The Biochemical Profile of Post-Mortem Brain from People Who Suffered from Epilepsy Reveals Novel Insights into the Etiopathogenesis of the Disease


 , and
, and 
Abstract
:
1. Introduction
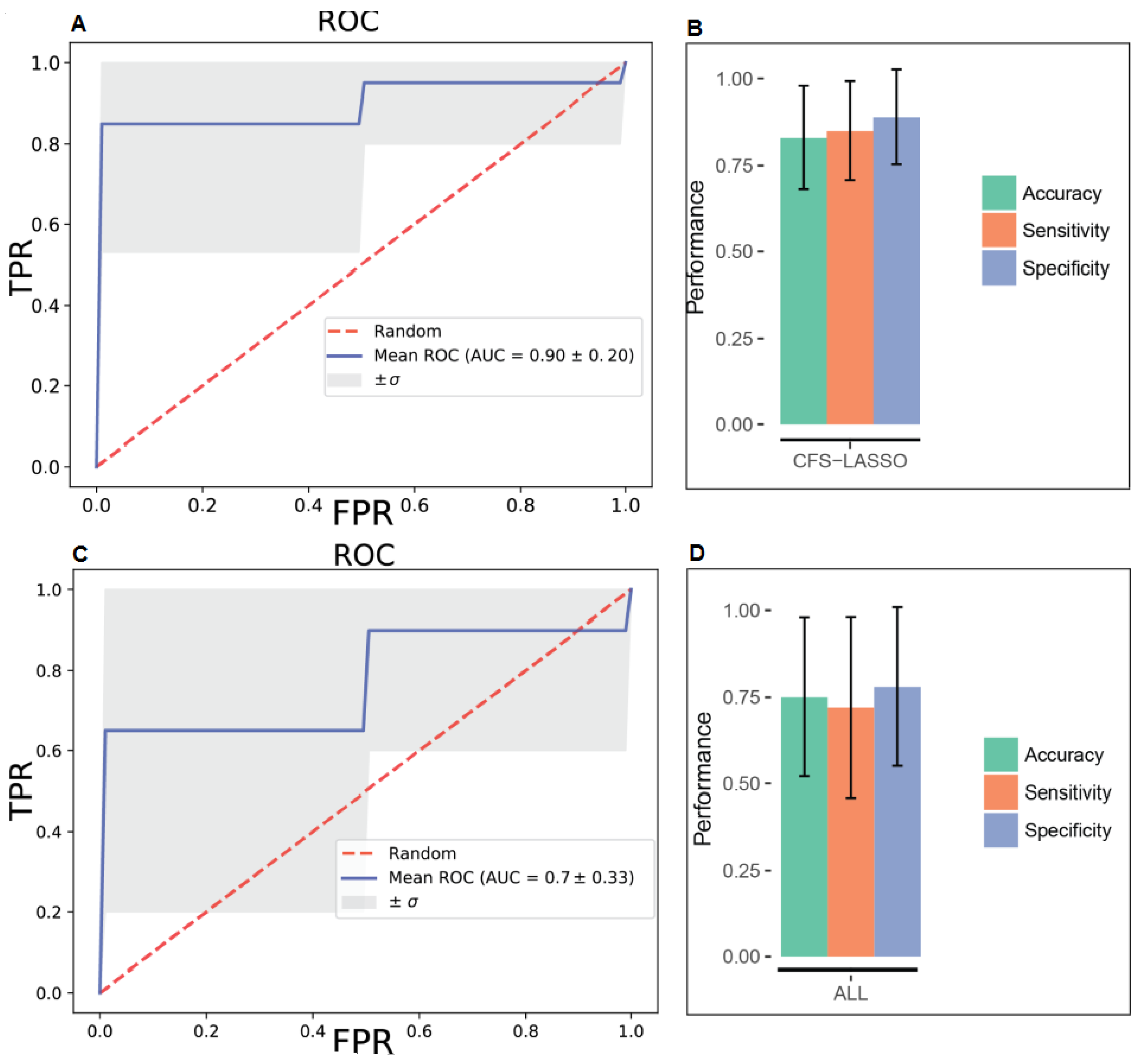
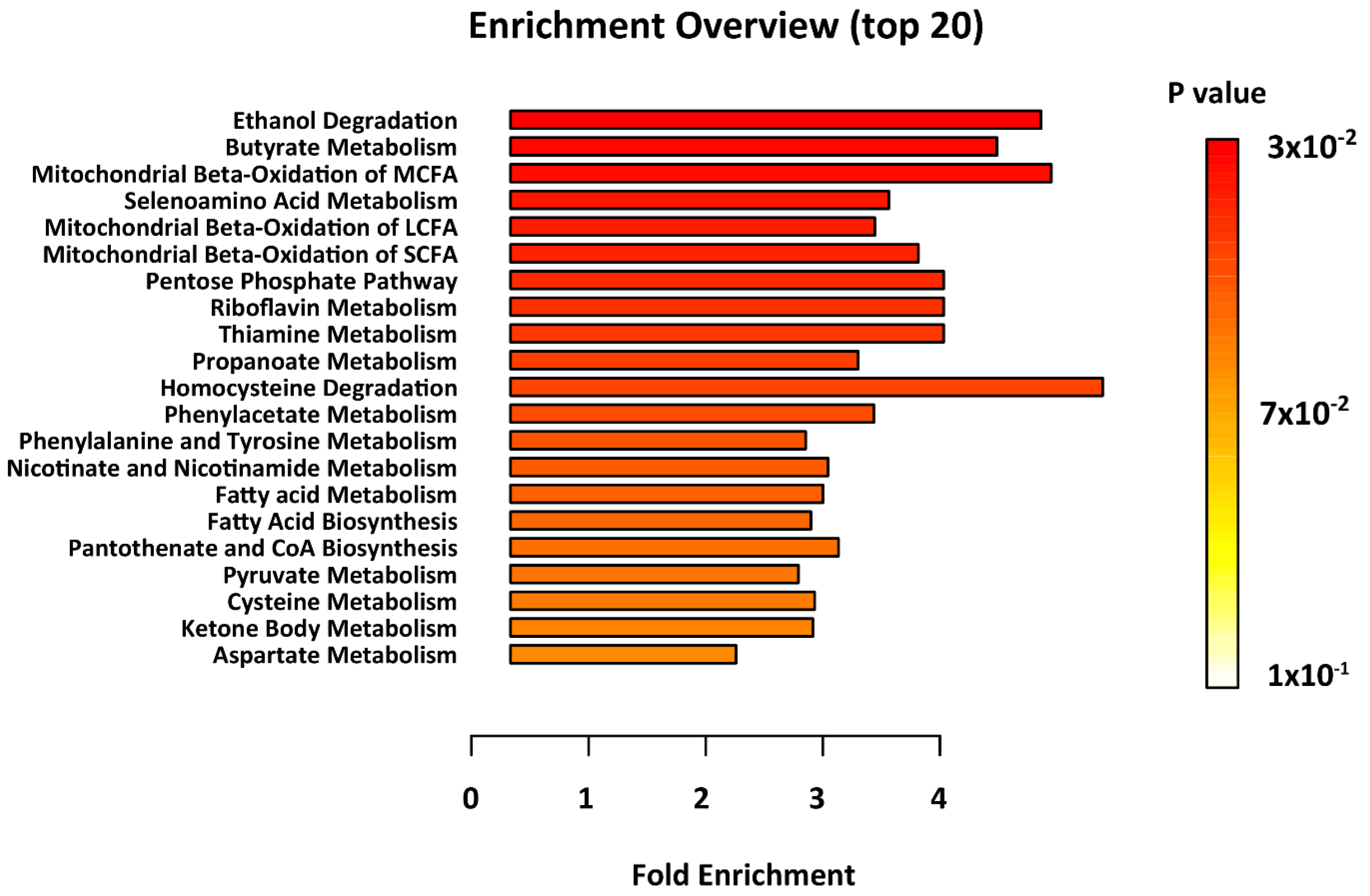
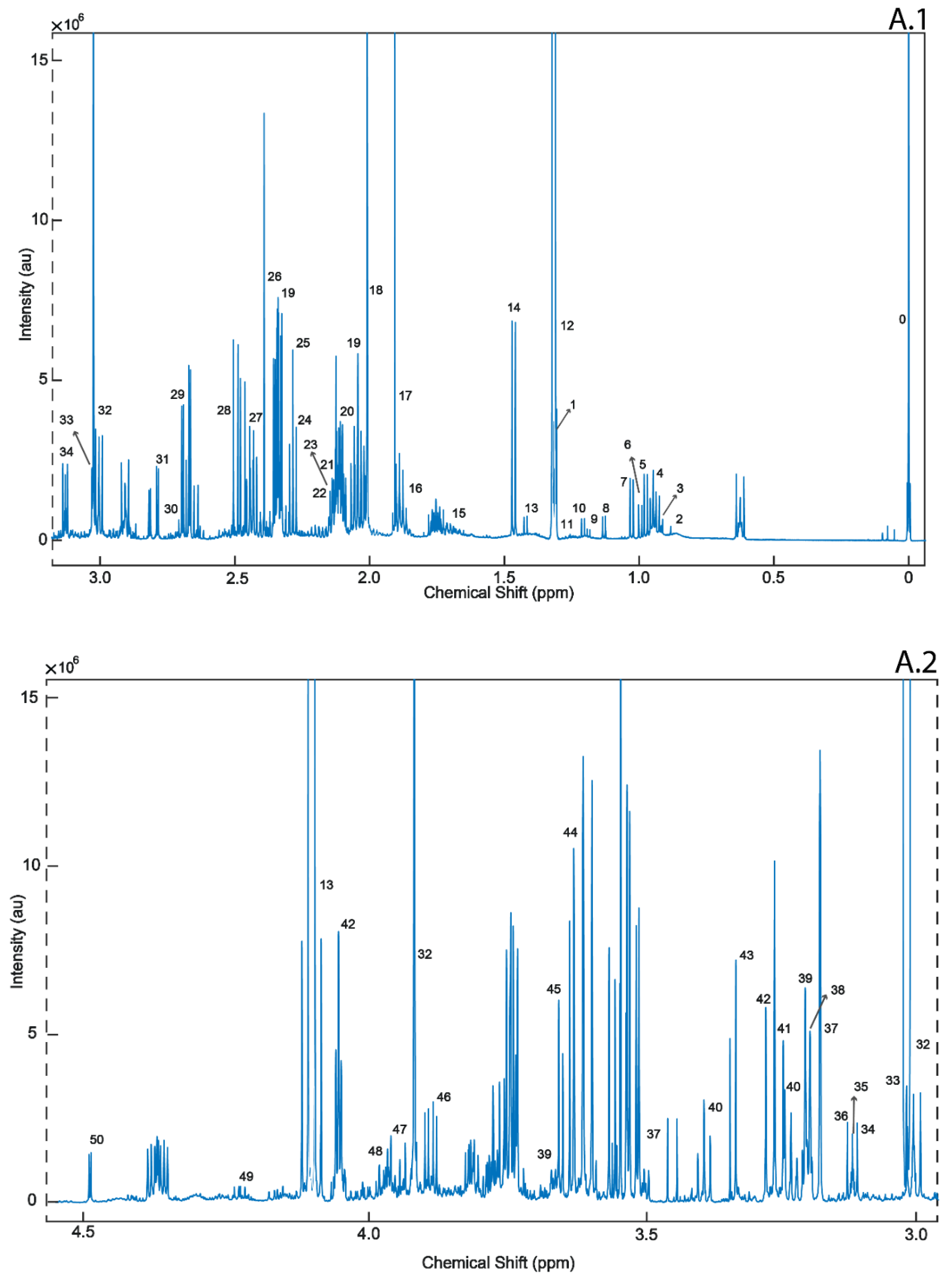
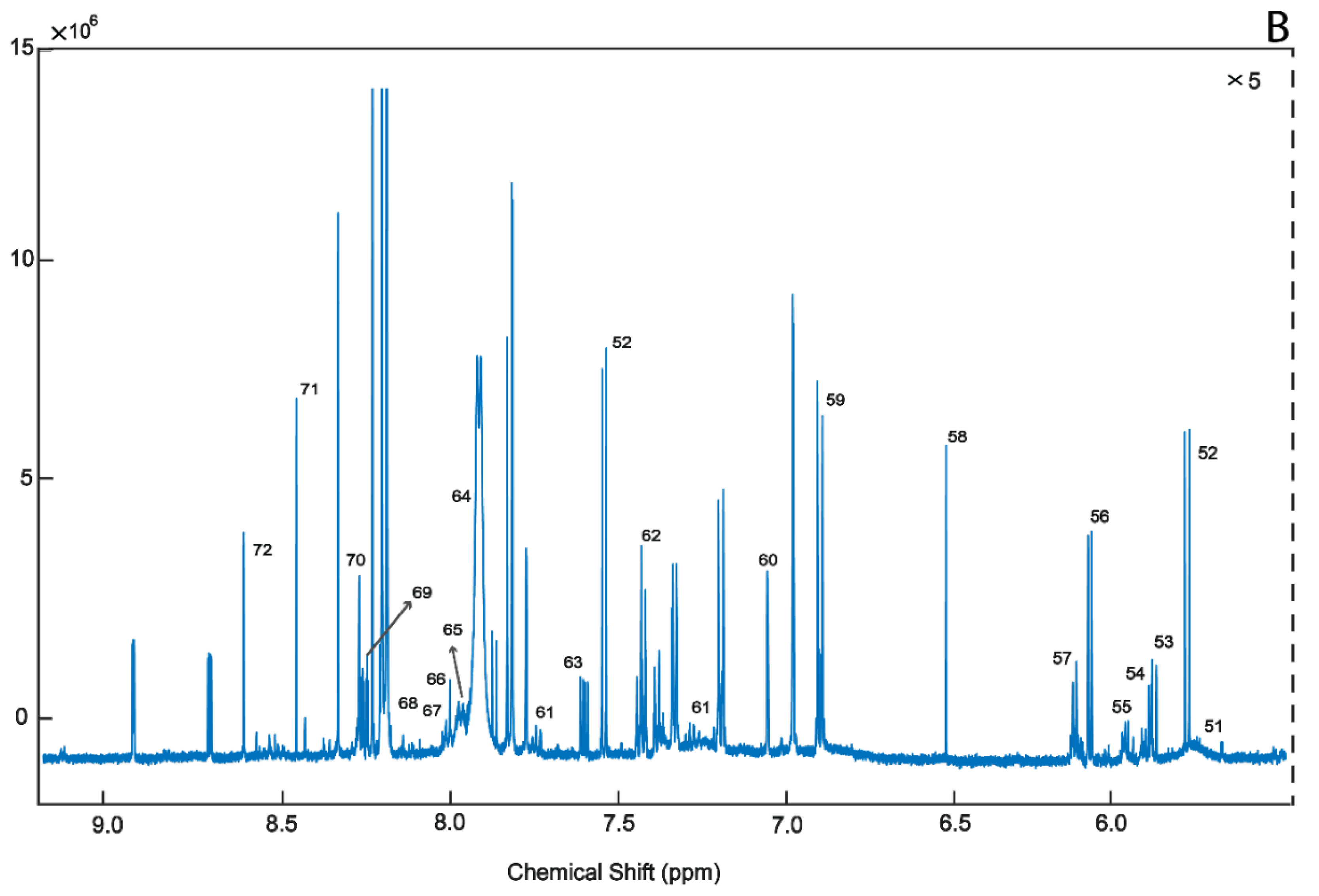
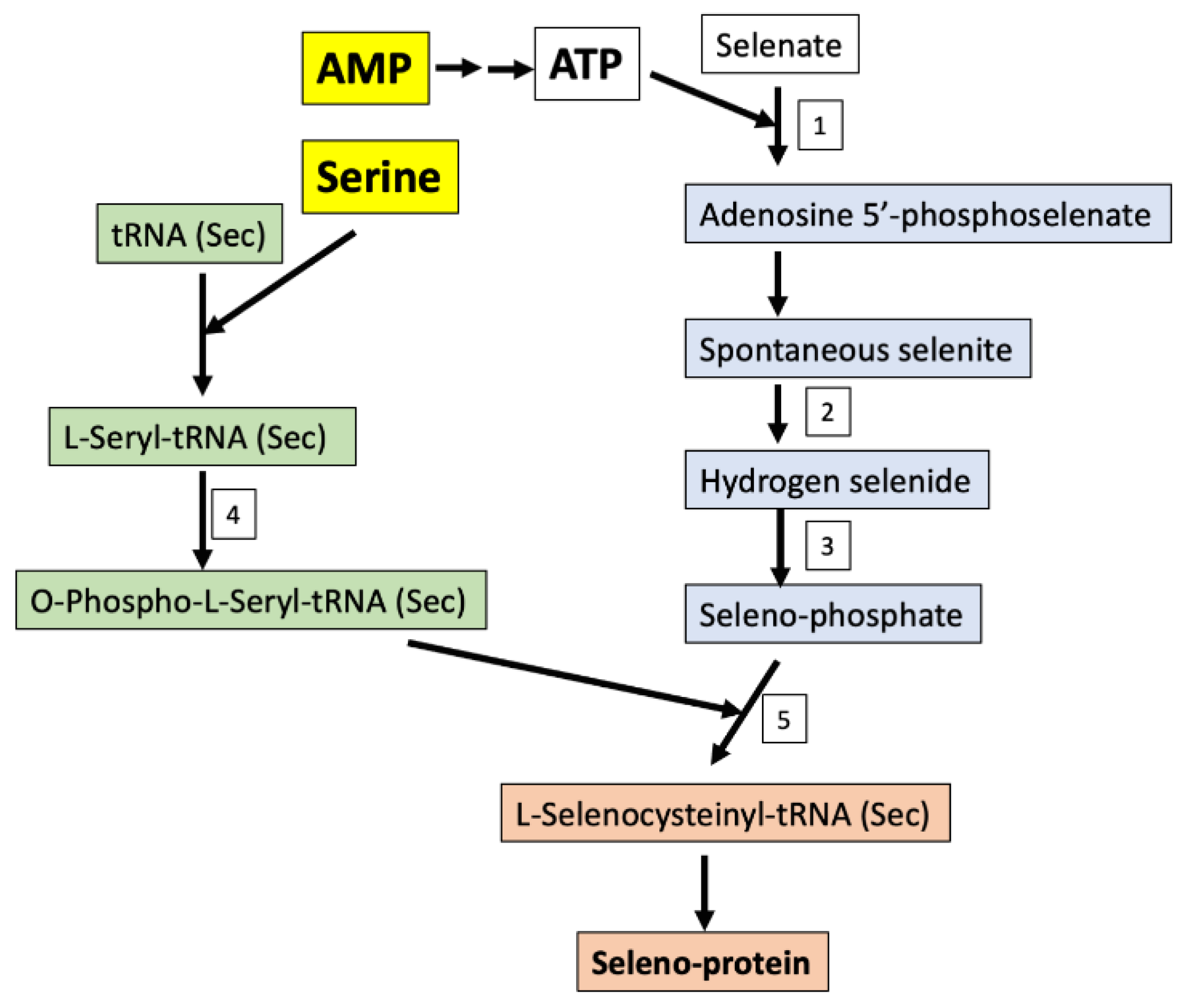
2. Results
3. Discussion
4. Materials and Methods
4.1. Tissue Samples
4.2. Sample Preparation
4.2.1. 1H-NMR Sample Preparation
4.2.2. DI/LC-MS/MS Sample Preparation
4.3. Data Collection and Metabolic Profiling
4.3.1. 1H-NMR Analysis
4.3.2. DI/LC-MS/MS Analysis
4.4. Statistical Analysis
4.4.1. Data Preprocessing
4.4.2. Univariate Analysis
4.4.3. Feature Selection
4.4.4. Predictive Models with Support Vector Machines
4.4.5. Model Evaluation
4.5. Metabolites Pathways Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Sirven, J.I. Epilepsy: A spectrum disorder. Cold Spring Harb. Perspect. Med. 2015, 5, a022848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- England, M.J.; Liverman, C.T.; Schultz, A.M.; Strawbridge, L.M. Summary: A reprint from epilepsy across the spectrum: Promoting health and understanding. Epilepsy Curr. 2012, 12, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Begley, C.E.; Durgin, T.L. The direct cost of epilepsy in the United States: A systematic review of estimates. Epilepsia 2015, 56, 1376–1387. [Google Scholar] [CrossRef] [PubMed]
