
Abstract
Parvoviridae, a diverse family of small single-stranded DNA viruses was established in 1975. It was divided into two subfamilies, Parvovirinae and Densovirinae, in 1993 to accommodate parvoviruses that infect vertebrate and invertebrate animals, respectively. This relatively straightforward segregation, using host association as the prime criterion for subfamily-level classification, has recently been challenged by the discovery of divergent, vertebrate-infecting parvoviruses, dubbed “chapparvoviruses”, which have proven to be more closely related to viruses in certain Densovirinae genera than to members of the Parvovirinae. Viruses belonging to these genera, namely Brevi-, Hepan- and Penstyldensovirus, are responsible for the unmatched heterogeneity of the subfamily Densovirinae when compared to the Parvovirinae in matters of genome organization, protein sequence homology, and phylogeny. Another genus of Densovirinae, Ambidensovirus, has challenged traditional parvovirus classification, as it includes all newly discovered densoviruses with an ambisense genome organization, which introduces genus-level paraphyly. Lastly, current taxon definition and virus inclusion criteria have significantly limited the classification of certain long-discovered parvoviruses and impedes the classification of some potential family members discovered using high-throughput sequencing methods. Here, we present a new and updated system for parvovirus classification, which includes the introduction of a third subfamily, Hamaparvovirinae, resolves the paraphyly within genus Ambidensovirus, and introduces new genera and species into the subfamily Parvovirinae. These proposals were accepted by the ICTV in 2020 March.
Similar content being viewed by others


Current challenges in parvovirus taxonomy
The Parvoviridae are a large and remarkably diverse family of viruses that have small, icosahedral, non-enveloped virions and single-stranded DNA (ssDNA) genomes. The parvoviral genome is linear, 3.9–6.3 kb in size, and the coding region is bracketed by terminal repeats that can fold into hairpin-like structures, which are either different (heterotelomeric) or identical (homotelomeric) [9]. The coding region of the genome contains two major expression cassettes, with open reading frames (ORFs) on the left-hand side giving rise to non-structural (NS) proteins, whereas mRNA populations responsible for translating structural proteins (VPs) are transcribed from the right-hand cassette. The largest of the NS proteins, traditionally designated NS1, is a multi-domain protein that contains a highly conserved helicase superfamily 3 (SF3) domain with helicase and ATPase activity, as well as further domains with endonuclease and sequence-specific DNA-binding activity, all of which are essential for viral replication [7, 23].
Parvoviruses have been found in almost all major vertebrate clades and in both proto- and deuterostome invertebrates. The family Parvoviridae was established in 1975 and divided into two subfamilies in 1993 to classify viruses that infect either vertebrate (Parvovirinae) or invertebrate (Densovirinae) hosts [9]. Initially, this host-based classification criterion was supported by phylogeny; indeed, parvoviruses of vertebrates and arthropods both had rather narrow host spectra, and it seemed unlikely that lineages spanning this robust, long-standing schism would ever be discovered.
The discovery of viruses currently classified in the genera Brevidensovirus, Penstyldensovirus and Hepandensovirus revealed sequence diversity among members of the Densovirinae that was inconsistent with the rather well-conserved nature of the other subfamily members [8]. Members of the Parvovirinae share NS1 as well as VP protein-coding genes of a clearly homologous nature, as indicated by their detectable sequence similarity, which allows the sequences of both proteins to be aligned confidently throughout their entire length. However, the highly variable N- and C-terminal regions of the NS1 as well as the sequences comprising the variable loops of the VP are exceptions. This is not the case with the Densovirinae, as its members are linked together only by the aforementioned short (approx. 200-aa-long) SF3 helicase domain. The helicase domain, however, is highly conserved throughout the entire family, suggesting that certain members of the subfamily Densovirinae, such as hepandensoviruses, are as closely related to certain densoviruses as they are to members of the subfamily Parvovirinae.
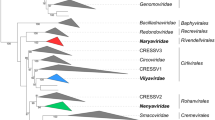
Since 2012, novel genomes from a divergent lineage designated “Chapparvovirus”, have been detected in kidney and liver tissue and in various fluids (such as blood) and excretions (such as feces) of vertebrates [11, 17, 24, 29, 30, 35, 43]. Recent phylogenetic analysis of the helicase domain, however, has revealed a close relationship between this lineage and some current densoviruses, while endogenous chapparvoviral sequences have been identified in several arthropod genomes [21, 25] (Fig. 1).

Bayesian inference of the tripartite helicase domain (167 aa), the only protein motif conserved throughout the family Parvoviridae, showing phylogenetic relationships at the genus level. Newly established taxa are indicated by an asterisk, while existing genera that have been re-classified are indicated by black dots. Background colors indicate the branches comprising the three subfamilies. The alignment was constructed as a consensus of results obtained using T-Coffee Expresso and T-Coffee PSI-Coffee. This calculation was carried out using BEAST v. 1.10.4, using the substitution model LG+I+G+F with a lognormal relaxed clock, based on a prior of Yule process, throughout 50 million generations. Tree diagnostics were carried out using Tracer v. 1.7.1. The size of node shapes is relative to the posterior probability value. The size of the triangles represents the distance from the most basal node to the branch peaks for each genus, which includes multiple species. Posterior probability values are shown at the nodes
Lastly, in 2014, the transcription-strategy-based approach for classifying ambisense densoviruses was revised, merging two established genera, Densovirus and Pefudensovirus, with the hypothetical genus “Cupidensovirus” to create the current genus Ambidensovirus [8]. Even at the time, this genus did not conform to the genus demarcation criteria, but the ambisense genome organization proved to be a strong enough argument to support the creation of the genus Ambidensovirus. Since then, however, multiple ambisense densoviruses have been described, eventually rendering Ambidensovirus paraphyletic.
