
Crosstalk Between Alpha-Synuclein and Other Human and Non-Human Amyloidogenic Proteins: Consequences for Amyloid Formation in Parkinson’s Disease
Abstract
It was recently shown (Sampson et al., Elife 9, 2020) that an amyloidogenic protein, CsgA, present in E. coli biofilms in the gut can trigger Parkinson’s disease in mice. This study emphasizes the possible role of the gut microbiome in modulation (and even initiation) of human neurodegenerative disorders, such as Parkinson’s disease. As the CsgA protein was found to accelerate alpha-synuclein (the key amyloidogenic protein in Parkinson’s disease) amyloid formation in vitro, this result suggests that also other amyloidogenic proteins from gut bacteria, and even from the diet (such as stable allergenic proteins), may be able to affect human protein conformations and thereby modulate amyloid-related diseases. In this review, we summarize what has been reported in terms of in vitro cross-reactivity studies between alpha-synuclein and other amyloidogenic human and non-human proteins. It becomes clear from the limited data that exist that there is a fine line between acceleration and inhibition, but that cross-reactivity is widespread, and it is more common for other proteins (among the studied cases) to accelerate alpha-synuclein amyloid formation than to block it. It is of high importance to expand investigations of cross-reactivity between amyloidogenic proteins to both reveal underlying mechanisms and links between human diseases, as well as to develop new treatments that may be based on an altered gut microbiome.
INTRODUCTION
The name amyloid was first used in medical literature by Rudolf Virchow in 1854 who used it to wrongly identify deposits in the nervous system as starch (amylum in Latin). For a while, the scientific community debated whether amyloid deposits contained fat or carbohydrates until it was shown in 1859 that they are, in fact, protein deposits. In 1907, Alois Alzheimer reported a patient with dementia who had plaques (now, known to be extracellular amyloids) but it took until 1960s before the cross-β-sheet structure of amyloids and experimental methods to detect and isolate amyloids were developed. In subsequent years many amyloid-related human diseases were discovered. Today, we know that the aberrant self-assembly of proteins into amyloid fibers in different organs and tissues is a unifying molecular event in up to 50 human diseases, including several neurodegenerative disorders (e.g., Alzheimer’s, Parkinson’s, Huntington’s and amyotrophic lateral sclerosis (ALS) diseases) as well as systemic (e.g., amyloid light chain (AL) and serum amyloid A (AA) amyloidosis) and local (e.g., type-2 diabetes) diseases [1]. Although proteins with different primary and secondary structures all adopt similar cross-β-sheet amyloid structures in the various disorders, mechanisms and triggers behind the self-assembly remain unknown and may vary from protein to protein.
Parkinson’s disease
Parkinson’s disease (PD) [2, 3], is the second most common neurodegenerative disorder after Alzheimer’s disease and the most common movement disorder. PD is characterized by widespread deterioration of subcortical structures of the brain, especially dopaminergic neurons in the substantia nigra [4]. These changes are coupled to bradykinesia, rigidity and tremor, resulting in difficulties in walking and abnormal gait in patients [4]. Assembly of the protein alpha-synuclein (aS) into amyloid fibrils is linked to the molecular pathology of PD, with aS amyloids being the major content of the pathological neuronal inclusions, Lewy bodies, found post-mortem in the brain of PD patients [5–7] and considered PD hallmarks. The number of patients with PD in the world is increasing but we have no cure: there are only symptomatic drugs in the form of L-DOPA (and derivatives thereof), which is a precursor of dopamine, and electrodes inserted into patients’ brains; both approaches have only temporary effects. In accord with the key role of aS in PD, duplications, triplications and point-mutations in the aS gene, enhancing concentration and aggregation propensity, are linked to familial PD cases [8]. It is thought that aS assembly to amyloid fibers via intermediate oligomers results in toxic gain-of-functions, that are coupled to mitochondrial dysfunction, oxidative stress, protein degradation failure, and eventually cell death [9]. Soluble aS oligomers have been proposed to be most toxic [10, 11], but work with pre-formed αS fibrils have demonstrated that the amyloid fibrils themselves are toxic and can be transmitted from cell to cell and are also able to cross the blood-brain barrier [12–14].
Functional amyloids
Despite the initial association of amyloids with proteins involved in neurodegenerative disorders, in the last two decades, proteins from all kingdoms of life have been reported to form functional amyloids [15]. For example, biofilms are structures used by bacteria to adhere to surfaces which contain amyloids such as curli proteins [16, 17]. Human functional amyloids include proteins in the signaling pathway leading to necrosis (Rip1-Rip3 co-assemblies) [18] and in melanosome ultra-structures (fragments of the pre-melanosome protein, PMEL) [19]. Recently, it was revealed that food allergens may adopt amyloid states that confer protection against the harsh conditions during gastrointestinal transit and allow for uptake in the blood. This was reported for allergenic food proteins such as whey and casein proteins in milk, ovalbumin and lysozyme in egg, and β-parvalbumin in fish [20–23]. In fact, test tube studies have shown that most proteins can form amyloids at certain (often extreme, structure-perturbing) solution conditions [24].
ALPHA-SYNUCLEIN PROPERTIES
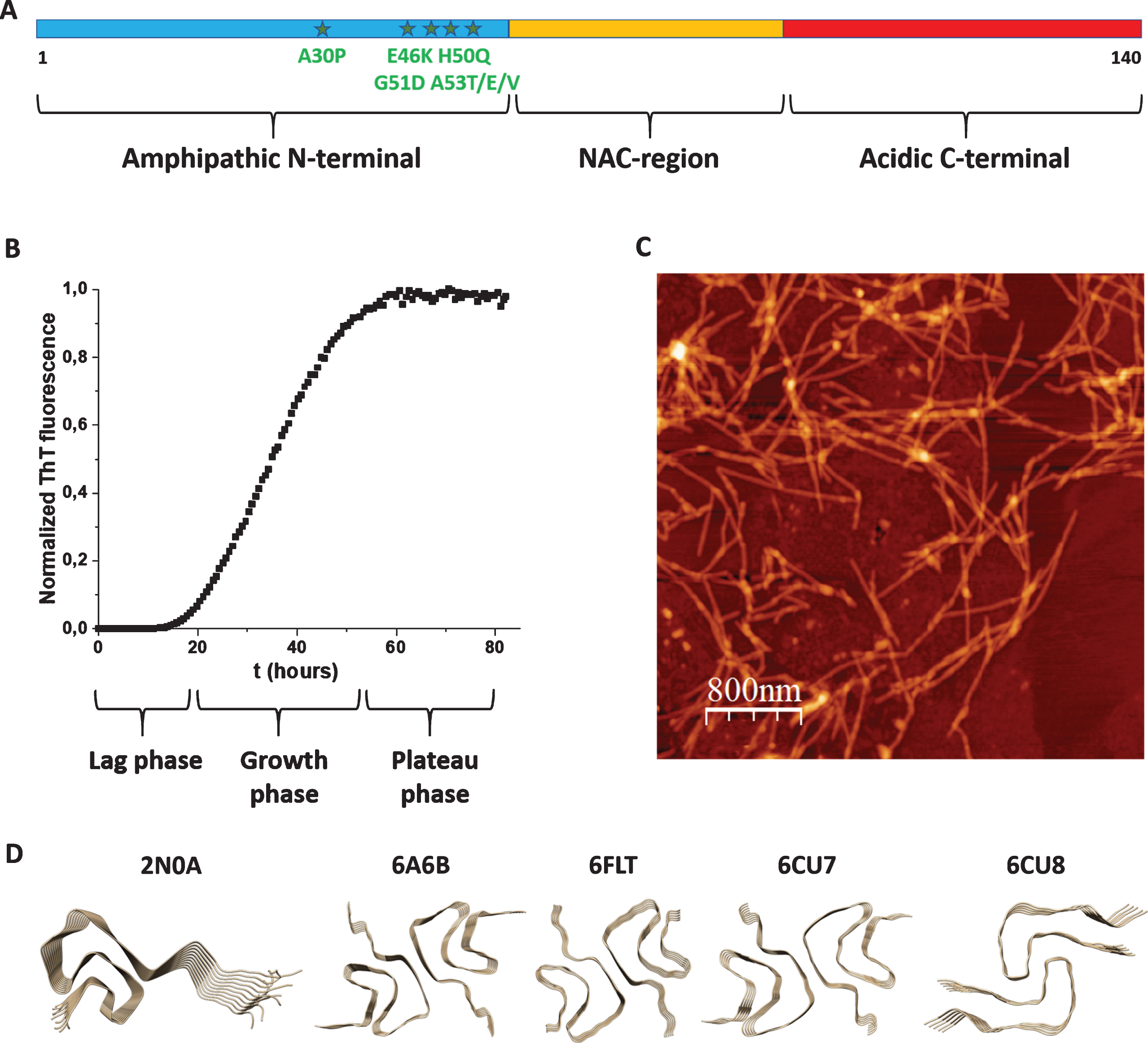
The aS gene was cloned in 1993 [25] and the connection of the protein with PD was made in 1997 [8]. aS is a 140-residue polypeptide that can be divided in three parts: N-terminal, NAC (Non-Amyloid-β Component) and C-terminal regions (Fig. 1A). Whereas the N-terminus (residues 1–60) is amphipathic with many basic residues, the C-terminus (residues 95–140) is acidic with many negatively charged residues. The NAC region (residues 61–94) is hydrophobic and contains several amino-acid repeats which constitute the core of the amyloid structures formed upon aS aggregation. Most PD cases are sporadic, but there are disease-causing mutations, all found in the N-terminal part (A30P, E46K, H50Q, G51D, A53T/E/V), that promote early-onset PD [9]. aS is acetylated at the N-terminus in vivo [26] and posttranslational modifications such as phosphorylation, ubiquitination, and C-terminal truncations are found in aS aggregate deposits [9].