- Roffman, J.L.; Stern, T.A. A complex presentation of complex partial seizures. Prim. Care Companion J. Clin. Psychiatry 2006, 8, 98–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J., Jr.; Forsgren, L.; French, J.A.; Glynn, M.; et al. Ilae official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.W.; Williamson, A.; Cavus, I.; Hetherington, H.P.; Zaveri, H.; Petroff, O.A.; Spencer, D.D. Neurometabolism in human epilepsy. Epilepsia 2008, 49 (Suppl. 3), 31–41. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Shen, Y.; Chen, H.; Yuan, Z.; Mao, S.; Gao, F. Clinical and molecular analysis of epilepsy-related genes in patients with dravet syndrome. Medicine 2018, 97, e13565. [Google Scholar] [CrossRef] [PubMed]
- Berkovic, S.F. Genetics of epilepsy in clinical practice. Epilepsy Curr. 2015, 15, 192–196. [Google Scholar] [CrossRef] [Green Version]
- Steinlein, O.K. Genetics and epilepsy. Dialogues Clin. Neurosci. 2008, 10, 29–38. [Google Scholar]
- Vadlamudi, L.; Milne, R.L.; Lawrence, K.; Heron, S.E.; Eckhaus, J.; Keay, D.; Connellan, M.; Torn-Broers, Y.; Howell, R.A.; Mulley, J.C.; et al. Genetics of epilepsy: The testimony of twins in the molecular era. Neurology 2014, 83, 1042–1048. [Google Scholar] [CrossRef] [Green Version]
- Clossen, B.L.; Reddy, D.S. Novel therapeutic approaches for disease-modification of epileptogenesis for curing epilepsy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1519–1538. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, M.M. Overview of drugs used for epilepsy and seizures: Etiology, diagnosis, and treatment. Pharm. Ther. 2010, 35, 392–415. [Google Scholar]
- Berg, A.T.; Levy, S.R.; Testa, F.M. Evolution and course of early life developmental encephalopathic epilepsies: Focus on lennox-gastaut syndrome. Epilepsia 2018, 59, 2096–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, S.D.; Younus, I.; Sridhar, V.; Reddy, D.S. Neuroimaging biomarkers of experimental epileptogenesis and refractory epilepsy. Int. J. Mol. Sci. 2019, 20, 220. [Google Scholar] [CrossRef] [Green Version]
