, lignocellulosic biomass is utilized. With annual production of dried biomass exceeding 220 billion tons, it provides us with an enormous amount of truly renewable raw materials (Huang and Fu 2013; Isikgor and Becer 2015).
Unlike the first-generation raw materials, lignocellulosic biomass consists of the following three different components: cellulose, hemicellulose, and lignin. Cellulose can be hydrolyzed to yield D-glucose. Fermentation of D-glucose to chemicals is a straightforward process because it is a universal substrate for most industrially relevant microorganisms. During degradation of the second carbohydrate polymer, hemicellulose will result in several pentose and hexose sugars, of which D-xylose and L-arabinose are two major constituents (Isikgor and Becer 2015). In contrast to D-glucose, these hemicellulose sugars are not easily fermentable. Several works have thus been focused on introducing pentose metabolism pathways in industrially relevant hosts in order to establish hexose and pentose co-fermentation and, therefore, to increase the feasibility of biomass utilization (Fernandes and Murray 2010; Chandel et al. 2011).
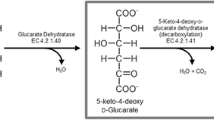
There are three major pathways found in nature that metabolize D-xylose and L-arabinose. The first pathway is the pentose phosphate pathway, which is accessed after isomerization of the pentose sugars from the aldolase to the ketose form. The second and third pathways are called non-phosphorylated oxidative pathways (Jagtap and Rao 2018; Valdehuesa et al. 2018). In the latter pathways, pentose sugars are oxidized to their respective sugar acids. Dehydration at position C2 and C3 results in 2-keto-3-deoxy-D-xylonate and 2-keto-3-deoxy-L-arabinonate, respectively. At this point, the two pathways are branched. In one pathway, aldolase will split the respective intermediates to pyruvate and glycoaldehyde. This pathway is known as the Dahms pathway (Stephen Dahms 1974). The alternative takes place via further dehydration of the 2-keto-3-deoxy-D-xylonate and 2-keto-3-deoxy-L-arabinonate at positions C4 and C5 by means of a dehydratase producing α-ketoglurate semialdehyde (KGSA). Oxidation of the terminal aldehyde of KGSA will yield α-ketoglutarate (α-KG), which is an intermediate of the TCA cycle. This pathway is known as the Weimberg pathway (Weimberg 1961). Because the intermediates at the branch point (2-keto-3-deoxy-D-xylonate and 2-keto-3-deoxy-L-arabinonate) lose their chirality at C2 and C3, all D-pentose sugars (D-xylose, D-ribose, D-arabinose, and D-lyxose) will result in the same intermediate after undergoing one oxidation followed up by the subsequent dehydration at C2 and C3. This is also true for their respective L-sugars. D and L functionality is maintained because it is dictated by the position of a hydroxyl group at C4. Therefore, 2-keto-3-deoxy-D-xylonate and 2-keto-3-deoxy-L-arabinonate are referred to herein as 2-keto-3-deoxy-D-pentonate (D-KDP) and 2-keto-3-deoxy-L-pentonate (L-KDP), respectively, as used in the nomenclature from a previous study (Watanabe et al. 2006). The enzymes that catalyze the dehydration reactions are thus called D-KdpD and L-KdpD, respectively.
Introduction of the Weimberg pathway in industrially relevant microorganisms will allow the production of α-KG and its subsequent derivatives in the TCA cycles from D-xylose and L-arabinose (Tai et al. 2016; McClintock et al. 2017). A-KG is a promising building block for the synthesis of heterocyclic compounds (Stottmeister et al. 2005). A-KG has also widely been used in animal feed and for humans as a food and medicine additive (Wu et al. 2016; Liu et al. 2018). The longevity effect of α-KG in Caenorhabditis elegans was also recently shown (Chin et al. 2014). The Weimberg pathway has also been explored further in recent years in order to determine its potential toward the synthesis of non-natural chemicals from the hemicellulose sugars. Several platform chemicals that have been produced from modified Weimberg pathways include 1,4-butanediol (BDO), mesaconate, glutamate, and succinate (Fig. 1) (Bai et al. 2016; Tai et al. 2016; McClintock et al. 2017). These are important platform chemicals for the production of several polymers. The dehydration of BDO results in tetrahydrofuran, which is a building block for Spandex and an industrial solvent. Decarboxylation of mesaconate will yield methacrylic acid, which is a precursor of vinyl ester resin (Yadav et al. 2018). Succinate has been utilized in the production of polyester, surfactants, detergent, food additives, and pharmaceuticals (Saxena et al. 2016).

The Weimberg pathway and its subsequent modification for the conversion of L-arabinose and D-xylose to added-value compounds/chemicals. The natural pathway is shown in black. The conversions catalyzed by L-KdpD and D-KdpD that are the focus of this study are highlighted by a green box. The pathways are adapted from previous studies (Bai et al. 2016; Tai et al. 2016)
In order to design efficient pathways in a microorganism, the kinetic characterization of responsible enzymes is indispensable. This parameter is equally important when an in vitro approach is preferred. In vitro approaches to producing chemicals have gained increasing attention in recent years as an alternative to in vivo (Guterl and Sieber 2013; Dudley et al. 2015; Claassens et al. 2019). In contrast to the other enzymes of the Weimberg pathway, D-KdpD and L-KdpD have only been characterized partially. It has been reported that Caulobacter crescentus and Paraburkholderia xenovorans are able to encode functional D-KdpD. However, their activities are rather low (kcat: 0.5 s−1 and KM: 1.9 mM for CcD-KdpD; kcat: 4.7 s−1 and KM: 9 mM for PxD-KdpD) (Tai et al. 2016). With regard to CcD-KdpD in particular, its activity seems to be much lower than the two preceding enzymes of the Weimberg pathway in C. crescentus (D-xylose dehydrogenase and D-xylonate dehydratase) (Andberg et al. 2016). Regarding L-KdpD, one elegant work from the 1960s previously characterized a functional enzyme from Pseudomonas saccharopila (also known as Pelomonas saccharophila) (Stoolmiller and Abeles 1966). Possible catalytic mechanisms have also been proposed on the basis of isotopes and inhibitor studies (Portsmouth et al. 1967). However, no protein sequence is available, which one would be necessary for easy heterologous protein expression. The protein sequence of a homologous enzyme from Azospirillum brasilense (AbL-KdpD) has been reported recently and exhibits an activity of 20 U/mg (kcat ~ 11 s−1) (Watanabe et al. 2006). Another homologous enzyme from Bukholderia multivorans (BmL-KdpD) has also been reported, but it exhibits considerably lower activity (kcat of 0.2 s−1) (Tai et al. 2016).
Therefore, several D-KdpDs and L-KdpDs have been characterized in detail in this study. CcD-KdpD and AbL-KdpD were used as guide sequences to mine homologous enzymes. A chemoenzymatic approach was applied to synthesize the substrates D-KDP and L-KDP from D-xylose and L-arabinose, respectively. In addition to kinetic parameters, pH optimum, temperature stability, and inhibitory effects of substrates and intermediates in the Weimberg pathway were determined. The best enzyme variants with respect to activity and stability were applied in the conversion of high substrate load (> 0.5 M).