Fig. 1
A) Scheme of aS’s polypeptide with N-terminal (blue), NAC (yellow) and C-terminal (red) parts indicated as well as known disease-causing mutations (stars). B) Typical aggregation curve of aS as probed by ThT fluorescence (lag, growth and plateau phases indicated). C) AFM image of aS amyloid fibers. D) High-resolution structures (2N0A, 6A6B, 6FLT, 6CU7, and 6CU8; Table 1) of aS amyloid fiber cores.
Biophysical characterization and biological functions
The aS monomer is intrinsically disordered in solution with some charge-charge interactions between N- and C-termini that shield the NAC region [27]. aS displays high affinity for negatively charged lipid vesicles: the N-terminal 100 residues adopt a helical structure when bound to vesicles. Chemical (e.g., lipid headgroup chemistry) and physical (e.g., curvature, rafts) properties of lipid vesicles appear to sensitively modulate the binding mode of aS and, at some conditions, formation of amyloids are triggered by vesicles [28]. aS appears to have many functions and interaction partners in human cells. A key activity of aS in brain cells is proposed to be modulation of synaptic-vesicle trafficking, fusion and release [29–31]. In accord, aS is often localized at presynaptic nerve terminals associated with synaptic vesicles [32–34] but it has also been found associated with mitochondrial membranes [35] and in the nucleus [36]. Outside the brain, aS is expressed in, for example, red blood cells, islets of the pancreas and enteroendocrine cells of the gut epithelium. The latter cells form a direct neural circuit from the gut to the brain [37]. This may be of high importance for spreading of PD, as it was recently speculated that the gut microbiome can initiate and modulate PD [38]. aS is annotated as a copper-binding protein in uniprot.org and copper (Cu) ions can bind readily in vitro; in cells, it appears that Cu can be delivered to aS via the copper chaperone Atox1 [39]. aS interaction with Cu may be of functional relevance as Cu ions like neurotransmitters are involved in synapse signaling and Cu ions are loaded in the synaptic vesicles that aS is believed to traffic in the presynapse [40, 41]. It remains unknown if initiation of PD involves aberrant aS functional interactions or whether there are external (not related to normal function) triggers of aS aggregation.
Purpose of this review
Since amyloid fibers have similar structures, there may be generic aspects of aggregation processes that allow for interactions between different amyloidogenic proteins. Cells are crowded with biomolecules [42]. In fact, up to 40% of the available volume in a cell is occupied by other macromolecules. The crowded environment results in many effects including amplified opportunities for non-specific inter-protein interactions [43–50]. In this review, we summarize what is known about how other amyloidogenic proteins affect aS amyloid formation kinetics. We focus on in vitro studies of cross-reactivity, as truly the use of purified proteins is the only way to probe effects of protein-protein interactions on a molecular level. Nonetheless, to be biologically relevant, biophysical studies should always be followed up by in vivo studies and we will mention cell/animal work when appropriate. For more comprehensive descriptions of co-aggregation and co-localization of amyloidogenic proteins in cells (not necessarily involving aS), see [51, 52]. After describing aS amyloid formation and how to detect assembly in vitro, we discuss data concerning the effects of first human amyloidogenic proteins and then non-human amyloidogenic proteins on aS amyloid formation, before ending with a concluding section.
ALPHA-SYNUCLEIN AMYLOID FORMATION IN VITRO
Aggregation of aS into amyloid fibers can be probed in vitro using monomeric aS protein (fresh from a gel filtration column) that is placed at 37° C, often with some shaking and glass-beads to speed up the reaction. At these conditions, the aS amyloid formation reaction, normally monitored in real time by Thioflavin T (ThT) fluorescence, shows a sigmoidal curve (Fig. 1B) in which there is first a lag time (no change in ThT fluorescence), followed by a growth phase (drastic increase in ThT fluorescence) and then a plateau is reached (no further change in ThT fluorescence). In the very beginning of amyloid formation (assuming a purely monomeric starting solution) the formation of the first small aggregates takes place via primary nucleation, which is commonly very slow. This is followed by polymerization/elongation, often involving secondary processes, such as secondary nucleation on amyloid fiber surfaces and fiber fragmentation (creating new nuclei). Most often, both primary and secondary pathways play roles in the overall reaction scheme [53]. With careful kinetic data at a range of protein concentrations and conditions, aspects of the underlying molecular mechanisms can be deduced from global fitting procedures [54]. It is reported that for aS, primary processes dominate at physiological pH, whereas at lower pH, secondary processes become more important [55].
Experimental methods
Whereas ThT probe the appearance of amyloid fibers, other experimental methods can be used for complementary assessment. Far-UV circular dichroism (CD) provides information about aS secondary structure and can be used to follow the transition from random coil (start, monomer) to β-sheet (end, amyloid fiber) structures. To confirm that the ThT data truly reports on amyloid fiber formation, and to macroscopically characterize the formed amyloids, scientists turn to electron microscopy (EM) and atomic force microscopy (AFM) analyses (Fig. 1C). Although the fibers need to be attached to surfaces before analyses, and the methods are not quantitative, many properties of the amyloids can be revealed, such as cross-section size, length, straight vs. curly, presence of branching, etc. To bypass aS’s slow primary nucleation, and sometimes used to get more reproducible kinetic data, one can add aS seeds (i.e., preformed aS amyloid fibers; most often sonicated samples) to the aS monomer solution. Also the addition of lipid vesicles (at certain lipid to protein ratios) and metal ions can catalyze aS aggregation [28, 56]. Cross-reactivity with other amyloidogenic proteins is another (but less explored) way that aS amyloid formation kinetics can be perturbed.