- De Aguiar, K.; Franca, F.M.; Barbosa, V.C.; Teixeira, C.A. Early detection of epilepsy seizures based on a weightless neural network. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2015, 2015, 4470–4474. [Google Scholar] [PubMed]
- Jouny, C.C.; Franaszczuk, P.J.; Bergey, G.K. Improving early seizure detection. Epilepsy Behav. 2011, 22 (Suppl. 1), S44–S48. [Google Scholar] [CrossRef] [Green Version]
- Mbuba, C.K.; Ngugi, A.K.; Newton, C.R.; Carter, J.A. The epilepsy treatment gap in developing countries: A systematic review of the magnitude, causes, and intervention strategies. Epilepsia 2008, 49, 1491–1503. [Google Scholar] [CrossRef] [Green Version]
- Sugano, H.; Arai, H. Epilepsy surgery for pediatric epilepsy: Optimal timing of surgical intervention. Neurol. Med.-Chir. 2015, 55, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Freitag, H.; Tuxhorn, I. Cognitive function in preschool children after epilepsy surgery: Rationale for early intervention. Epilepsia 2005, 46, 561–567. [Google Scholar] [CrossRef]
- Voskuyl, R.A.; Clinckers, R. Antiepileptic drugs | pharmacological approaches for the assessment of antiepileptic drug efficacy in experimental animal models. In Encyclopedia of Basic Epilepsy Research; Schwartzkroin, P.A., Ed.; Academic Press: Oxford, UK, 2009; pp. 90–97. [Google Scholar]
- Stafstrom, C.E.; Carmant, L. Seizures and epilepsy: An overview for neuroscientists. Cold Spring Harb. Perspect. Med. 2015, 5, a022426. [Google Scholar] [CrossRef]