Materials and methods
Cloning, expression, and enzyme purification
Caulobacter crescentus (DSM 4727), Pseudomonas putida KT 2440 (DSM 6125), and Cupriavidus necator H16 (DSM 428) were purchased from DSMZ (Germany). All cells were grown according to the protocol described by DSMZ for isolating their gDNA. The isolation of gDNA was performed using the DNeasy UltraClean Microbial Kit (Qiagen, Germany). gDNA of Herbaspirillum seropedicae Z67 (DSM 6445) was purchased directly from DSMZ. D-kdpD and L-kdpD genes were amplified from the corresponding gDNAs using a two-step PCR protocol (PCR without annealing step). In brief, the PCR mix consisted of the following: Phusion high-fidelity polymerase 1 U (NEB, Germany), 2 mM of dNTPs (VWR; Germany), 0.5 mM of forward and reverse primers, 25 ng of gDNA, 1x GC buffer (NEB, Germany), and ddH2O up to 50 μL. The PCR reactions were as follows: 98 °C for 30 s, 30 cycles at 98 °C for 10 s, 72 °C for 1 min, and 72 °C for 5 min, and an indefinite hold at 16 °C. Upon PCR, the products obtained were separated using agarose gel electrophoresis. Bands with correct sizes were excised and purified using the NucleoSpin Gel and PCR Clean up kit (Machery and Nagel, Germany). Purified DNAs were then digested and ligated into either pET28a or pET24a (Invitrogen, Germany) to give corresponding N- or C-terminal hexa-histidine tags. Another kdpD1 gene from Caulobacter crescentus (CcD-kdpD1), D-kdpD gene from Paraburkholderia xenovorans (PxD-kdpD), as well as L-kdpD genes from Azospirillum brasilense (AbL-kdpD) were ordered from ATG Biosynthesis (Germany) as optimized codons for expression in E. coli. The CcDkdpD1 gene was synthesized according to the sequence described in the previous study (Tai et al. 2016). These genes were also cloned to either pET28a or pET24a using appropriate restriction enzymes. The ligated plasmids were used to transform E. coli BL21 (DE3) (Invitrogen, Germany). Forward and reverse primers used to amplify the genes as well as restriction enzymes used for cloning to pET vectors are presented in Table S1 in the Supporting Information.
To express the enzymes, a single colony of E. coli BL21 (DE3) possessing the correct gene was grown overnight in 10 mL LB medium supplemented with 100 μg/mL kanamycin in a 100 mL baffled flask at 37 °C, 150 rpm. The overnight culture was then transferred to a 2 L baffled flask containing 500 mL autoinduction media supplemented with 100 μg/mL kanamycin (Studier 2005). The culture was incubated at 30 °C, 120 rpm for 16 h. The cells were pelleted by centrifugation at 4000 ×g for 15 min. After decanting the supernatant, the cell pellet was transferred to a 50 mL falcon tube for subsequent purification.
A binding buffer (50 mM KPi pH 8, 20 mM imidazole, 500 mM NaCl, and 10 vol% glycerin) was added up to 40 mL in the 50 mL falcon tube. A total of 5 μg/mL DNase (AppliChem) and 2 mM MgCl2 were added to the cell suspension. Cells were disrupted by sonication (80% and cycle 0.5 s) in ice for 20 min. The solution was then cleared by centrifugation at 20,000 ×g for 30 min. The supernatant was filtered through a 0.45 μm cellulose filter (VWR, Germany) before application to an Äkta purifier (GE Healthcare, Germany). A 5 mL HisTrap FF Crude (GE Healthcare, Germany) was used for purification of His-tagged proteins. The column was washed with 30 mL binding buffer to remove E. coli proteins. An elution buffer (50 mM KPi, pH 8, 500 mM imidazole, 500 mM NaCl, and 10 vol% glycerin) was used to elute the His-tagged protein from the column. The buffer was exchanged using a HiPrep desalting column (GE Healthcare, Germany). A total of 50 mM HEPES pH 7.5 was used as the final buffer for all enzymes in this study. After the buffer exchange, all enzymes were flash-frozen in liquid nitrogen and stored at − 80 °C until further use. The purity and size of the enzymes were analyzed via SDS-PAGE.
Synthesis of 2-keto-3-deoxy-L-pentonate and 2-keto-3-deoxy-D-pentonate
A chemoenzymatic approach was applied for synthesizing the substrates (L-KDP and D-KDP) from their respective aldoses. This approach was modified from previously published work (Sperl et al. 2016). L-Arabinose and D-xylose were oxidized separately using a 0.5% gold catalyst (Evonik, Germany) in the presence of saturated oxygen. Oxidation was performed in an automatic titrator (SI Analytics, Germany). The pH and temperature were set to 8 and 50 °C, respectively. The gold catalyst was used at a ratio of 0.033 g/g sugar. The oxygen flow rate used was 40 mL/min, and the reaction was mixed using the magnetic stirrer of the automatic titrator. The oxidation was started by the addition of the gold catalyst. After the oxidation was finished (yield > 97% for L-arabinose and D-xylose), the catalyst was pelleted by centrifugation at 4000 ×g for 15 min. The sugar acid (L-arabinonate and D-xylonate) solutions were filtered through a 0.45 μm cellulose filter.
Dehydration of the sugar acids was performed as follows. CcXylDHT was used to dehydrate D-xylonate and RlAraDHT to dehydrate L-arabinonate (Andberg et al. 2016). The expression of each enzyme was performed as described previously (Sutiono et al. 2020). Dehydration of 37.5 mmol of the sugar acids was achieved by using 5 mg of each enzyme. Magnesium chloride (5 mM) was added to the reaction as a cofactor for both enzymes. No buffer was used in the reaction. After an overnight reaction, > 99% conversion of D-xylonate and L-arabinonate was achieved. Given that this was an HPLC analysis and no sugar acid or other peaks were observed beyond a single product peak, it was assumed that > 99% conversion of the sugar acids resulted only in 2-keto-3-deoxy-D-pentonate and 2-keto-3-deoxy-L-pentonate. The final product of the dehydration reaction was a clear solution for both KDPs. The solutions were filtered using an Amicon Ultra Centrifugal Filters 10K (Sigma Aldrich, Germany) to remove the enzymes. The solutions were stored at − 20 °C until further use. No apparent substrate decomposition after being stored > 3 months was observed by HPLC. The standard HPLC method to detect sugar acids and their subsequent dehydrated product was described in earlier research (Guterl et al. 2012).