Cross-reactivity and amyloid strains
With cross-reactivity we mean how another amyloidogenic protein (in monomer or amyloid form) may affect aS amyloid formation via direct protein-protein interactions. Another amyloidogenic protein, when added as a monomer, may co-aggregate with aS to speed up amyloid formation or it may interact with aS monomers to block further aS amyloid formation. In similarity, for pre-formed amyloid fibers of other amyloidogenic proteins, they may either seed aS monomers to aggregate faster (heterologous seeding) or they may inhibit aS monomers to form amyloids. In all the cases, this requires interactions between different amyloidogenic proteins and such are proposed to be governed by ‘conformational selection and population shift’, meaning that one or both proteins need to be flexible and able to adjust their conformations to compatible binding states [57]. Using high-resolution methods (solid-state NMR and cryo-EM), aS amyloid fibers have been found to adopt different morphologies (sometimes called strains) depending on the aggregation conditions. In fact, the amyloid folds adopted by different amyloidogenic proteins, including aS, are unexpectedly diverse and complex [58]. In Table 1, we summarize high-resolution structures of aS amyloids reported in the Protein Data Bank (deposited up to February 2020), and the first five structures are illustrated in Fig. 1D. It is not clear how amyloid-core structural differences relate to aS amyloid toxicity and PD propagation, but one may speculate that, in addition to solution conditions, interactions with other amyloidgenic proteins may induce different final amyloid structures. However, all reported high-resolution structures have been generated by aS polypeptides alone, expect the three last entries (still unpublished studies) in Table 1 that are sarkosyl-insoluble fractions from the putamen of multiple system atrophy brains.
Table 1
High-resolution aS amyloid structures reported in PDB; from the first one (by solid-state NMR) published in 2016 and subsequent ones (by cryo-EM) until February 2020, along with comments in some cases. The first five structures are shown in Fig. 1D
| PDB file | Year | Reference | Comments |
| 2N0A | 2016 | [87] | Solid state NMR |
| 6A6B | 2018 | [88] | |
| 6FLT | 2018 | [89] | |
| 6CU7 | 2018 | [90] | Rod |
| 6CU8 | 2018 | [90] | Twister |
| 6SSX | 2019 | [91] | Polymorph 2A |
| 6SST | 2019 | [91] | Polymorph 2B |
| 6OSL | 2019 | [92] | 1–122, acetylated |
| 6OSM | 2019 | [92] | 1–103, acetylated |
| 6OSJ | 2019 | [92] | 1–140, acetylated |
| 6PEO | 2019 | [93] | H50Q narrow fibril |
| 6PES | 2019 | [93] | H50Q wide fibril |
| 6UFR | 2020 | [94] | E46K |
| 6LRQ | 2020 | [95] | A53T |
| 6XYQ | 2020 | not yet published | Multiple system atrophy, type II-2 |
| 6XYO | 2020 | not yet published | Multiple system atrophy, type I |
| 6XYP | 2020 | not yet published | Multiple system atrophy, type II-1 |
CROSS-REACTIVITY WITH HUMAN AMYLOIDOGENIC PROTEINS
IAPP and pro-IAPP
In type-2 diabetes, the hormone amylin (islet amyloid polypeptide, IAPP), made from the longer unprocessed peptide pro-IAPP, forms amyloids in pancreatic β-cells. This leads to β-cell dysfunction, cell death and development of diabetes. Since it was found that upon deletion of the gene Ide (coding for the insulin degrading enzyme, IDE) in mice, the animals developed diabetes and the levels of both IAPP and aS in the pancreas increased [59, 60], the idea of cross-reactivity between IAPP and aS arose. In addition to pancreatic cells, IAPP is also found in the brain and there has been reports suggesting that type-2 diabetes patients are predisposed to get PD but underlying molecular mechanisms are not known [61]. When purified IAPP and pro-IAPP peptides were tested in aS amyloid formation reactions in vitro, it was found that whereas IAPP pre-formed amyloid seeds speed up aS amyloid formation, pro-IAPP amyloid seeds instead reduce aS amyloid formation (Table 2). When monomers of IAPP was mixed with monomers of aS, aggregation was accelerated (faster than either protein alone) and immunogold staining of the resulting amyloid fibers revealed that the amyloids contained both peptides [62]. In contrast, pro-IAPP monomers mixed with aS monomers resulted in aggregation that was slower than that for aS alone. Taken together, the cross-reactivity between both monomers and amyloid fibers of IAPP in aS aggregation hints to a possible explanation for the predisposition of type-2 diabetes patients towards PD.
Table 2
Effects of human and non-human amyloidogenic proteins (in monomer and amyloid forms) on aS amyloid formation kinetics reaction in vitro
| Amyloidogenic protein | Effect on aS amyloid formation reaction | References | |
| In monomer form | In amyloid form | ||
| Human: | |||
| IAPP | Acceleration + coaggregation | Acceleration | [62] |
| Pro-IAPP | Inhibition | Inhibition | [62] |
| S100A9 | Acceleration | No effect | [69] |
| Tau (K19, tau23) | Acceleration + coaggregation | Acceleration | [68] |
| Amyloid-β (1–40, 1–42, 3–42) | Acceleration + coaggregation | Acceleration | [63, 64] |
| Non-human: | |||
| Parvalbumin fish | No effect | Inhibition | [96] |
| CsgA bacteria | Acceleration | Acceleration | [78] |
| FapC bacteria | Inhibition (when using less amyloidogenic variant) | No effect | [80] |
| Insulin bovine | – | Acceleration | [83] |
| Lysozyme hen | – | Acceleration | [83] |
| GroES bacteria | – | Acceleration | [83] |
Amyloid-β
Alzheimer’s disease is characterized by accumulation of extracellular amyloid-β plaque and intracellular amyloids (so-called tangles) of another protein, tau. Interestingly, more than half of Alzheimer’s disease cases exhibit Lewy body pathology, in addition to amyloid-β plaques and tau tangles, and patients with dementia with Lewy bodies often also exhibit amyloid-β plaques. Thus, interactions between amyloid-β and aS may be involved in promoting pathology. In vitro experiments showed that amyloid fiber seeds of amyloid-β speed up amyloid formation of aS (but not as much as amyloid seeds of aS itself) [63]. Also cross-linked oligomers of amyloid-β were tested as seeds and were found to promote aS aggregation, but these were not as potent as the amyloid-β fiber seeds [63]. In another study [64], it was found that when monomeric amyloid-β was added to monomeric aS in a sub-stoichiometric molar ratio, coaggregation was observed and the kinetics became much faster (Table 2). Immunogold staining of the resulting amyloids demonstrated that amyloid-β was incorporated in the aS amyloids. The amyloid fibers formed in the presence of monomeric amyloid-β appeared thicker than aS-only fibers [64]. In these experiments, no aggregation kinetics of amyloid-β alone was shown as a control and, thus, it is unclear if the added small amount of monomeric amyloid-β first formed homogeneous amyloid-β fibers that then seeded aS amyloid formation (and got incorporated in the aS amyloids), or if there was true coaggregation of protein monomers to mixed-protein amyloids.
Tau
Tau is an intrinsically disordered microtubule-binding protein that is abundant in the central nervous system. As mentioned above, intracellular deposits of tau amyloids (tangles) is a hallmark of Alzheimer’s disease along with extracellular amyloid-β plaques. However, in the tau amyloid deposits, aS can sometimes be found, and tau amyloids have been detected in some PD patients. Since tau and aS appeared to cross-react in neuronal cell experiments [65], in vitro assessment of direct protein-protein interactions, and consequences, were performed [66]. Tau alone does not aggregate unless a trigger is added, such as heparin. However, when monomeric tau (in the absence of heparin) was added to aS, at a condition at which aS alone did not aggregate (i.e., no shaking, low concentration), it was found that aS amyloid formation was stimulated (Table 2). The resulting amyloids (likely containing both proteins) were morphologically different from amyloids of the individual proteins; the amyloid fibers from the co-incubated sample showed distinct twists and were thicker [67]. Also tau pre-formed amyloid fibers (unclear how these were made; there was no mentioning of heparin) added to aS monomers in vitro accelerated aS amyloid formation [68]. Tau monomers were found to interact with the C-terminus of aS monomers, and it was proposed that electrostatic attraction between the positive central part of tau and aS’s negatively-charged C-terminus would ‘open up’ the aS monomer conformation, exposing the NAC domain [67]. In accord, tau did not affect aggregation of an aS variant in which the C-terminus had been removed [68].