- Poduri, A.; Lowenstein, D. Epilepsy genetics—Past, present, and future. Curr. Opin. Genet. Dev. 2011, 21, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Su, X.; Klein, M.S.; Lewis, I.A.; Fiehn, O.; Rabinowitz, J.D. Metabolite measurement: Pitfalls to avoid and practices to follow. Annu. Rev. Biochem. 2017, 86, 277–304. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K. Recent advances in targeted and untargeted metabolomics by nmr and ms/nmr methods. High Throughput 2018, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Di Guida, R.; Engel, J.; Allwood, J.W.; Weber, R.J.M.; Jones, M.R.; Sommer, U.; Viant, M.R.; Dunn, W.B. Non-targeted uhplc-ms metabolomic data processing methods: A comparative investigation of normalisation, missing value imputation, transformation and scaling. Off. J. Metab. Soc. 2016, 12, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Locasale, J.W. Metabolomics: A primer. Trends Biochem. Sci. 2017, 42, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigmarsdottir, T.; McGarrity, S.; Rolfsson, O.; Yurkovich, J.T.; Sigurjonsson, O.E. Current status and future prospects of genome-scale metabolic modeling to optimize the use of mesenchymal stem cells in regenerative medicine. Front. Bioeng. Biotechnol. 2020, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chen, C.; Zhu, Y.; Shang, E.; Zhao, M.; Guo, S.; Guo, J.; Qian, D.; Tang, Z.; Yan, H.; et al. Exploratory cortex metabolic profiling revealed the sedative effect of amber in pentylenetetrazole-induced epilepsy-like mice. Molecules 2019, 24, 460. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, X.; Kong, J.; Wu, J.; Lai, M. Gc–ms–based metabolomics discovers a shared serum metabolic characteristic among three types of epileptic seizures. Epilepsy Res. 2016, 126, 83–89. [Google Scholar] [CrossRef]