Kinetic characterization of the dehydratases
To determine dehydratase activity, a coupled-assay with PpKGSADH as auxiliary enzyme was used to link the oxidation of KGSA to α-KG using NAD+, as was described in a previous study (Beer et al. 2017). The measurement was performed in a 96-well F-bottom plate (Greiner Bio-One, Germany). 20 μL of a diluted enzyme was added to a 96-well plate. A total of 180 μL of reaction solution containing different concentrations of either D-KDP or L-KDP with 0.5 U/mL PpKGSADH, 2 mM NAD+, 5 mM MgCl2, 50 mM HEPES pH 7.5 (end concentration) were added subsequently. The measurement was performed with a Multiskan spectrophotometer (Thermo Scientific, Germany) by measuring the development of NADH at 340 nm at 25 °C. Enzyme activity (kcat) was defined as the number of NADH molecules formed per molecule of enzyme per second. Measurements were performed in triplicate.
Effect of Mg2+ on activity, kinetic, and thermodynamic stability
Two different enzyme stock solutions were used to determine activities. The first stock was a standard enzyme solution, whereas the second stock was an enzyme solution that had been pre-incubated with 5 mM EDTA at 25 °C for 1 h. A total of 20 μL of a diluted enzyme solution from the first stock was added to a 96-well F-bottom plate. A total of 180 μl of reaction mix containing 5 mM of each substrate (L-KDP for CnL-KdpD and HsL-KdpD; D-KDP for PpD-KdpD and HsD-KdpD), 0.5 U/mL PpKGSADH, 2 mM NAD+, and 50 mM HEPES pH 7.5, without and with 5 mM MgCl2 was added. The development of NADH was monitored at 340 nm using Multiskan spectrophotometry. The same reaction mixture without MgCl2 was also used to measure activity of the second enzyme stock. All measurements were performed at 25 °C.
To measure T501h (temperature at which an enzyme loses 50% of its initial activity after 1 h incubation), 20 μL of enzyme solution was transferred to a 96-PCR plate (Brand, Germany). The enzyme solution contained 1 mg/mL enzyme in 50 mM HEPES pH 7.5 with 5 mM EDTA, with or without 5 mM MgCl2. The PCR plate was incubated in a thermal cycler (MyCycler, Bio-Rad, Germany) at gradient temperatures from 30 to 55 °C. The incubation was performed for 1 h. Following this heating step, the enzyme solution was diluted accordingly with 50 mM HEPES pH 7.5. A total of 20 μL of diluted solution was transferred to a 96-well plate. A total of 180 μL of reaction solution containing 5 mM of each substrate (L-KDP for CnL-KdpD and HsL-KdpD and D-KDP for PpD-KdpD and HsD-KdpD), 0.5 U/mL PpKGSADH, 2 mM NAD+, 2.5 mM MgCl2, 50 mM HEPES pH 7.5 (end concentration) was added. The development of NADH was monitored at 340 nm using a Multiskan spectrophotometer. The activities obtained were normalized to the highest value. Three independent repeats were performed.
The thermodynamic stability is displayed as the melting temperature (Tm) and was analyzed by the Thermofluor assay (Boivin et al. 2013; Sutiono et al. 2018). The buffer used in the assay was 50 mM HEPES pH 7.5 with 5 mM EDTA, with or without 5 mM MgCl2.
Determination of pH optimum
A total of 20 μL of diluted enzyme (CnL-KdpD with N-terminal His-tag or PpD-KdpD) was added to a 96-well plate. A total of 180 μL reaction solution containing 5 mM of each substrate (L-KDP for CnL-KdpD and D-KDP for PpD-KdpD), 0.5 U/mL PpKGSADH, 2 mM NAD+, 5 mM MgCl2, and 50 mM of buffer with corresponding pH (end concentration) was added. The reaction was monitored at 340 nm using Multiskan spectrophotometer to detect NADH formation. For pH 6 to 8, KPi buffer and for pH 7 to 9, Tris-HCl was used. Activities obtained were normalized to the highest value. Measurements were performed in triplicate. PpKGSADH was confirmed to be active in the entire pH range tested (data not shown).
Effect of substrates and selected intermediates of the Weimberg pathway
D-Xylose and D-xylonate were used to check their inhibitory effects on PpD-KdpD. L-Arabinose and L-arabinonate were used for CnL-KdpD with a N-terminal fused His-tag. The experiments were performed as follows. 20 μL of diluted enzyme (PpD-KdpD and CnL-KdpD) was added to a 96-well plate along with 180 μL of reaction solution containing 5 mM of each substrate (L-KDP for CnL-KdpD and D-KDP for PpD-KdpD), 0.5 U/mL PpKGSADH, 2 mM NAD+, 5 mM MgCl2, 50 mM HEPES pH 7.5, and increasing concentration of the respective inhibitors (end concentration). The formation of NADH was monitored at 340 nm using a Multiskan spectrophotometer. Measurements were performed in triplicate. The lack of inhibition of PpKGSADH by the highest compound concentration tested (100 mM) was confirmed.
One-step conversion of high substrate loads
Regarding the CnL-KdpD with a N-terminal His-tag and PpD-KdpD, this was analyzed if high substrate load (L-KDP and D-KDP) concentrations would have an inhibitory effect. Each enzyme was used to convert each substrate in a reaction mix. The reaction mix contained 0.1 mg/mL enzyme, 5 mM MgCl2, 500 mM of respective substrate in 50 mM HEPES pH 7.5. The reaction mix was incubated in a thermoshaker (ThermoMixer C, Eppendorf, Germany) at 300 rpm and 25 °C. At certain times, 5 μL of the solution was removed from the reaction mix and diluted using 995 μL of ddH2O. A total of 500 μL of the diluted solution was transferred to a 10 KDa centrifugal filter (VWR, Germany), and the filter column was centrifuged at 12,000 ×g for 2 min. This step was used to stop the reaction by removing protein from the solution. The filtrate was used to analyze the formation of KGSA from either D-KDP or L-KDP by a previously established method (Beer et al. 2017). In brief, Metrosep A Supp 16–250 was used as a stationary phase, and 30 mM ammonium bicarbonate pH 10.4 was used as the mobile phase. The isocratic flow was set to 0.2 mL/min, and D-KDP, L-KDP, and KGSA were monitored by a UV detector at 210 nm. In this system, D-KDP and L-KDP showed the same retention time. KGSA eluted directly after D-KDP and L-KDP (Fig. S4 in the Supporting Information).
Results
Cloning, expression, and purification of the dehydratases
The position of a hexa-histidine tag (His-tag) sometimes has an effect on enzyme yield, stability, and activity. Therefore, CnL-KdpD was cloned as an N-terminally as well as a C-terminally fused His-tag protein. After expression and purification, the protein yield of the N-Histag variant was almost 3 times higher (data not shown). In subsequent L-KdpDs, all variants were therefore cloned using N-terminally fused His-tags (Table 1). All variants were expressed using autoinduction media. A total of 20 to 50 mg of protein were able to be obtained from the 500 mL culture (except for AbL-KdpD). To improve expression of AbL-KdpD, a more enriched media (Terrific broth media) was used (Watanabe et al. 2006). From 500 mL culture, only 5 mg of this enzyme having lower protein purity were able to be obtained (Fig. S1). As a result, due to the difficulty with expression and the other L-KdpDs demonstrated having a similar magnitude of activity (Table 2), this enzyme would be of less interest with regard to application.