S100A9
Neuroinflammation, which is linked to PD and other neurodegenerative disorders, is associated with increased levels of pro-inflammatory factors in the brain. One such pro-inflammatory protein is S100A9 that belongs to the large family of calcium-binding S100 proteins that are involved in many inflammatory, cancer and neurodegenerative conditions [69], and are proposed to have intrinsic amyloid-forming capacity [70]. S100A9 is reported to be highly amyloidogenic: it was found in plaque from Alzheimer’s disease patients and it could modulate amyloid-β aggregation in vitro. Since S100A9 was also found to co-localize with aS in Lewy bodies, the possibility of cross-reactivity with aS was investigated in vitro [69]. It was first shown that S100A9 monomers interacted with the C-terminus of aS monomers. When mixed in an equimolar ratio as monomers, the two proteins co-aggregated in a reaction that was faster than those of the individual proteins. The resulting aggregates also appeared larger than amyloids formed from any of the proteins by themselves (Table 2). Also, preformed amyloid fibers of S100A9 (not sonicated; but prepared with agitation and glass bead so short) could speed up aS amyloid formation, but the effect was less dramatic than upon addition of S100A9 monomers. As S100A9 becomes abundant in the brain during neuroinflammation, it was suggested to act as a common denominator in many inflammation-dependent amyloid diseases. Interestingly, oligomers formed by S100A9-aS mixtures were less toxic than oligomers of S100A9 or aS alone [69].
CROSS-REACTIVITY WITH NON-HUMAN AMYLOIDOGENIC PROTEINS
CsgA
CsgA was the first functional amyloid described (by Chapman et al. in 2002 [72]) and is the main component of extracellular curli fibers in biofilms made by E. coli and other bacteria. E. coli possesses at least six proteins, encoded by the csgBA and csgDEFG operons, that are dedicated to curli biogenesis. CsgA is the major curli fiber subunit protein; it contains five imperfect repeating units (19–23 amino acids long), each predicted to form a β-strand-loop-β-strand motif, proposed to constitute the amyloid core of CsgA amyloid fibers [73]. The biofilm helps the organism to adhere to surfaces, for example in the gastrointestinal tract where humans host a vast number of microorganisms [74, 75]. Several studies have indicated roles for the gastrointestinal tract and the enteric nervous system (ENS) in PD. In fact, Lewy bodies are often observed in neurons of the ENS in early stages of PD and, the appendix has been suggested to play (conflicting) roles in PD initiation [76]. ENS neurons can be found in the wall of the gastrointestinal tract and connect directly with enteroendocrine cells which, in turn, communicate with the gut content. Thus, the microbiome may interact with the brain through the ENS. An initial study showed that when rats were fed curli-producing E. coli, there was an increase in aS aggregation as compared to animals that had been fed curli-deficient E. coli [77]. A subsequent study showed that the bacterial composition of the microbiome, or products produced by these bacteria, in aS-expressing mice can lead to increase in aS brain deposits, neuroinflammation and motor deficits [38]. From these studies, it was proposed that alterations in the human microbiome represent a risk factor for PD. To search for a possible molecular mechanism, in vitro experiments with purified CsgA and aS were recently reported [78]. Mixing of CsgA and aS, both in monomer forms, with CsgA at a sub-stoichiometric molar ratio of 1 : 25 CsgA:aS (and at such a low concentration that CsgA did not aggregate on its own) was found to accelerate aS amyloid formation in vitro (Table 2). Since the amount of added CsgA was sub-stoichiometric, a catalytic mechanism involving transient interactions were proposed and this conclusion agreed with lack of stable interactions found in surface plasmon resonance binding experiments [78]. Also, CsgA preformed amyloids accelerated aS amyloid formation in vitro (unpublished results), and the same has been reported for CsgA fibers added to amyloid-β monomers [79]. The cross-reactivity between CsgA and aS was shown to depend on CsgA’s amyloid-forming ability as aS-expressing mice mono-colonized with E. coli bacteria which produced an amyloid-deficient CsgA protein (or mice injected with a non-amyloidogenic mutant of CsgA) did not show as much aS aggregation and had less motor impairments as compared to mice in corresponding experiments with wild-type CsgA [78].
FapC
FapC is a functional amyloid protein produced mainly by members of the Pseudomonas genus. Like CsgA, FapC amyloids provide structural stability to bacterial biofilms, but has fewer amyloid-promoting repeating units in the polypeptide than CsgA (three versus five). To elucidate the mechanism of FapC amyloid formation and the role of the amyloidogenic repeating units, various variants of FapC were characterized in vitro. It was found that upon removal of all repeats, the protein still aggregated, albeit slower, and this delay in amyloid formation allowed for disulfide-bond formation between cysteine residues in monomers, which further delayed the aggregation [80]. Since Pseudomonas is relatively abundant in the gut [81], the authors decided to test for cross-reactivity between FapC and aS in vitro. Surprisingly, the slower-aggregating variant of FapC was found to delay aS aggregation, whereas wild-type FapC showed slightly accelerating or no effect on aS amyloid formation kinetics (Table 2). The interaction between the FapC mutant and aS resulted in small (dead-end) oligomers containing both proteins in vitro. In contrast, pre-formed amyloids of either wild-type or mutant FapC did not affect aS amyloid formation kinetics [80].
Parvalbumin
The most common allergen in fish, the highly abundant protein β-parvalbumin (PV), forms amyloids that escape gastrointestinal degradation and instead transits to the blood [20, 22, 82]. PV is a small, calcium-binding protein with a helical structure that can transform to amyloids upon calcium removal. Since fish is considered beneficial against several age-related diseases including dementia and Alzheimer’s disease, we speculated that protein components in the fish, e.g., PV known to be taken up in human blood [20], could provide benefits perhaps via cross-reactivity with human amyloidogenic proteins resulting in aggregation inhibition. In vitro, PV starts to form amyloids as soon as calcium is removed with EDTA. Mixtures of PV and aS monomers resulted in aggregation reactions that where identical to PV-only reactions. This implied that PV formed amyloids, and this process blocked aS from amyloid formation but did not affect PV’s intrinsic aggregation kinetics. At these conditions, PV aggregated faster than aS alone and subsequent experiments with pre-formed PV amyloids, showed that it is PV amyloids that inhibit aS aggregation (Table 2). It was proposed that PV amyloids, perhaps via their protruding negatively charged calcium-binding loops, scavenged aS monomers to the surface. In accord, immunogold staining experiments showed PV amyloids to be covered with aS but upon calcium addition, aS was released and started to aggregate. This study also provided some hints about the aggregation mechanism of PV itself. As binding of aS to the PV amyloid surface had no effect on PV aggregation kinetics, this suggests that only primary (no secondary, including secondary nucleation and fragmentation) processes are involved in PV amyloid formation. In addition, inhibition of amyloid formation via scavenging of monomers to a pre-formed amyloid (of a different protein) is a new inhibition mechanism not reported before. It remains unclear if PV meets aS in humans after eating fish, although one can speculate this to be possible in the blood as well as in gut enteroendocrine cells. In confocal microscopy experiments, fluorescently labeled PV amyloids are found to enter enteroendocrine cells (unpublished results).
Others
The possibility that amyloid fibers generated from unrelated proteins affect aS amyloid fiber formation kinetics has been addressed in one study [83]. Here amyloid fibers of the E. coli chaperonin GroES, hen lysozyme, and bovine insulin were added to aS aggregation reactions. Notably, all three amyloids could accelerate aS amyloid formation dramatically in vitro (Table 2). Immunogold staining revealed that the added non-aS amyloid seeds were incorporated in the aS amyloids, which appeared able to grow bi-directionally from the initial seed amyloid [83].