- Kobylarek, D.; Iwanowski, P.; Lewandowska, Z.; Limphaibool, N.; Szafranek, S.; Labrzycka, A.; Kozubski, W. Advances in the potential biomarkers of epilepsy. Front. Neurol. 2019, 10, 685. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Enright, N.; Simonato, M.; Henshall, D.C. Discovery and validation of blood micrornas as molecular biomarkers of epilepsy: Ways to close current knowledge gaps. Epilepsia Open 2018, 3, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Raoof, R.; Bauer, S.; El Naggar, H.; Connolly, N.M.C.; Brennan, G.P.; Brindley, E.; Hill, T.; McArdle, H.; Spain, E.; Forster, R.J.; et al. Dual-center, dual-platform microrna profiling identifies potential plasma biomarkers of adult temporal lobe epilepsy. EBioMedicine 2018, 38, 127–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The human serum metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonini, F.; McGonigal, A.; Trebuchon, A.; Gavaret, M.; Bartolomei, F.; Giusiano, B.; Chauvel, P. Frontal lobe seizures: From clinical semiology to localization. Epilepsia 2014, 55, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Entz, L.; Tóth, E.; Keller, C.J.; Bickel, S.; Groppe, D.M.; Fabó, D.; Kozák, L.R.; Erőss, L.; Ulbert, I.; Mehta, A.D. Evoked effective connectivity of the human neocortex. Hum. Brain Mapp. 2014, 35, 5736–5753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skene, N.G.; Grant, S.G.N. Identification of vulnerable cell types in major brain disorders using single cell transcriptomes and expression weighted cell type enrichment. Front. Neurosci. 2016, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.C.; Dachet, F.; Ghoddoussi, F.; Bagla, S.; Fuerst, D.; Stanley, J.A.; Galloway, M.P.; Loeb, J.A. Altered metabolomic-genomic signature: A potential noninvasive biomarker of epilepsy. Epilepsia 2017, 58, 1626–1636. [Google Scholar] [CrossRef] [Green Version]