CcD-KdpD1 was used as a template for analyzing which His-tag variant (N- or C-terminally fused His-tag) would be better with respect to activity and stability of D-KdpDs. Both His-tag variants of CcD-KdpD1 were able to be expressed and purified, and similar protein yields were also obtained. However, the N-terminal variant precipitated rather quickly during enzyme characterization. With respect to all D-KdpDs, all variants therefore were cloned using C-terminally fused His-tags (Table 1). We could not obtain reasonable yield for VpD-KdpD, therefore this variant was not pursued further.
Kinetic characterization of the respective dehydratases
Regarding the enzymes purified, L-KdpD showed higher a preference for L-KDP than for D-KDP by at least 45-fold (Table 2). The activity (kcat) and KM-values of all L-KdpDs are also of the same magnitude as reported for the first L-KdpD from Pseudomonas saccharophila published in the late 1960’s (Stoolmiller and Abeles 1966). Regarding D-KdpDs, all enzymes exhibited a higher a preference for D-KDP over L-KDP by over 60-fold, except for CnD-KdpD, which showed only a 35-fold higher preference. The activities of CcD-KdpD1 and PxD-KdpD were considerably higher than reported in earlier research. The significant difference is presumably due to the fact that the authors in the previous study expressed CcD-KdpD1 and PxD-KdpD with a N-terminally fused His-tag (Tai et al. 2016). The in vivo production of chemicals using CcD-KdpD1 and PxD-KdpD from D-xylose has been demonstrated and no accumulation of D-KDP was reported. This could indicate that the enzyme introduced in E. coli did not bear any His-tag. Two enzymes from each class showing the highest activity and catalytic efficiency were then selected for further studies. CnL-KdpD (Nhis) and HsL-KdpD were chosen for L-KdpD and PpD-KdpD and HsKdpD for D-KdpD conversion.
Effect of magnesium on activity, and the kinetic and thermodynamic stabilities of the most promising L-KdpDs and D-KdpDs
Magnesium has been reported as playing a role in D-KdpD from Sulfolobus solfataricus (SsD-KdpD) (Brouns et al. 2006; Brouns et al. 2008). The cation is also observed in the crystal structure of SsD-KdpD. However, PsL-KdpD, which is the enzyme catalyzing the respective dehydration of the L-stereoisomer, was reported to be active without the addition of magnesium ions (Stoolmiller and Abeles 1966; Portsmouth et al. 1967). Magnesium ions have also been reported for the activity and stability of several other lyases, e.g., Fe-S dependent dehydratases and decarboxylases (De La Plaza et al. 2004; Andberg et al. 2016). This work studied the effect of magnesium ions on the most promising L-KdpDs and D-KdpDs. There was no apparent effect of magnesium on the activity of CnL-KdpD and HsL-KdpD (Fig. 2a). Nor did pretreatment of the enzymes with EDTA affect their activity. A different result was observed for PpD-KdpD and HsD-KdpD. When magnesium ions were not present during the activity measurement, a significant reduction of activity was observed (Fig. 2b), and the initial activity also seemed to be delayed (Fig. S3). Pretreatment with EDTA further decreased the initial activity.

Influence of MgCl2 and EDTA on the activity, kinetic, and thermodynamic stabilities of L-KdpDs and D-KdpDs. L-KdpDs (a) and D-KdpDs (b) were pre-incubated with 5 mM EDTA (labeled with EDTA) prior to activity measurement using the coupled enzyme assay in the absence of MgCl2. In addition, activities were also determined without EDTA pre-incubation in the absence (labeled with “No”) and presence of 5 mM MgCl2. The kinetic stability of L-KdpDs (c) and D-KdpDs (d) was assayed as T501h (temperature at which 50% of kinetic activity is lost after 1 h of incubation). The thermodynamic stability of L-KdpDs (e) and D-KdpDs (f) was determined using Thermofluor and is represented as the melting temperature (Tm). The effect of the addition of EDTA (5 mM), the absence of MgCl2, and the addition of MgCl2 (5 mM) in the enzyme solution was investigated for both enzymes. The numerical values are presented in Table S3
Kinetic stability is also an important parameter in determining the applicability of an enzyme. In this study, T501h was used as an indicator of kinetic stability. This value can be used to reliably compare kinetic stability of enzymes given the same experimental conditions. CnL-KdpD appeared to have higher kinetic stability (T501h of 53.2 °C) in comparison to HsL-KdpD (T501h of 47.1 °C) (Fig. 2c). Again, it was shown that the absence of magnesium had barely any effect in regard to their kinetic stability. The presence of 5 mM metal chelator EDTA (> 250 equivalent) also did not decrease nor increase the kinetic stability of the two L-KdpDs. Regarding the two D-KdpDs, PpD-KdpD revealed a T501h of 48.0 °C in the presence of magnesium compared to a T501h of 42.5 °C for HsD-KdpD. The absence of magnesium did not significantly affect the kinetic stability of PpD-KdpD. However, in the presence of 5 mM EDTA a slight decrease in kinetic stability (T501h lowered by 2.5 °C) was observed. The T501h of HsD-KdpD was decreased by 1.3 °C and 2.4 °C in the absence of magnesium and presence of 5 mM EDTA, respectively.
In the present study, the thermodynamic stability is represented by the melting temperature (Tm) and was determined by a thermal shift assay using Sypro orange dye. No apparent effect of magnesium in either of the representative L-KdpDs was found. The presence of 5 mM EDTA (>2500 equivalent) did not alter the Tm of CnL-KdpD significantly, but it slightly increased the Tm of HsL-KdpD (Fig. 2e). CnL-KdpD and HsL-KdpD appeared to have similar Tm, although CnL-Kdpd showed a higher T501h. The stabilizing effect of magnesium was observed for the representative D-KdpDs. An addition of 5 mM magnesium increased the Tm of PpD-KdpD and HsD-KdpD by 5.5 and 5.1 °C, respectively. The presence of 5 mM EDTA (>2500 equivalent) decreased the Tm of both enzymes by about 2 °C, further supporting the effect of magnesium on their thermodynamic stability. Given that kinetic stability is a more relevant parameter in practical enzyme application in vivo and in vitro than thermodynamic stability, this parameter was used to further select the most promising enzyme variant from each dehydratase class. CnL-KdpD and PpD-KdpD were chosen for the determination of their pH profiles, the effect of selected intermediates of the Weimberg pathway, as well as suitability for high substrate conversion.