These anomalies have prompted us to revisit the classification criteria for parvoviruses, and here we present the revision of the current Parvoviridae taxonomy. We submitted a taxonomic proposal [2019; proposal code 2019.010D.A.v2.] that has been approved by the Executive Committee of the International Committee on Taxonomy of Viruses (ICTV) and has been ratified by the ICTV membership. Here, we seek to introduce the revision, which aims to (i) provide a suitable classification for the large number of characterized parvoviruses that are currently described under an unofficial umbrella term “Chapparvovirus”, (ii) establish a new subfamily to resolve the issue of polyphyly among the current Densovirinae members and abandon the host-association-based classification, (iii) resolve the paraphyly of the current genus Ambidensovirus by creation of new, phylogeny-supported genera, (iv) update the criteria for the classification of viruses within the family Parvoviridae to accommodate previously discovered but hitherto unclassified parvoviruses, (v) introduce two new genera into the subfamily Parvovirinae to accommodate two hitherto unclassified parvoviruses; and (vi) classify several currently unclassified viruses within established genera of the subfamily Parvovirinae. All changes contained in this proposal are summarized in Table 1. A phylogenetic tree, summarizing evolutionary relationships at the genus level for the revised taxonomy, is shown in Figure 1. As a result of another proposal, introducing a megataxonomic framework [2019; proposal code 2019.005G.N.v5.], the family Parvoviridae has been classified in the realm Monodnaviria, kingdom Shotokuvirae, phylum Cossaviricota, class Quintoviricetes, order Piccovirales, based on its realtions to other DNA virus taxa.
Updated parvovirus definition and taxon demarcation criteria
In order for a virus to be classified as a member of the family Parvoviridae, it must be identified to be an authentic parvovirus on the basis of having been isolated and sequenced or, failing this, on the basis of having been sequenced from tissue samples, secretions, or excretions of its likely host and reported in a credible peer-reviewed publication. Insights on its biology, such as genome organisation, transcription strategy, epidemiology, serology, structure, trafficking, replication and evolution are strongly encouraged. The sequence must be contiguous and contain the complete coding region of the large nonstructural protein (NS1), which must possess an SF3 helicase domain in its protein sequence, as well as the capsid protein (VP) coding regions. Furthermore, it must meet the size constraints (approx. 4–6 kb) and motif patterns typical of the family. Upon receiving a proposal, the Parvoviridae Study Group is tasked to individually verify the integrity of the suggested virus sequence and approve the proposed classification.
Parvoviruses can be considered members of the same species if their NS1 proteins share more than 85% amino acid sequence identity, and they can be classified in the same genus if their protein sequences cluster as a robust monophyletic lineage based on their complete NS1 protein sequence at the subfamily level and on their SF3 helicase domains at the family level. Additionally, NS1 proteins of members of the same genus should share at least 35-40% amino acid sequence identity with a coverage of >80% between any two members. Failing the sequence-identity-based criteria, common genus affiliation can also be justified based on a similar genome organization, i.e., presence or absence of certain auxiliary-protein-encoding genes, genome length, and/or transcription strategy.
Splitting the subfamily Densovirinae and introducing the subfamily Hamaparvovirinae
To date, the subfamily Densovirinae has served as a holding taxon for all invertebrate-infecting parvoviruses. The subfamily is very heterogenous, and according to phylogenetic inference, some novel vertebrate-infecting parvoviruses have been found to cluster with these viruses (Fig. 2). Our aim was to split the subfamily Densovirinae into two less-heterogenous subfamilies, both with better phylogenetic and biological support. To this end, we introduced the changes detailed below.

Phylogeny of the complete NS1 protein sequence (486 aa) of viruses belonging to the revised subfamily Densovirinae. The Bayesian inference and maximum-likelihood phylogenetic reconstructions were congruent and are represented as one phylogeny rooted with the NS1 protein sequences of two vertebrate-infecting viruses. Newly-introduced taxa are shown in red. Branches presented in the same color and indicated by various symbols representing the demarcations of the newly established genera in the subfamily Densovirinae that replace the former genus Ambidensovirus. The alignment was made as detailed in Fig. 1. These calculations were carried out using BEAST v. 1.10.4. as described in Fig. 1 for Bayesian inference and PhyML v3.3 for maximum-likelihood phylogenetic reconstruction, using the substitution model LG+I+G+F. Posterior probability support values and bootstrap values for 100 iterations are shown at the nodes. The names of viruses and taxa established for the first time are underlined
Current genera Ambidensovirus and Iteradensovirus to be assigned to the subfamily Densovirinae
Although the ambidensoviruses are distinguished by their ambisense genome organization, as opposed to the exclusively unisense genome organization of members of the genus Iteradensovirus, the two genera cluster together as a well-supported monophyletic clade in both NS1-based and helicase domain-based amino acid phylogenies (Fig. 1 and 2). Furthermore, the NS1 proteins of any iteradensoviruses or ambidensoviruses share at least 32% amino acid sequence identity, and their VP proteins, although sharing limited sequence similarity, appear to be derived from a common ancestral VP protein gene. This is not true for members of the three remaining genera in the current subfamily Densovirinae, which share no detectable protein sequence similarity to the ambidensovirus or iteradensovirus NS1s or VPs, except for the aforementioned helicase domain. Lastly, both ambidensovirus and iteradensoviruses have a conserved phospholipase A2 (PLA2) domain in the N-terminus of their minor capsid protein VP1, as is found in most members of the Parvovirinae, but not in the brevidensoviruses, hepandensoviruses, or penstyldensoviruses [9, 21]. Therefore, we assigned the genera Ambidensovirus and Iteradensovirus to a separate subfamily. Since Galleria mellonella densovirus, which currently belongs to the species Lepidopteran ambidensovirus 1 in the genus Ambidensovirus, was the very first identified member of the Densovirinae [20], we propose to keep the Densovirinae designation for this redefined subfamily. The genus Iteradensovirus would keep its current name and affiliation, while the genus-level changes concerning the genus Ambidensovirus are detailed below.
Splitting the genus Ambidensovirus to resolve paraphyly
After investigating the phylogenetic relationships of viral proteins in the revised subfamily Densovirinae by both Bayesian and maximum-likelihood (ML) methods, we established that there are seven well-supported lineages with an ambisense genome organization (plus iteradensoviruses) that represents basis for the newly proposed classification scheme (Fig. 2). Interestingly, members of some of these newly established genera share more NS1 amino acid identity with members of genus Iteradensovirus than they do with viruses of other ambisense genera. Of the seven proposed genera, six are composed of members of the current genus Ambidensovirus, whereas one new genus will accommodate currently unclassified ambisense densoviruses. To indicate the ambisense nature of these viruses, we will keep the “ambi” prefix even in the newly introduced genus names.