CONCLUDING REMARKS
Neurodegenerative disorder cases are increasing in the world, mainly because the population is getting older. This is expected to become a huge burden for healthcare systems in the future as we have no cures for neither PD nor other neurodegenerative diseases. In this review, we have focused on aS and PD, and summarized cross-reactivity data (acceleration or inhibition) between aS and other amyloidogenic proteins (in monomer or amyloid states), see Table 2 and Fig. 2. Human amyloidogenic proteins such as amyloid-β, tau and S100A9 can often be found together with aS in amyloid deposits in PD patients. This suggests a link between these amyloidogenic proteins on a molecular level. In accord, in vitro experiments show that amyloid-β, tau, S100A9, IAPP and pro-IAPP all can modulate aS amyloid formation kinetics in either or both of monomer and amyloid states. There are many more putative cross-reactivity reactions between aS and human amyloidogenic proteins that await to be mechanistically assessed in vitro. For example, expression of a mutant SOD1 (amyloidogenic protein in ALS) in cells promoted cellular aS aggregation [84] and inoculation of infectious prions into aS-expressing transgenic mice promoted extensive aS aggregate deposits and exacerbated aS pathology [85]. These studies hint to possible cross-reactivity on the molecular level such that, in both cases, interaction with the other protein (as monomer or amyloid) would accelerate aS aggregation.
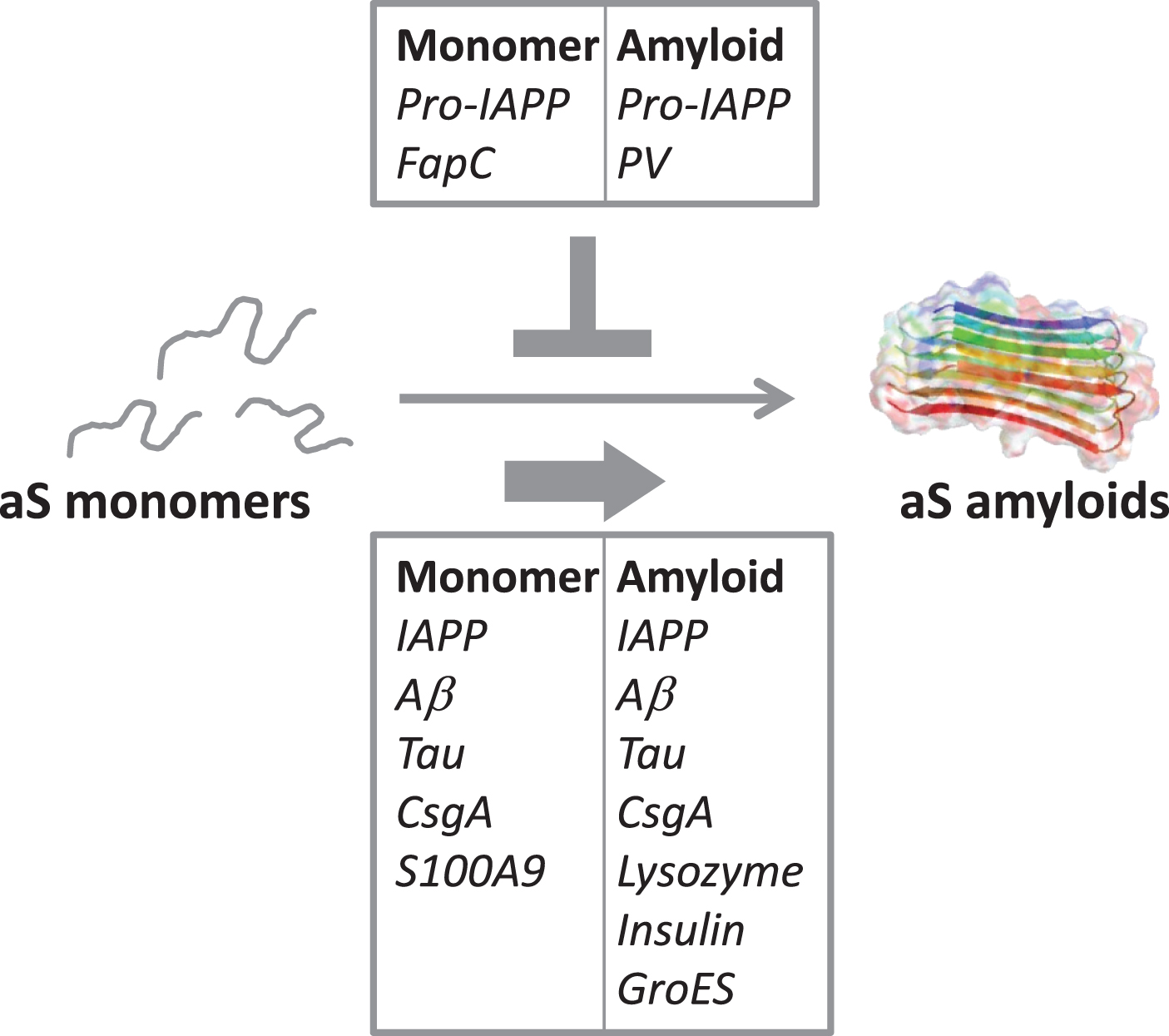
Fig. 2
Illustration of the cross-reactivity results reported in Table 2, indicating the amyloidogenic proteins (as monomers or amyloids) that accelerate aS amyloid formation (bottom box) and the ones that reduce/block aS aggregation (top box).
Today, it is evident that the human microbiome, and even some food proteins, form amyloids [21]. In addition to Escherichia and Pseudomonas already mentioned above, Streptoccocus, Staphylococcus, Salmonella, Mycobacteria, Klebsiella, Citrobacter and Bacillus species are found in the gut and make extracellular amyloids. However, little is known about the abundance of different bacterial amyloids due to the large number of microbes (10-100 trillion in each individual) and extensive variability of composition from person to person. Further studies of bacterial amyloids and amyloid-forming proteins in the diet, that may escape the gastrointestinal tract (such as PV, and perhaps other allergenic proteins), should be pursued. We may find amyloidogenic proteins in food that both accelerate and inhibit human amyloidogenic proteins and these may also cross-react with amyloidogenic proteins in the gut microbiome. Regardless of escaping the gastrointestinal tract or not, food amyloids may modulate the gut microbiome via cross-reactivity with bacterial amyloids. In this context, it is important to note that scientists are looking into ways to force food proteins towards amyloid states as a method to improve food texture; this is done because amyloids have favorable foaming, emulsifying and gel properties [86]. Although most proteins within our diet may be digested, even if in an amyloid form, and levels of individual proteins may be very low [21], one must be careful before using amyloids in food and first assess possible toxicity in a case-by-case basis. When duck or goose foie gras was injected into transgenic mice, amyloids in the foie gras promoted AA amyloidosis in the mice [23]. Although this was an extreme experiment, the results emphasize that amyloid-containing food products may hasten aggregation of host proteins.
To understand the complex interplay between neurodegeneration and diet as well as microbes, many scientific disciplines must come together. Controlled in vitro biophysical experiments with purified proteins can act as the basis (providing intrinsic protein properties) for subsequent interpretations of in vivo observations, where admittedly the situation is more complex. From the limited molecular in vitro data that has been published on cross-reactivity between aS and other amyloidogenic proteins, it emerges that interactions often are observed and both inhibition and acceleration of aS amyloid formation can result, with acceleration appearing more common than inhibition (Table 2, Fig. 2). In fact, of the human amyloidogenic proteins tested for cross-reactivity in vitro, all accelerate aS amyloid formation except pro-IAPP. One possibility is that the net effect in each case depends on the intrinsic aggregation speed of the interacting protein; in accord, pro-IAPP and the FapC variant both aggregate slower than aS when alone, and both inhibit aS amyloid formation. In contrast, most of the proteins reported to accelerate aS amyloid formation, aggregates faster than aS individually. In addition, conformational plasticity (allowing for compatibility despite differences in sequence and structure among interacting proteins) may play a key role [57]. To conclude, it is desired that the scientific community collects more fundamental knowledge of how other human and non-human amyloidogenic proteins modulate aS amyloid formation: both to reveal underlying (causative) reasons for disease onset and as novel avenues towards the use of gut bacteria (and perhaps diet) to counteract PD.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We thank the Knut and Alice Wallenberg Foundation, the Swedish Research Council and the Chalmers Foundation for funding.