Optimum pH for PpD-KdpD and CnL-KdpD
The pH profile of an enzyme is an important parameter for its application in vitro and in vivo. In particular, the pH of a reaction can be adjusted easily in a cell-free approach. Both enzymes, CnL-KdpD and PpD-KdpD, showed broad pH activity (Fig. 3). However, the main difference was that CnL-KdpD showed higher activity under acidic conditions (optimum pH at 6.5 to 7) and showed significantly reduced activity (> 50% reduction) at pH 9. PpD-KdpD, on the other hand, showed a preference for alkaline conditions (optimum pH at 8 to 9), and its activity was significantly reduced at pH 6 (70% reduction). There was no apparent effect on either enzyme activity by the buffer used, KPi or TRIS. A slightly acidic pH optimum was previously reported for PsL-KdpD (Stoolmiller and Abeles 1966).

Optimum pH of CnL-KDAD and PpD-KdpD. CnL-KDAD revealed an optimum pH of between 6.5 and 7 (darker blue), whereas PpD-KdpD possessed an optimum pH of between 8 and 9 (darker green). Error bars represent the standard deviation of the three replicates
Effect of upstream intermediates of the Weimberg pathways on CnL-KdpD and PpD-KdpD
In a multi-enzymatic reaction, intermediates can be accumulated depending on varying enzyme activities in a cascade reaction. Therefore, it would be important to determine whether the substrates or intermediates of the Weimberg pathway would have an inhibitory effect on CnKdpD and PpD-KdpD. L-arabinose and L-arabinonate were selected for the oxidative L-arabinose pathway and D-xylose and D-xylonate were selected for the oxidative D-xylose pathway. KGSA and α-KG were excluded in this study because the activities of CnKdpD and PpD-KdpD were determined by a coupled assay using PpKGSADH as auxiliary enzyme to oxidize the formed KGSA to α-KG in the presence of NAD+. As for α-KG, not all modified pathways of D-xylose and L-arabinose will end or go via α-KG, e.g., toward 1,4-BDO formation (Fig. 1). Therefore, the effect of α-KG and other intermediates in the synthetic pathways would need to be determined later based on the desired target products.
CnLKdpD demonstrated no inhibitory effects in the presence of L-arabinose and L-arabinonate up to 100 mM (Fig. 4). These characteristics would make CnLKdpD a suitable enzyme for application in the non-phosphorylative oxidative conversion of L-arabinose (McClintock et al. 2017). As for PpD-KdpD, the enzyme was inhibited in the presence of D-xylonate with an I50 value (the concentration of a compound that gives 50% inhibition to the initial activity of an enzyme) of 75 mM, whereas the PpD-KdpD was not inhibited by D-xylose (Fig. 4). PpKGSADH, the auxiliary enzyme in the coupled assay, was also confirmed not to be inhibited by D-xylonate. Therefore, the inhibition effect shown was only for PpD-KdpD.

Effect of substrates and intermediates of the Weimberg pathway on the activity of CnL-KdpD and PpD-KdpD. There was no inhibitory effect of D-xylose up to 100 mM on the activity of PpD-KdpD, as well as L-arabinose and L-arabinonate on the activity of CnL-KdpD. D-xylonate inhibited PpD-KdpD activity at an I50-value of 75 mM
One-step conversion of L-KDP and D-KDP to KGSA using CnL-KdpD and PpKdpD
A high substrate load is often desirable for the biotransformation of chemicals. To determine the feasibility of both enzymes respecting high substrate load conversion, each enzyme was used to transform 500 mM D-KDP and L-KDP. A previously established HPLC analysis was utilized for this experiment (Beer et al. 2017). For the conversion of D-KDP, PpD-KdpD was able to reach 90% conversion to KGSA after 4 h. For L-KDP, CnL-KdpD reached > 95% conversion after only 2 h (Fig. 5a). In the first 10 min, PpD-KdpD showed a higher turnover rate than CnL-KdpD, but the conversion of PpD-KdpD slowed over time. When the concentration of KGSA reached 300 mM, the activity of PpD-KdpD was only 17 U/mg, representing only 12% of its initial activity (in the first 10 min). As for CnL-KdpD, its activity was 38 U/mg in the presence of 300 mM KGSA, representing 45% of its initial activity. A combination of both enzymes was also able to convert a 500 mM racemic mixture of D,L-KDP (250 mM of each isomer). In this experimental set-up, > 80% conversion was observed after 4 h (Fig. 4b). The concentrations of KGSA decreased after 8 h, and a further decrease after 24 h of incubation was noticed in both set-ups (Fig. 4a and b). The degradation of KGSA was likely due to interaction of its terminal aldehyde group with amino acids on the protein surface (Robert and Penaranda 1954; Uchida 2003; Grimsrud et al. 2008). This is further supported by the yellow color of the reaction solutions observed after 24 h of reaction, which is indicative of a typical aldehyde protein interaction.

Time-dependent conversion of L-KDP and D-KDP to KGSA using CnL-KdpD and PpD-KdpD. A total of 0.1 mg/mL of each enzyme was used to convert 500 mM of the respective KDP (a,c). A combination of CnL-KdpD and PpD-KdpD (0.05 mg/mL each) was used to convert 500 mM of a racemic mixture of D,L-KDP (b). After each enzyme was used to convert the non-preferred isomer for 21 h (in panel c), 100 μL of each solution was transferred to 100 μL with 500 mM of the respective natural isomer (labeled with bold) to see if both enzymes were still able to convert their natural substrate (d). All experiments were performed in HEPES pH 7.5 at 25 °C
During the kinetic characterization, it was shown that both enzymes were also reactive toward the other non-preferred stereoisomer (PpD-KdpD toward L-KDP and CnL-KdpD toward D-KDP) although at a much lower efficiency. A similar finding was previously reported for the D-KdpD from Herbaspirillum huttiense and L-KdpD from Paraburkholderia mimosarum (Watanabe et al. 2019). The application of only one enzyme for the conversion of both stereoisomers would be of interest for practical applications such as the conversion of mixture of sugars, e.g., L-arabinose and D-xylose, into certain chemicals. Therefore, in order to confirm their promiscuity, each enzyme was used to convert 500 mM of the non-preferred stereoisomer. As shown in Fig. 4c, both enzymes were active toward the other non-preferred stereoisomer. However, the reaction appeared to have halted rather quickly. After 21 and 45 h of reaction, only a slightly higher formation of KGSA was observed for both enzymes. HPLC analysis indicated that the product formed was indeed KGSA because the same retention time was observed (Fig. S4). When the solution of each enzyme after 21 h reaction with the other non-preferred isomer (Fig. 5c) was transferred to a new solution containing 500 mM of their natural isomer, the formation of KGSA was immediately initiated (Fig. 5d). This indicates that the presence of high concentration of the non-preferred isomer did not have any negative effect for both enzymes. CnL-KdpD reached 40% of the theoretical yield of KGSA (80% if calculated from L-KDP only), whereas PpD-KdpD reached 33% of the theoretical yield of KGSA (66% from D-KdpD) after 3 h.