Genus Miniambidensovirus
Only one species, Orthopteran miniambidensovirus 1, the type species of the genus, have been assigned to this genus. This species contains a single reported virus, Acheta domestica mini ambidensovirus (AdMDV), which has the smallest genome in the subfamily thus far, at 4.9 kb, as mirrored in its name. This virus infects common house crickets (Acheta domestica) with high mortality rates following infection. AdMDV has a unique, split NS1-encoding ORF, suggesting that splicing may be a prominent feature of its yet unresolved transcription strategy [26]. The NS1 of AdMDV shares less than 27% amino acid sequence identity with the NS1 of any other parvovirus.
Genus Aquambidensovirus
Two species have been assigned to this genus, Decapod aquambidensovirus 1 and Asteroid aquambidensovirus 1. The members of both of these species are known to infect only aquatic hosts, which the genus name reflects. Members of the proposed genus share about 70% NS1 amino acid sequence identity with each other and ~30% with other members of the proposed subfamily Densovirinae. Decapod aquambidensovirus 1 is the type species of the genus, with only one virus, Cherax quadricarinatus densovirus (CqDV), which was identified in an Australian freshwater crayfish (Cherax quadricarinatus). CqDV has one of the largest genomes in the family (6.3 kb) [3].
The species Asteroid aquambidensovirus 1 comprises three viruses identified in members of the deuterostome invertebrate phylum Echinodermata. All of these are highly pathogenic, but sea star-associated densovirus is the only one classified to date [13].
Genus Scindoambidensovirus
Three species have been assigned to this new genus, Orthopteran scindoambidensovirus 1, Hymenopteran scindoambidensovirus 1, and Hemipteran scindoambidensovirus 1, with viruses sharing 40-43% NS1 amino acid sequence identity with each other and <30% with other densoviruses. Members of this genus are also characterized by a split VP-encoding ORF, which gives rise to the VP1 minor capsid protein via a spliced transcript as well as another minor capsid protein (VP2) possessing a unique N-terminal region, which has not been observed in any other parvoviruses to date [37]. The name “Scindo”, from “split” or “cut” in Latin, refers to this split VP gene. The type species is Orthopteran scindoambidensovirus 1, and includes another virus, Acheta domestica densovirus (AdDV). AdDV is known for causing widespread mortality in common house crickets reared on large farms in Europe and North America [19]. The species Hymenopteran scindoambidensovirus 1 includes Solenopsis invicta densovirus, a pathogen of red imported fire ants (Solenopsis invicta) [38]. The third species, Hemipteran scindoambidensovirus 1, accommodates Planococcus citri densovirus, a pathogen of citrus mealybugs [36].
Genus Protoambidensovirus
Two species have been assigned to this genus, Lepidopteran protoambidensovirus 1 and Dipteran protoambidensovirus 1. Since Galleria mellonella densovirus was the first member of the entire subfamily to be discovered, the genus is designated “Proto” after “first” in Latin. Members of the proposed genus share approximately 50% NS1 amino acid sequence identity with each other and <35% with other densoviruses. Galleria mellonella densovirus and four other densoviruses that infect members of the order Lepidoptera are classified in the species Lepidopteran protoambidensovirus 1, the type species of the genus. The structural and trafficking properties of Galleria mellonella densovirus and Junonia coenia densovirus are relatively well characterized, which is rare among the invertebrate parvoviruses [22, 33]. The species Dipteran protoambidensovirus 1 includes Culex pipiens densovirus, which infects mosquitoes of the species Culex pipiens pallens. This virus is known for its complicated NS splicing pattern, which is an unusual characteristic of members of this subfamily [2].
Genus Hemiambidensovirus
This genus includes two species, Hemipteran hemiambidensovirus 1 and Hemipteran hemiambidensovirus 2, both including viruses that are pathogens of hemipteran hosts, namely aphids. The NS1 amino acid sequence identity within the genus is 62%, whereas it is only 35% with members of other genera. Although members of both species display a split VP ORF, the transcription strategy of these viruses is currently unknown. Both their phylogenetic relationship and the lack of significant sequence similarity suggest that this VP expression strategy probably evolved independently from that of viruses in the genus Scindoambidensovirus. Assigned species include Hemipteran hemiambidensovirus 1, the type species, with one virus, Dysaphis plantaginea densovirus, which infects rosy aphids (Dysaphis plantaginea). This virus is capable of stimulating the aphids to transform into the migratory, winged morph of the species [31]. The other species in this genus, Hemipteran hemiambidensovirus 2, includes Myzus persicae densovirus, which infects green peach aphids (Myzus persicae) [39].
Genus Pefuambidensovirus
This new genus comprises only one species, Blattodean pefuambidensovirus 1, and one virus, Periplaneta fuliginosa densovirus, which infects smoky brown cockroaches (Periplaneta fuliginosa). The NS1 amino acid sequence of this virus has a maximum of 35% identity to that of other members of this subfamily; hence, it can be classified as a member of a monotypic genus on its own. Although its genome also displays a split VP coding gene, there are two spliced transcripts derived from these, encoding both the VP1 and VP2 minor structural proteins [12]. The sequence divergence, phylogenetic relationship, and unique splicing pattern of the structural ORFs suggest that Periplaneta fuliginosa densovirus is the first member of a third lineage of ambisense densoviruses possessing a split VP gene.
Genus Blattambidensovirus
This is another monotypic genus, as its only member is Blattella germanica densovirus 1, species Blattodean blattambidensovirus 1, which infects German cockroaches (Blattella germanica). It shares less than 33% NS1 amino acid sequence identity with any officially classified densoviruses. Its transcription strategy suggests, however, that the genus Blattambidensovirus represent the fourth lineage whose members construct their minor capsid proteins by splicing two VP genes together [15]. Recently, a densovirus-like virus was obtained from the lung tissue of a great tit (Parus major) and proven to be infectious in vertebrate cell lines [46]. Although its host association has yet to be clarified, the NS1 of this virus shares 56% amino acid sequence identity with that of Blattella germanica densovirus, suggesting it is a candidate member of a possible second species in this genus.