REFERENCES
[1] | Chiti F , Dobson CM ((2017) ) Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Ann Rev Biochem 86: , 27–68. |
[2] | John A , van der Pluijm W ((2018) ) The global prevalence of Parkinson’s disease over the next ten years. Ann Neurol 84: , S219–S219. |
[3] | Elkouzi A , Vedam-Mai V , Eisinger RS , Okun MS ((2019) ) Emerging therapies in Parkinson disease - repurposed drugs and new approaches. Nat Rev Neurol 15: , 204–223. |
[4] | Chen AY , Wilburn P , Hao X , Tully T ((2014) ) Walking deficits and centrophobism in an alpha-synuclein fly model of Parkinson’s disease. Genes Brain Behav 13: , 812–820. |
[5] | Goldberg MS , Lansbury PT ((2000) ) Is there a cause-and-effect relationship between alpha-synuclein fibrillization and Parkinson’s disease? Nat Cell Biol 2: , E115–E119. |
[6] | Spillantini MG , Schmidt ML , Lee VMY , Trojanowski JQ , Jakes R , Goedert M ((1997) ) α-Synuclein in Lewy bodies. Nature 388: , 839–840. |
[7] | Uversky VN ((2007) ) Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J Neurochem 103: , 17–37. |
[8] | Polymeropoulos MH , Lavedan C , Leroy E , Ide SE , Dehejia A , Dutra A , Pike B , Root H , Rubenstein J , Boyer R , Stenroos ES , Chandrasekharappa S , Athanassiadou A , Papapetropoulos T , Johnson WG , Lazzarini AM , Duvoisin RC , Di Iorio G , Golbe LI , Nussbaum RL ((1997) ) Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276: , 2045–2047. |
[9] | De Mattos EP , Wentink A , Nussbaum-Krammer C , Hansen C , Bergink S , Melki R , Kampinga HH ((2020) ) Protein quality control pathways at the crossroad of synucleinopathies. J Parkinsons Dis 10: , 369–382. |
[10] | Xu J , Kao SY , Lee FJ , Song W , Jin LW , Yankner BA ((2002) ) Dopamine-dependent neurotoxicity of alpha-synuclein: A mechanism for selective neurodegeneration in Parkinson disease. Nat Med 8: , 600–606. |
[11] | Gosavi N , Lee HJ , Lee JS , Patel S , Lee SJ ((2002) ) Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J Biol Chem 277: , 48984–48992. |
[12] | Peelaerts W , Bousset L , Van der Perren A , Moskalyuk A , Pulizzi R , Giugliano M , Van den Haute C , Melki R , Baekelandt V ((2015) ) alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522: , 340–344. |
[13] | Luk KC , Kehm V , Carroll J , Zhang B , O’Brien P , Trojanowski JQ , Lee VM ((2012) ) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338: , 949–953. |
[14] | Paumier KL , Luk KC , Manfredsson FP , Kanaan NM , Lipton JW , Collier TJ , Steece-Collier K , Kemp CJ , Celano S , Schulz E , Sandoval IM , Fleming S , Dirr E , Polinski NK , Trojanowski JQ , Lee VM , Sortwell CE ((2015) ) Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis 82: , 185–199. |
[15] | Otzen D ((2010) ) Functional amyloid. Prion 4: , 256–264. |
[16] | Evans ML , Chorell E , Taylor JD , Aden J , Gotheson A , Li F , Koch M , Sefer L , Matthews SJ , Wittung-Stafshede P , Almqvist F , Chapman MR ((2015) ) The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Mol Cell 57: , 445–455. |
[17] | Andersson EK , Bengtsson C , Evans ML , Chorell E , Sellstedt M , Lindgren AE , Hufnagel DA , Bhattacharya M , Tessier PM , Wittung-Stafshede P , Almqvist F , Chapman MR ((2013) ) Modulation of curli assembly and pellicle biofilm formation by chemical and protein chaperones. Chem Biol 20: , 1245–1254. |
[18] | Li J , McQuade T , Siemer Ansgar B , Napetschnig J , Moriwaki K , Hsiao Y-S , Damko E , Moquin D , Walz T , McDermott A , Chan Francis K-M , Wu H ((2012) ) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150: , 339–350. |
[19] | Dean DN , Lee JC ((2019) ) pH-Dependent fibril maturation of a Pmel17 repeat domain isoform revealed by tryptophan fluorescence. Biochim Biophys Acta Proteins Proteom 1867: , 961–969. |
[20] | Sanchez R , Martinez J , Castro A , Pedrosa M , Quirce S , Rodriguez-Perez R , Gasset M ((2016) ) The amyloid fold of Gad m 1 epitopes governs IgE binding. Sci Rep 6: , 32801. |
[21] | Cao Y , Mezzenga R ((2019) ) Food protein amyloid fibrils: Origin, structure, formation, characterization, applications and health implications. Adv Colloid Interface Sci 269: , 334–356. |
[22] | Scheers N , Lindqvist H , Langkilde AM , Undeland I , Sandberg AS ((2014) ) Vitamin B12 as a potential compliance marker for fish intake. Eur J Nutr 53: , 1327–1333. |
[23] | Solomon A , Richey T , Murphy CL , Weiss DT , Wall JS , Westermark GT , Westermark P ((2007) ) Amyloidogenic potential of foie gras. Proc Natl Acad Sci U S A 104: , 10998–11001. |
[24] | Chiti F , Webster P , Taddei N , Clark A , Stefani M , Ramponi G , Dobson CM ((1999) ) Designing conditions for in vitro formation of amyloid protofilaments and fibrils [see comments]. Proc Natl Acad Sci U S A 96: , 3590–3594. |
[25] | Ueda K , Fukushima H , Masliah E , Xia Y , Iwai A , Yoshimoto M , Otero DA , Kondo J , Ihara Y , Saitoh T ((1993) ) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 90: , 11282–11286. |
[26] | Kang L , Moriarty GM , Woods LA , Ashcroft AE , Radford SE , Baum J ((2012) ) N-terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci 21: , 911–917. |
[27] | Murray IV , Giasson BI , Quinn SM , Koppaka V , Axelsen PH , Ischiropoulos H , Trojanowski JQ , Lee VM ((2003) ) Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 42: , 8530–8540. |
[28] | Kiskis J , Horvath I , Wittung-Stafshede P , Rocha S ((2017) ) Unraveling amyloid formation paths of Parkinson’s disease protein α-synuclein triggered by anionic vesicles. Q Rev Biophys 50: , e3. |
[29] | Dev KK , Hofele K , Barbieri S , Buchman VL , van der Putten H ((2003) ) Part II: Alpha-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology 45: , 14–44. |
[30] | Lassen LB , Reimer L , Ferreira N , Betzer C , Jensen PH ((2016) ) Protein partners of alpha-synuclein in health and disease. Brain Pathol 26: , 389–397. |
[31] | Fusco G , Pape T , Stephens AD , Mahou P , Costa AR , Kaminski CF , Kaminski Schierle GS , Vendruscolo M , Veglia G , Dobson CM , De Simone A ((2016) ) Structural basis of synaptic vesicle assembly promoted by α-synuclein. Nat Commun 7: , 12563. |
[32] | Eliezer D , Kutluay E , Bussell R , Browne G ((2001) ) Conformational properties of α-synuclein in its free and lipid-associated states. J Mol Biol 307: , 1061–1073. |
[33] | Iwai A , Masliah E , Yoshimoto M , Ge N , Flanagan L , Rohan de Silva HA , Kittel A , Saitoh T ((1995) ) The precursor protein of non-Aβ component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 14: , 467–475. |
[34] | Maroteaux L , Campanelli J , Scheller R ((1988) ) Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 8: , 2804–2815. |
[35] | Faustini G , Marchesan E , Zonta L , Bono F , Bottani E , Longhena F , Ziviani E , Valerio A , Bellucci A ((2019) ) Alpha-synuclein preserves mitochondrial fusion and function in neuronal cells. Oxid Med Cell Longev 2019: , 4246350. |