Discussion
D-xylose and L-arabinose are the two most abundant pentose sugars in hemicellulose. Utilization of these sugars to produce platform chemicals would allow valorization of lignocellulose biomass. The enzymatic cascade reactions of the Weimberg pathways have been used to transform these pentose sugars into several chemicals, e.g., BDO, α-KG, succinate, glutamate, and mesaconate (Fig. 1). These chemicals are still industrially produced or derived from petrochemicals. Enzymes for the key steps of the Weimberg pathway, specifically L-KdpD and D-KdpD, have only been partially characterized with major focus on their kinetic properties. These specific enzymes catalyze the dehydration of L-KDP and D-KDP to KGSA.
In this study, several L-KdpDs and D-KdpDs were cloned and heterologously expressed in E. coli. For the CnL-KdpD, the N-terminally fused His-tag resulted in higher protein yields compared to the C-histag variant. For D-KdpD, however, the C-histag was the better variant. The fact of the N-terminally fused variant of CcD-KdpD1 quickly precipitating and the higher activity of CcD-KdpD2, which is a homologous enzyme of CcKdpD1 (58 amino acids shorter at the N-terminus, Fig. S1) as compared to CcD-KdpD1, could suggest that the N-terminus for D-KdpD is important to enzyme activity and/or stability, and that the addition of further amino acids N-terminally would impede either. However, in a recent study, N-terminally fused His-tag of CcKdpD2 (bearing additional 20 amino acids in N-terminus in comparison to C-histag of CcKdpD2 in this study) showed a magnitude of activity similar to CcKdpD2 (Shen et al. 2020). Further analysis using BLAST searches in the NCBI and UniProt database suggest that the previously cloned and characterized CcD-KdpD1 might be cloned with the wrong start codon (Tai et al. 2016), since most of the homologs do not show the 58 amino acids extension at the N-terminus. The amino acid downstream of the current start codon (the 59th amino acid encoded by GTG) is a second possible start site. Therefore, future studies have to be completed in order to confirm whether the truncated version is more stable and/or active and to decide whether the length of the additional amino acids is also an important factor.
All L-KdpDs revealed a high catalytic efficiency (> 100-fold) with L-KDP, whereas all D-KdpDs showed high efficiency (> 150-fold) with D-KDP. The catalytic efficiency of the KdpDs is an important parameter for in vitro and in vivo pathway design. In particular, the catalytic efficiency for D-KdpD measured in vitro has been shown to have a strong correlation with enzyme efficiency in vivo (Tai et al. 2016). In the previous study, it was shown that the E. coli mutant expressing PxD-KdpD produced BDO faster than the E. coli mutant expressing CcD-KdpD1 (Tai et al. 2016). The authors (as well as this study) have demonstrated a higher catalytic efficiency for PxD-KdpD than for CcD-KdpD1 (Table 2). Furthermore, in the recent study combining enzymatic and modeling approach, the authors were able to predict and simulate the activity of all five Weimberg enzymes, including CcD-KdpD2 in cell-free extracts based on their kinetic model (Shen et al. 2020).
A study on the effect of magnesium on activity, kinetic, and thermodynamic stability revealed that this cation showed effects on members of the D-KdpD class, while no apparent effects were observed on the enzymes of the L-KdpD class. The presence of magnesium increased the activity and kinetic (T501h) and thermodynamic (Tm) stabilities of PpD-KdpD and HsD-KdpD. No significant difference was observed when comparing the thermodynamic stability of CnL-KdpD and HsL-KdpD, although the kinetic stability suggested otherwise. Apparently, the relationships between the kinetic and thermodynamic stabilities were not able to be observed for L-KdpD, which stands in contrast to our previous studies of two α-keto acid decarboxylases (Sutiono et al. 2018; Sutiono et al. 2019).
The detailed characterization with respect to pH profiles revealed an optimum pH at 6.5 to 7 for CnL-KdpD, whereas PpD-KdpD showed the highest activity at pH 8 to 9. Together with the differing effect of magnesium toward L- and D-KdpD, these results suggest that L-KdpD and D-KdpD may adopt different catalytic mechanisms for performing the dehydration at position C4 and C5 of L-KDP and D-KDP, respectively. This outcome could also imply that different amino acid residues are acting during the catalysis, since the pH level would determine the protonation states of charged amino acids. This hypothesis is further supported by the fact that, from the automated gene annotation in these two enzyme classes, although they catalyze similar reactions, they belong to different enzyme families. All D-KdpDs are annotated as members of the fumarylacetoacetate hydrolase (FAH) family, whereas all L-KdpDs are annotated as members of the dihydrodipicolinate synthase (DHDPS) family. The relative similarity of each dehydratase in their corresponding family is presented in Fig. S5.
The effects of upstream substrates and intermediates in the Weimberg pathway on each enzyme were also studied. L-arabinose and L-arabinonate up to 100 mM did not show any inhibitory effect on CnL-KdpD. Nor did the same concentration of D-xylose inhibit the activity of Pp-KdpD. However, D-xylonate showed a significant inhibition of Pp-KdpD at an I50-value of 75 mM. This concentration seems to be quite high (the higher the I50, the less significant the inhibitory effect). However, due to the low activity of D-xylonate dehydratase (Fig. 1), D-xylonate could accumulate to this relevant range. The [Fe-S]-dependent sugar acid dehydratase has been reported as being one of the major bottlenecks in in vitro and in vivo utilization of the Weimberg pathway (Salusjärvi et al. 2017; Boer et al. 2019; Shen et al. 2020). Therefore, a more active D-xylonate dehydratase would be needed to avoid an accumulation of D-xylonate that would inhibit PpD-KdpD. In addition, due to the high kcat value exhibited by PpD-KdpD (75 s−1), it would be unlikely for this enzyme to be the bottleneck in the non-phosphorylative oxidative conversion of D-xylose (Fig. 1). A stronger inhibition effect of D-xylonate was reported for CcD-KdpD2 (Shen et al. 2020).
In the one-step conversion, CnL-KdpD was able to convert 500 mM of L-KDP to KGSA, reaching > 95% yield after only 2 h. When the same amount of PpD-KdpD (higher initial activity) was used to convert D-KDP, the enzyme needed 4 h to reach > 90% yield. This indicates that the KGSA formed has a higher inhibitory effect on PpD-KdpD than on CnL-KdpD. The combination of both enzymes can convert 500 mM of a racemic mixture of D,L-KDP, reaching 83% yield after 4 h. An initial activity test of both enzymes suggests that each enzyme could convert the non-preferred stereoisomer of its natural substrate (Table S2). However, time-based experiments of the non-preferred isomer (Fig. 4c) reveal that both enzymes were only able to convert < 5%, even after 45 h of reaction. This could indicate that the non-preferred isomers inhibit each of the two enzymes. Another, more plausible, explanation is that most (if not all) of the KGSA was formed due to contamination with the natural stereoisomer. An impurity could be derived from the initial substrates used, e.g., aldoses or an unwanted epimerization at C4 which could occur during preparation of the D- and L-KDP. Further experiments, in which the non-preferred stereoisomer was titrated to each enzyme reaction, demonstrated that a greater substrate addition leads to a greater formation of KGSA (Fig. S6). This result suggests that each enzyme is somewhat more specific to the respective stereoisomer than appeared to be the case from the initial activity measurement (Table S2) (Stoolmiller and Abeles 1966). Apparently, the initial activity measurements are not enough to determine the promiscuity of these enzyme classes.