The newly established subfamily Hamaparvovirinae comprises the current genera Hepandensovirus, Penstyldensovirus, and Brevidensovirus, together with the formerly unclassified “chapparvoviruses”
Although this subfamily is less well-supported, its members still cluster as a supported monophyletic lineage based on the helicase phylogeny. The 340 aa of their NS1 proteins can be aligned with an average of 30% amino acid sequence identity. In contrast, they only share the helicase domain with other parvoviruses, showing less than 20% amino acid sequence identity, which is limited to the three highly-conserved Walker domains, which are protein motifs with highly conserved three-dimensional structures [14]. Moreover, all members of this proposed subfamily lack the conserved PLA2 domain [47] in their VP proteins.
The name of this new subfamily is “Hamaparvovirinae” to reflect their nature of members infecting both vertebrate and invertebrate hosts, after the ancient Greek word meaning “together”. The following genera, previously members of the subfamily Densovirinae, have been reclassified into the new subfamily Hamaparvovirinae (and renamed accordingly):
Genus Hepanhamaparvovirus
This genus has one species, Decapod hepanhamaparvovirus 1, which is also the type species of its former genus named Hepandensovirus (Fig. 3).

Phylogeny based on the NS1 protein sequence (340 aa) of viruses belonging to the newly established subfamily Hamaparvovirinae. The Bayesian inference and maximum-likelihood topologies were congruent. Viruses and taxa established in the present taxonomy proposal are shown in red and are underlined. The alignment was made as detailed in Fig. 1. The phylogenetic reconstructions were carried out using BEAST v. 1.10.4. as described in Fig. 1 and PhyML v3.3, using the substitution model LG+I+G. Posterior probability support values and bootstrap values for 100 iterations are shown at the nodes
Genus Penstylhamaparvovirus
This genus has one species, Decapod penstylhamaparvovirus 1, which is also the type species of its former genus, named Penstyldensovirus (Fig. 3).
Genus Brevihamaparvovirus
Formerly known as the genus Brevidensovirus, this taxon has two species, Dipteran brevihamaparvovirus 1 and 2, with Dipteran brevihamaparvovirus 1 as the type species.
Previously unclassified parvoviruses that have commonly been referred to as “chapparvoviruses” are assigned to two new genera in the subfamily Hamaparvovirinae. In contrast to the other three genera, NS1 proteins of “chapparvoviruses” share a significant similarity (30-37% identity) over a longer stretch of sequence (approx. 500 aa). Their VP proteins also share detectable similarity, suggesting their origin from a single ancestral capsid protein [25]. Genome organization, phylogeny and identity scores, however, separate “chapparvoviruses” into two distinct groups corresponding to two newly established genera: Ichthamaparvovirus and Chaphamaparvovirus (Fig. 3).
Genus Ichthamaparvovirus
This genus has one species, Syngnathid ichthamaparvovirus 1, which is also the type species. The only virus in this species is Syngnathus scovelli chapparvovirus, which was identified in homogenized gill, muscle and male brood pouch tissue of a gulf pipefish (Syngnathus scovelli). This virus shares approx. 30% NS1 amino acid sequence identity with other chapparvoviruses. In another syngnathid fish, the tiger seahorse (Hippocampus comes), a closely related endogenous viral element (EVE) has been detected with 70% NS1 amino acid sequence identity that also spans the nucleoprotein (np) gene [25]. Syngnathus scovelli chapparvovirus has partially sequenced hairpins, suggesting that the genus is heterotelomeric.
Genus Chaphamaparvovirus
This genus has eight new species, and its members share >37% NS1 amino acid sequence identity. Future detection and characterization of new viruses related to current members of this proposed taxon may eventually result in splitting the genus into more genera. Currently, however, their clustering as a single genus is the only common node characterized by significant topology support by both Bayesian and ML-based inferences (Fig. 3).
The type species of this genus will be Rodent chaphamaparvovirus 1, which includes two viruses (mouse kidney parvovirus and murine chapparvovirus), that share 98.5% NS1 amino acid sequence identity. Mouse kidney parvovirus is the best-characterized chapparvovirus to date. It contains putative telomeres, and its transcription strategy has been elucidated. Mouse kidney parvovirus has been suggested to be associated with chronic kidney infection in immunosuppressed laboratory mice [30]. The published information suggests that mouse kidney parvovirus could be heterotelomeric. Murine chapparvovirus has been shown to be prevalent in both feces and liver tissue of mice in New York City [43].
Another rodent-infecting virus, rat parvovirus 2, is the only known member of the species Rodent chaphamaparvovirus 2. This virus has been found to be prevalent in the feces of wild rats in China [45].
Porcine parvovirus 7, the sole member of the species Ungulate chaphamaparvovirus 1, appears to contain an additional ORF, which probably codes for a non-structural protein analogous to those of other amniote vertebrate-infecting “chapparvoviruses” [25]. This virus shares approx. 37% NS1 amino acid sequence identity with chaphamaparvoviruses and has been abundantly detected in feces and rectal swabs from diarrheic piglets and young pigs on 41 different occasions. There has been one coding-complete sequence published to date [24].
The species Chiropteran chaphamaparvovirus 1 includes one virus, Desmodus rotondus chapparvovirus, which was identified in the kidney tissue of a common vampire bat (Desmodus rotondus) [35].
The species Carnivore chaphamaparvovirus 1 includes two viruses, both of which were derived from dogs with diarrhea, namely cachavirus 1A and cachavirus 1B [11].
The species Galliform chaphamaparvovirus 1 includes a single virus, turkey parvovirus 2, which was detected in the feces of domestic turkeys with high prevalence [29].
The species Galliform chaphamaparvovirus 2 will include chicken chapparvovirus 2, which was obtained from the intestines of broiler chickens, where it could not be associated with clinical signs despite its high prevalence [17].