- Zarrouk, A.; Vejux, A.; Nury, T.; El Hajj, H.I.; Haddad, M.; Cherkaoui-Malki, M.; Riedinger, J.M.; Hammami, M.; Lizard, G. Induction of mitochondrial changes associated with oxidative stress on very long chain fatty acids (c22:0, c24:0, or c26:0)-treated human neuronal cells (sk-nb-e). Oxid. Med. Cell. Longev. 2012, 2012, 623257. [Google Scholar] [CrossRef] [Green Version]
- Schonfeld, P.; Wojtczak, L. Short- and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. Chapter three—The role of short-chain fatty acids in health and disease. In Advances in Immunology; Alt, F.W., Ed.; Academic Press: Oxford, UK, 2014; Volume 121, pp. 91–119. [Google Scholar]
- Sakakibara, I.; Fujino, T.; Ishii, M.; Tanaka, T.; Shimosawa, T.; Miura, S.; Zhang, W.; Tokutake, Y.; Yamamoto, J.; Awano, M.; et al. Fasting-induced hypothermia and reduced energy production in mice lacking acetyl-coa synthetase 2. Cell Metab. 2009, 9, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, M.K.; Osborne, B.; Brown, S.H.; Small, L.; Mitchell, T.W.; Cooney, G.J.; Turner, N. Contrasting metabolic effects of medium- versus long-chain fatty acids in skeletal muscle. J. Lipid Res. 2013, 54, 3322–3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-D.; Yw Chang, A.; Chuang, Y.-C. The potential role of mitochondrial dysfunction in seizure-associated cell death in the hippocampus and epileptogenesis. J. Bioenerg. Biomembr. 