In conclusion, we have cloned, expressed, and characterized a number of L-KdpDs and D-KdpDs. On the basis of catalytic efficiency, activity, and kinetic stability, we demonstrated that CnL-KdpD and PpD-KdpD are the most promising variants from each enzyme class. Both enzymes possess a broad pH range. However, although CnL-KdpD revealed the highest activity at a slightly acidic pH, PpD-KdpD was more active at an alkaline pH. Both enzymes are suitable for high-substrate load conversion.
References
Andberg M, Aro-Kärkkäinen N, Carlson P, Oja M, Bozonnet S, Toivari M, Hakulinen N, Donohue MO (2016) Characterization and mutagenesis of two novel iron–sulphur cluster pentonate dehydratases. Appl Microbiol Biotechnol 100:7549–7563. https://doi.org/10.1007/s00253-016-7530-8
Bai W, Tai YS, Wang J, Wang J, Jambunathan P, Fox KJ, Zhang K (2016) Engineering nonphosphorylative metabolism to synthesize mesaconate from lignocellulosic sugars in Escherichia coli. Metab Eng 38:285–292. https://doi.org/10.1016/j.ymben.2016.09.007
Beer B, Pick A, Sieber V (2017) In vitro metabolic engineering for the production of α-ketoglutarate. Metab Eng 40:5–13
Boer H, Andberg M, Pylkkänen R, Maaheimo H, Koivula A (2019) In vitro reconstitution and characterisation of the oxidative d-xylose pathway for production of organic acids and alcohols. AMB Express 9:48. https://doi.org/10.1186/s13568-019-0768-7
Boivin S, Kozak S, Meijers R (2013) Optimization of protein purification and characterization using Thermofluor screens. Protein Expr Purif 91:192–206. https://doi.org/10.1016/j.pep.2013.08.002
Brouns SJJ, Walther J, Snijders APL, Van De Werken HJG, Willemen HLDM, Worm P, De Vos MGJ, Andersson A, Lundgren M, Mazon HFM, Van Den Heuvel RHH, Nilsson P, Salmon L, De Vos WM, Wright PC, Bernander R, Van Der Oost J (2006) Identification of the missing links in prokaryotic pentose oxidation pathways: evidence for enzyme recruitment. J Biol Chem 281:27378–27388. https://doi.org/10.1074/jbc.M605549200
Brouns SJJ, Barends TRM, Worm P, Akerboom J, Turnbull AP, Salmon L, van der Oost J (2008) Structural insight into substrate binding and catalysis of a novel 2-keto-3-deoxy-d-arabinonate dehydratase illustrates common mechanistic features of the FAH superfamily. J Mol Biol 379:357–371. https://doi.org/10.1016/j.jmb.2008.03.064
Chandel AK, Chandrasekhar G, Radhika K, Ravinder R, Ravindra P (2011) Bioconversion of pentose sugars into ethanol: a review and future directions. Biotechnol Mol Biol Rev 6:8–20. https://doi.org/10.1079/9781845936662.0101
Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, Hu E, Whelan SA, Wang JX, Jung G, Solis GM, Fazlollahi F, Kaweeteerawat C, Quach A, Nili M, Krall AS, Godwin HA, Chang HR, Faull KF, Guo F, Jiang M, Trauger SA, Saghatelian A, Braas D, Christofk HR, Clarke CF, Teitell MA, Petrascheck M, Reue K, Jung ME, Frand AR, Huang J (2014) The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 510:397–401. https://doi.org/10.1038/nature13264
Claassens NJ, Burgener S, Vo B, Erb TJ, Bar-even A (2019) A critical comparison of cellular and cell-free bioproduction systems. Curr Opin Biotechnol 60:221–229. https://doi.org/10.1016/j.copbio.2019.05.003
De La Plaza M, Fernández De Palencia P, Peláez C, Requena T (2004) Biochemical and molecular characterization of α-ketoisovalerate decarboxylase, an enzyme involved in the formation of aldehydes from amino acids by Lactococcus lactis. FEMS Microbiol Lett 238:367–374. https://doi.org/10.1016/j.femsle.2004.07.057
Dudley QM, Karim AS, Jewett MC (2015) Cell-free metabolic engineering: biomanufacturing beyond the cell. Biotechnol J 10:69–82. https://doi.org/10.1002/biot.201400330
Fernandes S, Murray P (2010) Metabolic engineering for improved microbial pentose fermentation. Bioeng Bugs 1:424–428. https://doi.org/10.4161/bbug.1.6.12724
Grimsrud PA, Xie H, Griffin TJ, Bernlohr DA (2008) Oxidative stress and covalent modification of protein with bioactive aldehydes. J Biol Chem 283:21837–21841. https://doi.org/10.1074/jbc.R700019200
Guterl J, Sieber V (2013) Biosynthesis “debugged”: novel bioproduction strategies. Eng Life Sci 13:4–18. https://doi.org/10.1002/elsc.201100231
Guterl JK, Garbe D, Carsten J, Steffler F, Sommer B, Reiße S, Philipp A, Haack M, Rühmann B, Koltermann A, Kettling U, Brück T, Sieber V (2012) Cell-free metabolic engineering: production of chemicals by minimized reaction cascades. ChemSusChem 5:2165–2172. https://doi.org/10.1002/cssc.201200365
Huang Y-B, Fu Y (2013) Hydrolysis of cellulose to glucose by solid acid catalysts. Green Chem 15:1095–1111. https://doi.org/10.1039/c3gc40136g
Isikgor FH, Becer CR (2015) Lignocellulosic biomass: a sustainable platform for the production of bio-based chemicals and polymers. Polym Chem 6:4497–4559. https://doi.org/10.1039/c5py00263j
Jagtap SS, Rao CV (2018) Microbial conversion of xylose into useful bioproducts. Appl Microbiol Biotechnol 102:9015–9036. https://doi.org/10.1007/s00253-018-9294-9
Liu S, He L, Yao K (2018) The antioxidative function of alpha-ketoglutarate and its applications. Biomed Res Int 2018:1–6. https://doi.org/10.1155/2018/3408467
McClintock MK, Wang J, Zhang K (2017) Application of nonphosphorylative metabolism as an alternative for utilization of lignocellulosic biomass. Front Microbiol 8:1–6. https://doi.org/10.3389/fmicb.2017.02310
Mohr A, Raman S (2015) Lessons from first generation biofuels and implications for the sustainability appraisal of second generation biofuels. Effic Sustain Biofuel Prod Environ Land-Use Res 63:281–310. https://doi.org/10.1016/j.enpol.2013.08.033
Naik SN, Goud VV, Rout PK, Dalai AK (2010) Production of first and second generation biofuels: a comprehensive review. Renew Sust Energ Rev 14:578–597. https://doi.org/10.1016/j.rser.2009.10.003
Portsmouth D, Toolmiller AC, Abeles RH (1967) Studies on the mechanism of action of 2-keto-3-deoxy-l-arabonate dehydratase. J Biol Chem 242:2751–2759. https://doi.org/10.1094/phyto-62-239