The species Galliform chaphamaparvovirus 3 has two potential members, namely, chicken chapparvovirus HK and chicken chapparvovirus 1. Chicken chapparvovirus 1 is only available as a partial sequence with a complete NS1. It was identified during the same study as chicken chapparvovirus 2, having 75% NS1 amino acid sequence identity [17]. Chicken chapparvovirus HK has a fully determined coding sequence and its NS1 shares 99% identity with that of chicken chapparvovirus 1.
Creating two new genera in the subfamily Parvovirinae
Since 2011, two parvoviruses that could not be assigned to any of the previously existing genera have been characterized. Moreover, the previously valid virus classification criteria made it impossible for one of these two unique vertebrate-infecting parvoviruses to be classified. However, using the updated, newly established criteria, both are eligible for classification, allowing the creation of two new genera in the subfamily Parvovirinae (Fig. 4).

Phylogeny based on the complete NS1 protein sequence of viruses belonging to the subfamily Parvovirinae (460 aa). The Bayesian and maximum-likelihood-inferred topologies were congruent. The alignment was made as detailed in Fig. 1. The phylogenetic reconstructions were carried out using BEAST v. 1.10.4 as described in Fig. 1 and PhyML v3.3. Posterior probability support and bootstrap values are shown at the nodes. The names of the newly classified viruses and taxa established in the 2019 taxonomy proposal are in red and underlined
Genus Artiparvovirus
Genus Artiparvovirus will initially be monotypic, comprising a single species, Chiropteran artiparvovirus 1, including one virus, Artibeus jamaicensis parvovirus, which was reported in leaf-nosed fruit bats in Panama [5]. The complete genome sequence of this parvovirus has been determined, and its NS1 shares 38% amino acid sequence identity with other parvoviral NS1s in the GenBank database. This, as well as its divergent phylogenetic position (Fig. 4), justifies its classification in its own monotypic genus.
Genus Loriparvovirus
The genus Loriparvovirus, another newly-established monotypic genus in the subfamily Parvovirinae, comprises one species, Primate loriparvovirus 1. Slow loris parvovirus, the only parvovirus to be assigned to this species, was detected in a slow loris (Nycticebus coucang) with diffuse histiocytic sarcoma. However, despite its persistence over many years, it is still unclear if this virus is oncogenic or oncolytic [6]. The complete genome of this virus has been sequenced, and its NS1 protein shares 38% amino acid sequence identity with that of other parvoviruses. Both its identity values and its phylogenetic relationship qualify slow loris parvovirus to be classified in a separate genus within the subfamily Parvovirinae (Fig. 4).
Assigning previously unclassified parvoviruses into already established genera of the subfamily Parvovirinae
Establishing a new species within the genus Amdoparvovirus
We will assign red panda amdoparvovirus to the new species Carnivore amdoparvovirus 5 in the genus Amdoparvovirus. This virus has been detected in both tissue and feces samples of endangered red pandas (Ailurus fulgens) and clusters with members of the genus Amdoparvovirus, which are already-established carnivore-infecting parvoviruses [1]. Red panda amdoparvovirus NS1 shares approximately 75% sequence identity with that of other amdoparvoviruses.
Establishing a new species within the genus Aveparvovirus
Red-crowned crane parvovirus is assigned into the new species Gruiform aveparvovirus 1 in the genus Aveparvovirus, where it robustly clusters with the members of a single currently recognized species, Galliform aveparvovirus 1. Its NS1 protein sequence shares 57-58% identity with that of previously assigned members of the genus Aveparvovirus. Red-crowned crane parvovirus was discovered in a study of the fecal virome of highly endangered red-crowned cranes (Grus japonensis) in China [42].
Establishing four new species within the genus Bocaparvovirus
Four parvoviruses that cluster robustly with previously recognized bocaparvoviruses have been assigned to four new species in the genus Bocaparvovirus.
Dromedary camel bocaparvovirus 1, which was identified in the feces of multiple dromedary camels (Camelus dromedaries) in Dubai [44], is the founding member of the species Ungulate bocaparvovirus 7. Its NS1 shares 40-60% amino acid sequence identity to other bocaparvovirus NS1 proteins.
Dromedary camel bocaparvovirus 2 is assigned to the species Ungulate bocaparvovirus 8, as the NS1 protein of this virus shares 41% amino acid sequence identity with that of other bocaparvoviruses, including dromedary camel bocaparvovirus 1. Like dromedary camel bocaparvovirus 1, this virus has been detected in the feces of multiple dromedary camels in Dubai [44].
Rat bocavirus is assigned to the species Rodent bocaparvovirus 1, as the NS1 protein of this virus shares 46-56% amino acid sequence identity with those of other bocaparvoviruses. The virus has been detected in multiple organs and feces of Norwegian rats (Rattus norvegicus) in China [16].
Murine bocavirus is assigned to the species Rodent bocaparvovirus 2, as the murine bocavirus NS1 shares 48-55% amino acid sequence identity with its counterparts in other bocaparvoviruses. This virus was detected in house mice of New York City, often simultaneously with murine chapparvovirus [43].
Establishing five new species within the genus Copiparvovirus
The following parvoviruses have been assigned to the genus Copiparvovirus, as they all cluster with the two recognized copiparvoviruses as a robustly supported monophyletic group (Fig. 4).
The species Ungulate copiparvovirus 3 includes roe deer copiparvovirus. The complete genome of this virus has been sequenced from deer ticks (Ixodes ricinus) feeding on a roe deer and from serum samples from the deer (Capreolus capreolus) [18]. Its NS1 protein shares 40-48% amino acid sequence identity with those of viruses of established copiparvovirus species and clusters reliably with members of the genus.
Porcine parvovirus 6 is assigned to the species Ungulate copiparvovirus 4, as its complete genome has been characterized and pathology has been associated with aborted porcine fetuses. Its NS1 shares 40-58% amino acid sequence identity with those of other copiparvoviruses.
Bosavirus is assigned to the species Ungulate copiparvovirus 5. Its NS1 shares 40% amino acid sequence identity with those of other copiparvoviruses and has been detected frequently in the calf serum virome [32].