[36] | Jiang K , Rocha S , Westling A , Kesarimangalam S , Dorfman KD , Wittung-Stafshede P , Westerlund F ((2018) ) Alpha-synuclein modulates the physical properties of DNA. Chemistry 24: , 15685–15690. |
[37] | Chandra R , Hiniker A , Kuo Y-M , Nussbaum RL , Liddle RA ((2017) ) α-Synuclein in gut endocrine cells and its implications for Parkinson’s disease. JCI Insight 2: , e92295. |
[38] | Sampson TR , Debelius JW , Thron T , Janssen S , Shastri GG , Ilhan ZE , Challis C , Schretter CE , Rocha S , Gradinaru V , Chesselet MF , Keshavarzian A , Shannon KM , Krajmalnik-Brown R , Wittung-Stafshede P , Knight R , Mazmanian SK ((2016) ) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167: , 1469–1480 e1412. |
[39] | Horvath I , Blockhuys S , Sulskis D , Holgersson S , Kumar R , Burmann BM , Wittung-Stafshede P ((2019) ) Interaction between copper chaperone Atox1 and Parkinson’s disease protein alpha-synuclein includes metal-binding sites and occurs in living cells. ACS Chem Neurosci 10: , 4659–4668. |
[40] | D’Ambrosi N , Rossi L ((2015) ) Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem Int 90: , 36–45. |
[41] | Gaier ED , Eipper BA , Mains RE ((2013) ) Copper signaling in the mammalian nervous system: Synaptic effects. J Neurosci Res 91: , 2–19. |
[42] | Ellis RJ , Minton AP ((2003) ) Cell biology: Join the crowd. Nature 425: , 27–28. |
[43] | Mikaelsson T , Aden J , Wittung-Stafshede P , Johansson LB ((2014) ) Macromolecular crowding effects on two homologs of ribosomal protein s16: Protein-dependent structural changes and local interactions. Biophys J 107: , 401–410. |
[44] | Mikaelsson T , Aden J , Johansson LB , Wittung-Stafshede P ((2013) ) Direct observation of protein unfolded state compaction in the presence of macromolecular crowding. Biophys J 104: , 694–704. |
[45] | Zhang DL , Wu LJ , Chen J , Liang Y ((2012) ) Effects of macromolecular crowding on the structural stability of human alpha-lactalbumin. Acta Biochim Biophys Sin (Shanghai) 44: , 703–711. |
[46] | Stagg L , Christiansen A , Wittung-Stafshede P ((2011) ) Macromolecular crowding tunes folding landscape of parallel alpha/beta protein, apoflavodoxin. J Am Chem Soc 133: , 646–648. |
[47] | Christiansen A , Wang Q , Samiotakis A , Cheung MS , Wittung-Stafshede P ((2010) ) Factors defining effects of macromolecular crowding on protein stability: An in vitro/in silico case study using cytochrome c. Biochemistry 49: , 6519–6530. |
[48] | Hall D , Dobson CM ((2006) ) Expanding to fill the gap: A possible role for inert biopolymers in regulating the extent of the ‘macromolecular crowding’ effect. FEBS Lett 580: , 2584–2590. |
[49] | Minton AP ((2005) ) Models for excluded volume interaction between an unfolded protein and rigid macromolecular cosolutes: Macromolecular crowding and protein stability revisited. Biophys J 88: , 971–985. |
[50] | Minton AP ((2005) ) Influence of macromolecular crowding upon the stability and state of association of proteins: Predictions and observations. J Pharm Sci 94: , 1668–1675. |
[51] | Sarell CJ , Stockley PG , Radford SE ((2013) ) Assessing the causes and consequences of co-polymerization in amyloid formation. Prion 7: , 359–368. |
[52] | Bondarev SA , Antonets KS , Kajava AV , Nizhnikov AA , Zhouravleva GA ((2018) ) Protein co-aggregation related to amyloids: Methods of investigation, diversity, and classification. Int J Mol Sci 19: , 2292. |
[53] | Cohen SI , Vendruscolo M , Dobson CM , Knowles TP ((2012) ) From macroscopic measurements to microscopic mechanisms of protein aggregation. J Mol Biol 421: , 160–171. |
[54] | Meisl G , Kirkegaard JB , Arosio P , Michaels TC , Vendruscolo M , Dobson CM , Linse S , Knowles TP ((2016) ) Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat Protoc 11: , 252–272. |
[55] | Buell AK , Galvagnion C , Gaspar R , Sparr E , Vendruscolo M , Knowles TP , Linse S , Dobson CM ((2014) ) Solution conditions determine the relative importance of nucleation and growth processes in alpha-synuclein aggregation. Proc Natl Acad Sci U S A 111: , 7671–7676. |
[56] | Lorentzon E , Kumar R , Horvath I , Wittung-Stafshede P ((2020) ) Differential effects of Cu(2+) and Fe(3+) ions on in vitro amyloid formation of biologically-relevant alpha-synuclein variants. Biometals, doi: 10.1007/s10534-020-00234-4. |
[57] | Ren B , Zhang Y , Zhang M , Liu Y , Zhang D , Gong X , Feng Z , Tang J , Chang Y , Zheng J ((2019) ) Fundamentals of cross-seeding of amyloid proteins: An introduction. J Mater Chem B 7: , 7267–7282. |
[58] | Gallardo R , Ranson NA , Radford SE ((2020) ) Amyloid structures: Much more than just a cross-beta fold. Curr Opin Struct Biol 60: , 7–16. |
[59] | Sharma SK , Chorell E , Steneberg P , Vernersson-Lindahl E , Edlund H , Wittung-Stafshede P ((2015) ) Insulin-degrading enzyme prevents alpha-synuclein fibril formation in a nonproteolytical manner. Sci Rep 5: , 12531. |
[60] | Steneberg P , Bernardo L , Edfalk S , Lundberg L , Backlund F , Ostenson CG , Edlund H ((2013) ) The type 2 diabetes-associated gene ide is required for insulin secretion and suppression of alpha-synuclein levels in beta-cells. Diabetes 62: , 2004–2014. |
[61] | Santiago JA , Potashkin JA ((2014) ) System-based approaches to decode the molecular links in Parkinson’s disease and diabetes. Neurobiol Dis 72 Pt A: , 84–91. |
[62] | Horvath I , Wittung-Stafshede P ((2016) ) Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson’s disease. Proc Natl Acad Sci U S A 113: , 12473–12477. |
[63] | Ono K , Takahashi R , Ikeda T , Yamada M ((2012) ) Cross-seeding effects of amyloid beta-protein and alpha-synuclein. J Neurochem 122: , 883–890. |
[64] | Koppen J , Schulze A , Machner L , Wermann M , Eichentopf R , Guthardt M , Hahnel A , Klehm J , Kriegeskorte MC , Hartlage-Rubsamen M , Morawski M , von Horsten S , Demuth HU , Rossner S , Schilling S ((2020) ) Amyloid-beta peptides trigger aggregation of alpha-synuclein in vitro. Molecules 25: , 580. |
[65] | Guo JL , Covell DJ , Daniels JP , Iba M , Stieber A , Zhang B , Riddle DM , Kwong LK , Xu Y , Trojanowski JQ , Lee VM ((2013) ) Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 154: , 103–117. |
[66] | Giasson BI , Forman MS , Higuchi M , Golbe LI , Graves CL , Kotzbauer PT , Trojanowski JQ , Lee VM ((2003) ) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300: , 636–640. |
[67] | Dasari AKR , Kayed R , Wi S , Lim KH ((2019) ) Tau interacts with the C-terminal region of alpha-synuclein, promoting formation of toxic aggregates with distinct molecular conformations. Biochemistry 58: , 2814–2821. |