Robert L, Penaranda FS (1954) Studies on aldehyde-protein interactions. I. Reaction of amino acids with lower aldehydes. J Polym Sci 12:337–350. https://doi.org/10.1002/pol.1954.120120128
Rulli MC, Bellomi D, Cazzoli A, De Carolis G, D’Odorico P (2016) The water-land-food nexus of first-generation biofuels. Sci Rep 6:1–10. https://doi.org/10.1038/srep22521
Salusjärvi L, Toivari M, Vehkomäki ML, Koivistoinen O, Mojzita D, Niemelä K, Penttilä M, Ruohonen L (2017) Production of ethylene glycol or glycolic acid from D-xylose in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 101:8151–8163. https://doi.org/10.1007/s00253-017-8547-3
Saxena RK, Saran S, Isar J, Kaushik R (2016) Production and applications of succinic acid. Curr Dev Biotechnol Bioeng Prod Isol Purif Ind Prod:601–630. https://doi.org/10.1016/B978-0-444-63662-1.00027-0
Shen L, Kohlhaas M, Enoki J, Meier R, Schönenberger B, Wohlgemuth R, Kourist R, Niemeyer F, van Niekerk D, Bräsen C, Niemeyer J, Snoep J, Siebers B (2020) A combined experimental and modelling approach for the Weimberg pathway optimisation. Nat Commun 11:1–13. https://doi.org/10.1038/s41467-020-14830-y
Sperl JM, Carsten JM, Guterl JK, Lommes P, Sieber V (2016) Reaction design for the compartmented combination of heterogeneous and enzyme catalysis. ACS Catal 6:6329–6334. https://doi.org/10.1021/acscatal.6b01276
Stephen Dahms A (1974) 3-Deoxy-D-pentulosonic acid aldolase and its role in a new pathway of D-xylose degradation. Biochem Biophys Res Commun 60:1433–1439. https://doi.org/10.1016/0006-291X(74)90358-1
Stoolmiller AC, Abeles RH (1966) Formation of α-ketoglutaric semialdehyde from L-2-Keto-3-deoxyarabonic acid and isolation of dehydratase from Pseudomonas saccharophila. J Biol Chem 241:5764–5771
Stottmeister U, Aurich A, Wilde H, Andersch J, Schmidt S, Sicker D (2005) White biotechnology for green chemistry: fermentative 2-oxocarboxylic acids as novel building blocks for subsequent chemical syntheses. J Ind Microbiol Biotechnol 32:651–664. https://doi.org/10.1007/s10295-005-0254-x
Studier FW (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41:207–234. https://doi.org/10.1016/j.pep.2005.01.016
Sutiono S, Carsten J, Sieber V (2018) Structure-guided engineering of α-keto acid decarboxylase for the production of higher alcohols at elevated temperature. ChemSusChem 11:3335–3344. https://doi.org/10.1002/cssc.201800944
Sutiono S, Satzinger K, Pick A, Carsten J, Sieber V (2019) To beat the heat—engineering of the most thermostable pyruvate decarboxylase to date †. RSC Adv 9:29743–29746. https://doi.org/10.1039/c9ra06251c
Sutiono S, Teshima M, Beer B, Schenk G, Sieber V (2020) Enabling the direct enzymatic dehydration of D-glycerate to pyruvate as the key step in synthetic enzyme cascades used in the cell-free production of fine chemicals. ACS Catal acscatal.9b05068. https://doi.org/10.1021/acscatal.9b05068
Tai YS, Xiong M, Jambunathan P, Wang J, Wang J, Stapleton C, Zhang K (2016) Engineering nonphosphorylative metabolism to generate lignocellulose-derived products. Nat Chem Biol 12:247–253. https://doi.org/10.1038/nchembio.2020
Uchida K (2003) Histidine and lysine as targets of oxidative modification. Amino Acids 25:249–257. https://doi.org/10.1007/s00726-003-0015-y
Valdehuesa KNG, Ramos KRM, Nisola GM, Bañares AB, Cabulong RB, Lee WK, Liu H, Chung WJ (2018) Everyone loves an underdog: metabolic engineering of the xylose oxidative pathway in recombinant microorganisms. Appl Microbiol Biotechnol 102:7703–7716. https://doi.org/10.1007/s00253-018-9186-z
Watanabe S, Shimada N, Tajima K, Kodaki T, Makino K (2006) Identification and characterization of L-arabonate dehydratase, L-2-keto-3-deoxyarabonate dehydratase, and L-arabinolactonase involved in an alternative pathway of L-arabinose metabolism: novel evolutionary insight into sugar metabolism. J Biol Chem 281:33521–33536. https://doi.org/10.1074/jbc.M606727200
Watanabe S, Fukumori F, Nishiwaki H, Sakurai Y, Tajima K, Watanabe Y (2019) Novel non-phosphorylative pathway of pentose metabolism from bacteria. Sci Rep 9:1–12. https://doi.org/10.1038/s41598-018-36774-6
Weimberg R (1961) Pentose oxidation by Pseudomonas fragi. J Biol Chem 236:629–635
Wu N, Yang M, Gaur U, Xu H, Yao Y, Li D (2016) Alpha-ketoglutarate: physiological functions and applications. Biomol Ther 24:1–8. https://doi.org/10.4062/biomolther.2015.078
Yadav SK, Schmalbach KM, Kinaci E, Stanzione JF, Palmese GR (2018) Recent advances in plant-based vinyl ester resins and reactive diluents. Eur Polym J 98:199–215. https://doi.org/10.1016/j.eurpolymj.2017.11.002
Acknowledgments
The authors would like to acknowledge funding by German Federal Ministry for Education and Research (BMBF) through HotSysAPP project (Grant Nos. 031L0078F, 031L0078A). The authors thank Prof. Dirk Tischler (Ruhr University Bochum) for providing Variovorax paradoxus (EPS) cells. The authors also wish to thank Alexander Benson for proofreading the manuscript.
Author contribution statement
SS and VS conceived and designed the study. SS performed the experiments and analyzed the data. SS, BS, and VS wrote the manuscript. All authors read and approved the final version of the manuscript.
Funding
Open Access funding provided by Projekt DEAL. This study was funded by German Federal Ministry for Education and Research (BMBF) through HotSysAPP project (Grant Nos. 031L0078F, 031L0078A).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 611 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sutiono, S., Siebers, B. & Sieber, V. Characterization of highly active 2-keto-3-deoxy-L-arabinonate and 2-keto-3-deoxy-D-xylonate dehydratases in terms of the biotransformation of hemicellulose sugars to chemicals. Appl Microbiol Biotechnol 104, 7023–7035 (2020). https://doi.org/10.1007/s00253-020-10742-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-020-10742-5