2010, 42, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H.; Young, M.E.; Lopaschuk, G.D.; Abel, E.D.; Brunengraber, H.; Darley-Usmar, V.; Rosiers, C.D.; Gerszten, R.; Glatz, J.F.; Griffin, J.L.; et al. Assessing cardiac metabolism: A scientific statement from the american heart association. Circ. Res. 2016, 118, 1659–1701. [Google Scholar] [CrossRef] [PubMed]
- Ingram, J.; Zhang, C.; Cressman, J.R.; Hazra, A.; Wei, Y.; Koo, Y.E.; Ziburkus, J.; Kopelman, R.; Xu, J.; Schiff, S.J. Oxygen and seizure dynamics: I. Experiments. J. Neurophysiol. 2014, 112, 205–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fradejas-Villar, N. Consequences of mutations and inborn errors of selenoprotein biosynthesis and functions. Free Radic. Biol. Med. 2018, 127, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Fairweather-Tait, S.J.; Bao, Y.; Broadley, M.R.; Collings, R.; Ford, D.; Hesketh, J.E.; Hurst, R. Selenium in human health and disease. Antioxid. Redox Signal. 2011, 14, 1337–1383. [Google Scholar] [CrossRef]
- Schibler, U. Selenium cysteine and epileptic seizures. Nat. Rev. Mol. Cell Biol. 2018, 19, 753. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium utilization by gpx4 is required to prevent hydroperoxide-induced ferroptosis. Cell 2018, 172, 409–422. [Google Scholar] [CrossRef] [Green Version]
- Martinc, B.; Grabnar, I.; Vovk, T. Antioxidants as a preventive treatment for epileptic process: A review of the current status. Curr. Neuropharmacol. 2014, 12, 527–550. [Google Scholar] [CrossRef] [Green Version]
- Kahn-Kirby, A.H.; Amagata, A.; Maeder, C.I.; Mei, J.J.; Sideris, S.; Kosaka, Y.; Hinman, A.; Malone, S.A.; Bruegger, J.J.; Wang, L.; et al. Targeting ferroptosis: A novel therapeutic strategy for the treatment of mitochondrial disease-related epilepsy. PLoS ONE 2019, 14, e0214250. [Google Scholar] [CrossRef] [Green Version]