[68] | Lu J , Zhang S , Ma X , Jia C , Liu Z , Huang C , Liu C , Li D ((2020) ) Structural basis of the interplay between α-synuclein and Tau in regulating pathological amyloid aggregation. J Biol Chem, doi: 10.1074/jbc.RA119.012284. |
[69] | Horvath I , Iashchishyn IA , Moskalenko RA , Wang C , Warmlander S , Wallin C , Graslund A , Kovacs GG , Morozova-Roche LA ((2018) ) Co-aggregation of pro-inflammatory S100A9 with alpha-synuclein in Parkinson’s disease: ex vivo and in vitro studies. J Neuroinflammation 15: , 172. |
[70] | Fritz G , Botelho HM , Morozova-Roche LA , Gomes CM ((2010) ) Natural and amyloid self-assembly of S100 proteins: Structural basis of functional diversity. FEBS J 277: , 4578–4590. |
[71] | Wang C , Klechikov AG , Gharibyan AL , Warmlander SK , Jarvet J , Zhao L , Jia X , Narayana VK , Shankar SK , Olofsson A , Brannstrom T , Mu Y , Graslund A , Morozova-Roche LA ((2014) ) The role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade. Acta Neuropathol 127: , 507–522. |
[72] | Chapman MR , Robinson LS , Pinkner JS , Roth R , Heuser J , Hammar M , Normark S , Hultgren SJ ((2002) ) Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295: , 851–855. |
[73] | Wang X , Hammer ND , Chapman MR ((2008) ) The molecular basis of functional bacterial amyloid polymerization and nucleation. J Biol Chem 283: , 21530–21539. |
[74] | Macfarlane S , Dillon JF ((2007) ) Microbial biofilms in the human gastrointestinal tract. J Appl Microbiol 102: , 1187–1196. |
[75] | Hori K , Matsumoto S ((2010) ) Bacterial adhesion: From mechanism to control. Biochem Eng J 48: , 424–434. |
[76] | Rubin R ((2019) ) Uncovering a link between the appendix and Parkinson disease risk. JAMA, doi: 10.1001/jama.2019.9041. |
[77] | Chen SG , Stribinskis V , Rane MJ , Demuth DR , Gozal E , Roberts AM , Jagadapillai R , Liu R , Choe K , Shivakumar B , Son F , Jin S , Kerber R , Adame A , Masliah E , Friedland RP ((2016) ) Exposure to the functional bacterial amyloid protein Curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans. Sci Rep 6: , 34477. |
[78] | Sampson TR , Challis C , Jain N , Moiseyenko A , Ladinsky MS , Shastri GG , Thron T , Needham BD , Horvath I , Debelius JW , Janssen S , Knight R , Wittung-Stafshede P , Gradinaru V , Chapman M , Mazmanian SK ((2020) ) A gut bacterial amyloid promotes alpha-synuclein aggregation and motor impairment in mice. Elife 9: , e53111. |
[79] | Perov S , Lidor O , Salinas N , Golan N , Tayeb-Fligelman E , Deshmukh M , Willbold D , Landau M ((2019) ) Structural insights into Curli CsgA cross-β fibril architecture inspire repurposing of anti-amyloid compounds as anti-biofilm agents. PLoS Pathogens 15: , e1007978. |
[80] | Christensen LFB , Jensen KF , Nielsen J , Vad BS , Christiansen G , Otzen DE ((2019) ) Reducing the amyloidogenicity of functional amyloid protein FapC increases its ability to inhibit alpha-synuclein fibrillation. ACS Omega 4: , 4029–4039. |
[81] | Monstein H-J , Tiveljung A , Kraft CH , Borch K , Jonasson J ((2000) ) Profiling of bacterial flora in gastric biopsies from patients with Helicobacter pylori-associated gastritis and histologically normal control individuals by temperature gradient gel electrophoresis and 16S rDNA sequence analysis. J Med Microbiol 49: , 817–822. |
[82] | Martinez J , Sanchez R , Castellanos M , Fernandez-Escamilla AM , Vazquez-Cortes S , Fernandez-Rivas M , Gasset M ((2015) ) Fish beta-parvalbumin acquires allergenic properties by amyloid assembly. Swiss Med Wkly 145: , w14128. |
[83] | Yagi H , Kusaka E , Hongo K , Mizobata T , Kawata Y ((2005) ) Amyloid fibril formation of alpha-synuclein is accelerated by preformed amyloid seeds of other proteins: Implications for the mechanism of transmissible conformational diseases. J Biol Chem 280: , 38609–38616. |
[84] | Helferich AM , Ruf WP , Grozdanov V , Freischmidt A , Feiler MS , Zondler L , Ludolph AC , McLean PJ , Weishaupt JH , Danzer KM ((2015) ) alpha-synuclein interacts with SOD1 and promotes its oligomerization. Mol Neurodegener 10: , 66. |
[85] | Masliah E , Rockenstein E , Inglis C , Adame A , Bett C , Lucero M , Sigurdson CJ ((2012) ) Prion infection promotes extensive accumulation of alpha-synuclein in aged human alpha-synuclein transgenic mice. Prion 6: , 184–190. |
[86] | Jansens KJA , Lambrecht MA , Rombouts I , Monge Morera M , Brijs K , Rousseau F , Schymkowitz J , Delcour JA ((2019) ) Conditions governing food protein amyloid fibril formation—Part I: Egg and cereal proteins. Compr Rev Food Sci Food Saf 18: , 1256–1276. |
[87] | Tuttle MD , Comellas G , Nieuwkoop AJ , Covell DJ , Berthold DA , Kloepper KD , Courtney JM , Kim JK , Barclay AM , Kendall A , Wan W , Stubbs G , Schwieters CD , Lee VM , George JM , Rienstra CM ((2016) ) Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol 23: , 409–415. |
[88] | Li Y , Zhao C , Luo F , Liu Z , Gui X , Luo Z , Zhang X , Li D , Liu C , Li X ((2018) ) Amyloid fibril structure of alpha-synuclein determined by cryo-electron microscopy. Cell Res 28: , 897–903. |
[89] | Guerrero-Ferreira R , Taylor NM , Mona D , Ringler P , Lauer ME , Riek R , Britschgi M , Stahlberg H ((2018) ) Cryo-EM structure of alpha-synuclein fibrils.e36402. Elife 7: . |
[90] | Li B , Ge P , Murray KA , Sheth P , Zhang M , Nair G , Sawaya MR , Shin WS , Boyer DR , Ye S , Eisenberg DS , Zhou ZH , Jiang L ((2018) ) Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun 9: , 3609. |
[91] | Guerrero-Ferreira R , Taylor NM , Arteni AA , Kumari P , Mona D , Ringler P , Britschgi M , Lauer ME , Makky A , Verasdonck J , Riek R , Melki R , Meier BH , Bockmann A , Bousset L , Stahlberg H ((2019) ) Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. Elife 8: , e48907. |
[92] | Ni X , McGlinchey RP , Jiang J , Lee JC ((2019) ) Structural insights into alpha-synuclein fibril polymorphism: Effects of Parkinson’s disease-related C-terminal truncations. J Mol Biol 431: , 3913–3919. |
[93] | Boyer DR , Li B , Sun C , Fan W , Sawaya MR , Jiang L , Eisenberg DS ((2019) ) Structures of fibrils formed by alpha-synuclein hereditary disease mutant H50Q reveal new polymorphs. Nat Struct Mol Biol 26: , 1044–1052. |
[94] | Boyer DR , Li B , Sun C , Fan W , Zhou K , Hughes MP , Sawaya MR , Jiang L , Eisenberg DS ((2020) ) The alpha-synuclein hereditary mutation E46K unlocks a more stable, pathogenic fibril structure. Proc Natl Acad Sci U S A 117: , 3592–3602. |
[95] | Sun Y , Hou S , Zhao K , Long H , Liu Z , Gao J , Zhang Y , Su XD , Li D , Liu C ((2020) ) Cryo-EM structure of full-length alpha-synuclein amyloid fibril with Parkinson’s disease familial A53T mutation. Cell Res 30: , 360–362. |
[96] | Werner T , Kumar R , Horvath I , Scheers N , Wittung-Stafshede P ((2018) ) Abundant fish protein inhibits alpha-synuclein amyloid formation. Sci Rep 8: , 5465. |