Equine hepatitis parvovirus is assigned to the species Ungulate copiparvovirus 6, as its complete genome has been characterized and the virus has been associated with severe pathology in horses [10]. Its NS1 shares 36-39% amino acid sequence identity with that of other copiparvoviruses and the organization of its viral genome is remarkably copiparvovirus-like, with a length of >5 kb and a cap gene capable of encoding a VP1 longer than 900 aa. Furthermore, phylogenetic inference clearly indicates that this virus clusters with other copiparvoviruses. Hence, assigning it into another genus would mean introducing paraphyly, or splitting the already established two species of this genus apart from each other (Fig. 4).
Sesavirus is assigned to the species Pinniped copiparvovirus 1. This virus has been detected only once—in a California sea lion pup (Zalophus californianus)—and its complete coding sequence has been determined [28]. Its NS1 shares 35% amino acid sequence identity with those of other copiparvoviruses, and its genome organization is remarkably copiparvovirus-like, with a genome length of >5 kb and a cap gene capable of encoding a VP longer than 900 aa. Moreover, it does not show more than 30% NS1 amino acid sequence identity to any other Parvovirinae members outside of the genus Copiparvovirus. Sesavirus will be the first non-ungulate copiparvovirus, which may provide another explanation for its divergent nature relative to other copiparvoviruses, which exclusively infect unglulates (Fig. 4).
Establishing two new species within the genus Dependoparvovirus
Two adeno-associated rodent parvoviruses, namely, murine adeno-associated virus 1 and 2, have been assigned to two new species in the genus Dependoparvovirus, Rodent dependoparvovirus 1 and 2, respectively. The large Rep proteins of both viruses share 50–57% identity with dependoparvoviral Rep proteins (equivalent of NS1). These two murine adeno-associated viruses, however, share 75% sequence identity in this protein. The phylogenetic position of both viruses and their identity scores suggest that they are divergent enough to represent two distinct new species. Both viruses were derived from mice living around New York City, often in coinfection with murine chapparvovirus and murine bocavirus, as mentioned above [43].
Establishing a new species within the genus Erythroparvovirus
Seal parvovirus was identified in the brain tissue of a stranded harbor seal (Phoca vitulina), and its complete coding sequence was determined [4]. Its NS1 protein sequence shares 36% amino acid sequence identity with those of other erythroparvoviruses, and only 30% with parvoviruses from other genera. Despite this low level of sequence identity, we still decided to assign this parvovirus to a species called Pinniped erythroparvovirus 1 in the genus Erythroparvovirus. This affiliation is supported by the phylogenetic position of seal parvovirus, as well as by the presence of a small ORF that overlaps the N-terminal coding region of VP1, which appears to be homologous to the X protein of human parvovirus B19 (Fig. 4).
Creation of two new species in the genus Protoparvovirus
We have introduced two new species into this genus.
Tusavirus was detected in the diarrheic stool of a child in Tunisia and characterized [27]. Later, anti-tusavirus antibodies were found in the serum of two more patients [40, 41]. Its NS1 shares 48% amino acid sequence identity with that of other protoparvoviruses, and it also clusters with these viruses in a phylogenetic tree (Fig. 4). Although its natural host species remains uncertain, tusavirus will be assigned to the species Primate protoparvovirus 4 in the genus Protoparvovirus.
Sea otter parvovirus clusters with protoparvoviruses designated as “bufaviruses” and its NS1 shares 45-65% amino acid sequence identity with those of other protoparvoviruses (Fig. 4). This virus was derived from stranded sea otters (Enhydra lutris) in coinfection with various other DNA viruses [34]. This virus will be assigned to the new species Carnivore protoparvovirus 2 in the genus Protoparvovirus.
Conclusions
With the introduction of the new classification system, as well as new virus and taxon definitions, we will be able to classify 30 previously unclassified parvoviruses into 29 new species, with 11 of these being placed into newly-established genera. Moreover, with this proposal we have also addressed long-standing issues of parvovirus taxonomy, such as the paraphyly of the genus Ambidensovirus. The family Parvoviridae is a diverse family of animal-infecting viruses with a host range extending from the invertebrate phyla that appeared for the first time in the early Cambrian period to modern vertebrates, including humans. The system proposed here was conceived with the aim of embracing this diversity. Hence, for the first time, we decided to classify parvoviruses based on common characteristics and phylogenetic relationships instead of solely on host association. Although the introduction of the subfamily Hamaparvovirinae provides a solution for accommodating divergent densoviruses together with vertebrate-infecting parvoviruses that evolved independently from members of the subfamily Parvovirinae, we have to point out that this new subfamily is still extremely heterogeneous. The discovery of “chapparvoviruses” revealed the existence of vertebrate parvoviruses that are more closely related to invertebrate-infecting parvoviruses than to members of the Parvovirinae, but this finding only emerged after decades of research in the field. This clearly illustrates that now we are only just beginning to scratch the surface of parvovirus diversity. Our proposal, nevertheless, is a first step toward introducing a new mindset in parvovirology that will be necessary to cope with the classification of both vertebrate- and invertebrate-infecting parvoviruses that are yet to be discovered.
References
Alex CE, Kubiski SV, Li L, Sadeghi M, Wack RF, McCarthy MA, Pesavento JB, Delwart E, Pesavento PA (2018) Amdoparvovirus infection in red pandas (Ailurus fulgens). Vet Pathol 55:552–561
Baquerizo-Audiot E, Abd-Alla A, Jousset FX, Cousserans F, Tijssen P, Bergoin M (2009) Structure and expession strategy of the genome of Culex pipiens densovirus, a mosquito densovirus with an ambisense organization. J Virol 83:6863–6873
Bochow S, Condon K, Elliman J, Owens L (2015) First complete genome of an Ambidensovirus; Cherax quadricarinatus densovirus, from freshwater crayfish Cherax quadricarinatus. Mar Genomics 3:305–312
Bodewes R, Rubio García A, Wiersma LC, Getu S, Beukers M, Schapendonk CM, van Run PR, van de Bildt MW, Poen MJ, Osinga N, Sánchez Contreras GJ, Kuiken T, Smits SL, Osterhaus AD (2013) Novel B19-like parvovirus in the brain of a harbor seal. PLoS ONE 5:79259
Canuti M, Eis-Huebinger AM, Deijs M, de Vries M, Drexler JF, Oppong SK, Müller MA, Klose SM, Wellinghausen N, Cottontail VM, Kalko EK, Drosten C, van der Hoek L (2011) Two novel parvoviruses in frugivorous New and Old World bats. PLoS ONE 6:29140
Canuti M, Williams CV, Gadi SR, Jebbink MF, Oude Munnink BB, Jazaeri Farsani SM, Cullen JM, van der Hoek L (2014) Persistent viremia by a novel parvovirus in a slow loris (Nycticebus coucang) with diffuse histiocytic sarcoma. Front Microbiol 1:655
Cotmore SF, Tattersall P (2005) A rolling-hairpin strategy: basic mechanisms of DNA replication in the parvoviruses. In: Kerr J, Cotmore SF, Bloom ME, Linden RM, Parrish CR (eds) Parvoviruses. Hodder Arnold, London, pp 171–181
Cotmore SF, Agbandje-McKenna M, Chiorini JA, Mukha DV, Pintel DJ, Qiu J, Soderlund-Venermo M, Tattersall P, Tijssen P, Gatherer D (2014) The family Parvoviridae. Arch Virol 159:1239–1247
Cotmore SF, Agbandje-McKenna M, Canuti M, Chiorini JA, Eis-Hubinger AM, Hughes J, Mietzsch M, Modha S, Ogliastro M, Penzes JJ, Pintel DJ, Qiu J, Soderlund-Venermo M, Tattersall P, Tijssen P (2019) ICTV virus taxonomy profile: Parvoviridae. J Gen Virol 100:367–368
Divers TJ, Tennant BC, Kumar A, McDonough S, Cullen J, Bhuva N, Jain K, Chauhan LS, Scheel TKH, Lipkin WI, Laverack M, Trivedi S, Srinivasa S, Beard L, Rice CM, Burbelo PD, Renshaw RW, Dubovi E, Kapoor A (2018) New parvovirus associated with serum hepatitis in horses after inoculation of common biological product. Emerg Infect Dis 24:303–310
Fahsbender E, Altan E, Seguin MA, Young P, Estrada M, Leutenegger C, Delwart E (2019) Chapparvovirus DNA found in 4% of dogs with diarrhea. Viruses 27:398
Guo H, Zhang J, Hu Y (2000) Complete sequence and organization of Periplaneta fuliginosa densovirus genome. Acta Virol 44:315–322
Hewson I, Button JB, Gudenkauf BM, Miner B, Newton AL, Gaydos JK, Wynne J, Groves CL, Hendler G, Murray M, Fradkin S, Breitbart M, Fahsbender E, Lafferty KD, Kilpatrick AM, Miner CM, Raimondi P, Lahner L, Friedman CS, Daniels S, Haulena M, Marliave J, Burge CA, Eisenlord ME, Harvell CD (2014) Densovirus associated with sea-star wasting disease and mass mortality. PNAS 111:17278–17283
Hickman AB, Dyda F (2005) Binding and unwinding: SF3 viral helicases. Curr Opin Struct Biol 15:77–85
Kapelinskaya TV, Martynova EU, Schal C, Mukha DV (2011) Expression strategy of densonucleosis virus from the German cockroach, Blattella germanica. J Virol 85:11855–11870
Lau SK, Yeung HC, Li KS, Lam CS, Cai JP, Yuen MC, Wang M, Zheng BJ, Woo PC, Yuen KY (2017) Identification and genomic characterization of a novel rat bocavirus from brown rats in China. Infect Genet Evol 47:68–76
Lima DA, Cibulski SP, Tochetto C, Varela APM, Finkler F, Teixeira TF, Loiko MR, Cerva C, Junqueira DM, Mayer FQ, Roehe PM (2019) The intestinal virome of malabsorption syndrome-affected and unaffected broilers through shotgun metagenomics. Virus Res 261:9–20
Linden A, Gilliaux G, Paternostre J, Benzarti E, Rivas JF, Desmecht D, Garigliany M (2019) A novel parvovirus, roe deer copiparvovirus, identified in Ixodes ricinus ticks. Virus Genes 55:425–428
Liu K, Li Y, Jousset FX, Zadori Z, Szelei J, Yu Q, Pham HT, Lépine F, Bergoin M, Tijssen P (2011) The Acheta domesticus densovirus, isolated from the European house cricket, has evolved an expression strategy unique among parvoviruses. J Virol 85:10069–10078
Meynadier G, Vago C, Plantevi G, Atger P (1964) Virose d’un type inhabituel chez le lépidoptère Galleria mellonella L. Rev Zoo Agric Appl 63:207–209
Mietzsch M, Pénzes JJ, Agbandje-McKenna M (2019) Twenty-five years of structural parvovirology. Viruses 11:362
Multeau C, Froissart R, Perrin A, Castelli I, Casartelli M, Ogliastro M (2012) Four amino acids of an insect densovirus capsid determine midgut tropism and virulence. J Virol 86:5937–5941
Nüesch JP, Tattersall P (1993) Nuclear targeting of the parvoviral replicator molecule NS1: evidence for self-association prior to nuclear transport. Virology 196:637–651
Palinski RM, Mitra N, Hause BM (2016) Discovery of a novel Parvovirinae virus, porcine parvovirus 7, by metagenomic sequencing of porcine rectal swabs. Virus Genes 52:564–567
Pénzes JJ, de Souza WM, Agbandje-McKenna M, Gifford RJ (2019) An ancient lineage of highly divergent parvoviruses infects both vertebrate and invertebrate hosts. Viruses 11:525
Pham HT, Yu Q, Bergoin M, Tijssen P (2013) A novel ambisense densovirus, Acheta domesticus mini ambidensovirus, from crickets. Genome Announc 7:1
Phan TG, Sdiri-Loulizi K, Aouni M, Ambert-Balay K, Pothier P, Deng X, Delwart E (2014) New parvovirus in child with unexplained diarrhea, Tunisia. Emerg Infect Dis 20:1911–1913
Phan TG, Gulland F, Simeone C, Deng X, Delwart E (2015) Sesavirus: prototype of a new parvovirus genus in feces of a sea lion. Virus Genes 50:134–136
Reuter G, Boros Á, Delwart E, Pankovics P (2014) Novel circular single-stranded DNA virus from turkey faeces. Arch Virol 159:2161–2164
Roediger B, Lee Q, Tikoo S, Cobbin JC, Henderson JM, Jormakka M, O’Rourke MB, Padula MP, Pinello N, Henry M et al (2018) An atypical parvovirus drives chronic tubulointerstitial nephropathy and kidney fibrosis. Cell 175:530–543
Ryabov EV, Keane G, Naish N, Evered C, Winstanley D (2009) Densovirus induces winged morphs in asexual clones of the rosy apple aphid, Dysaphis plantaginea. PNAS 106:8465–8470
Sadeghi M, Kapusinszky B, Yugo DM, Phan TG, Deng X, Kanevsky I, Opriessnig T, Woolums AR, Hurley DJ, Meng XJ, Delwart E (2017) Virome of US bovine calf serum. Biologicals 46:64–67
Simpson AA, Chipman PR, Baker TS, Tijssen P, Rossmann MG (1998) The structure of an insect parvovirus (Galleria mellonella densovirus) at 3.7 A resolution. Structure 6:1355–1367
Siqueira JD, Ng TF, Miller M, Li L, Deng X, Dodd E, Batac F, Delwart E (2017) Endemic infection of stranded southern sea otters (Enhydra lutris nereis) with novel parvovirus, poolyomavirus, and adenovirus. J Wildl Dis 53:532–542
Souza WM, Romeiro MF, Fumagalli MJ, Modha S, de Araujo J, Queiroz LH, Durigon EL, Figueiredo LT, Murcia PR, Gifford RJ (2017) Chapparvoviruses occur in at least three vertebrate classes and have a broad biogeographic distribution. J Gen Virol 98:225–229
Thao ML, Wineriter S, Buckingham G, Baumann P (2001) Genetic characterization of a putative Densovirus from the mealybug Planococcus citri. Curr Microbiol 43:457–458
Tijssen P, Pénzes JJ, Yu Q, Pham HT, Bergoin M (2016) Diversity of small, single-stranded DNA viruses of invertebrates and their chaotic evolutionary past. J Invertebr Pathol 140:83–96
Valles SM, Shoemaker D, Wurm Y, Strong CA, Varone L, Becnel JJ, Shirk PD (2013) Discovery and molecular characterization of an ambisense densovirus from South American populations of Solenopsis invicta. Biol Control 67:431–439
van Munster M, Dullemans AM, Verbeek M, van den Heuvel JF, Reinbold C, Brault V, Clérivet A, van der Wilk F (2003) A new virus infecting Myzus persicae has a genome organization similar to the species of the genus Densovirus. J Gen Virol 84:165–172
Väisänen E, Paloniemi M, Kuisma I, Lithovius V, Kumar A, Franssila R, Ahmed K, Delwart E, Vesikari T, Hedman K, Söderlund-Venermo M (2016) Epidemiology of two human protoparvoviruses, bufavirus and tusavirus. Sci Rep 6:39267
Väisänen E, Fu Y, Koskenmies S, Fyhrquist N, Wang Y, Keinonen A, Mäkisalo H, Väkevä L, Pitkänen S, Ranki A, Hedman K, Söderlund-Venermo M (2019) Cutavirus DNA in malignant and nonmalignant akin of cutaneous T-cell lymphoma and organ transplant patients but not of healthy adults. Clin Infect Dis 68:1904–1910
Wang Y, Yang S, Liu D, Zhou C, Li W, Lin Y, Wang X, Shen Q, Wang H, Li C, Zong M, Ding Y, Song Q, Deng X, Qi D, Zhang W, Delwart E (2019) The fecal virome of red-crowned cranes. Arch Virol 164:3–16
Williams SH, Che X, Garcia JA, Klena JD, Lee B, Muller D, Ulrich W, Corrigan RM, Nichol S, Jain K, Lipkin WI (2018) Viral diversity of house mice in New York city. MBio 9:01354
Woo PCY, Lau SKP, Tsoi HW, Patteril NG, Yeung HC, Joseph S, Wong EYM, Muhammed R, Chow FWN, Wernery U, Yuen KY (2017) Two novel dromedary camel bocaparvoviruses from dromedaries in the Middle East with unique genomic features. J Gen Virol 98:1349–1359
Yang S, Liu Z, Wang Y, Li W, Fu X, Lin Y, Shen Q, Wang X, Wang H, Zhang W (2016) A novel rodent Chapparvovirus in feces of wild rats. Virol J 29:133
Yang WT, Shi SH, Jiang YL, Zhao L, Chen HL, Huang KY, Yang GL, Wang CF (2016) Genetic characterization of a densovirus isolated from great tit (Parus major) in China. Infect Genet Evol 41:107–112
Zádori Z, Szelei J, Lacoste MC, Li Y, Gariépy S, Raymond P, Allaire M, Nabi IR, Tijssen P (2001) A viral phospholipase A2 is required for parvovirus infectivity. Dev Cell 1:291–302
Acknowledgements
MSV is funded by the Sigrid Jusélius Foundation and the Medical Foundation of Life and Health; MC is supported by funding from the Joint Mink Research Committee; JH is funded by Medical Research Council, grant number MC_UU_12014/12; BH is funded by the Hungarian Scientific Research Fund (OTKA NN128309). MSV is funded by the Sigrid Jusélius Foundation and the Medical Foundation of Life and Health; MC is supported by funding from the Joint Mink Research Committee; JH is funded by Medical Research Council, Grant number MC_UU_12014/12; BH is funded by the Hungarian Scientific Research Fund (OTKA NN128309).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Handling Editor: Sead Sabanadzovic.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pénzes, J.J., Söderlund-Venermo, M., Canuti, M. et al. Reorganizing the family Parvoviridae: a revised taxonomy independent of the canonical approach based on host association. Arch Virol 165, 2133–2146 (2020). https://doi.org/10.1007/s00705-020-04632-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-020-04632-4