- Pearson-Smith, J.N.; Patel, M. Metabolic dysfunction and oxidative stress in epilepsy. Int. J. Mol. Sci. 2017, 18, 2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldbaum, S.; Patel, M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 2010, 88, 23–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalf, J.L.; Song, S.J. Evaluating the impact of domestication and captivity on the horse gut microbiome. Sci. Rep. 2017, 7, 15497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, W.Y.; Herr, D.R.; Farooqui, T.; Ling, E.A.; Farooqui, A.A. Role of sphingomyelinases in neurological disorders. Expert Opin. Ther. Targets 2015, 19, 1725–1742. [Google Scholar] [CrossRef]
- Huang, N.-J.; Lin, Y.-C.; Lin, C.-Y.; Pishesha, N.; Lewis, C.A.; Freinkman, E.; Farquharson, C.; Millán, J.L.; Lodish, H. Enhanced phosphocholine metabolism is essential for terminal erythropoiesis. Blood 2018, 131, 2955–2966. [Google Scholar] [CrossRef] [Green Version]
- Yousf, S.; Sardesai, D.M.; Mathew, A.B.; Khandelwal, R.; Acharya, J.D.; Sharma, S. Metabolic signatures suggest o-phosphocholine to udp-n-acetylglucosamine ratio as a potential biomarker for high-glucose and/or palmitate exposure in pancreatic beta-cells. Off. J. Metab. Soc. 2019, 15, 55. [Google Scholar] [CrossRef]
- Ferreira, G.C.; McKenna, M.C. l-carnitine and acetyl-l-carnitine roles and neuroprotection in developing brain. Neurochem. Res. 2017, 42, 1661–1675. [Google Scholar] [CrossRef] [PubMed]
- LIU, J.; HEAD, E.; KURATSUNE, H.; COTMAN, C.W.; AMES, B.N. Comparison of the effects of l-carnitine and acetyl-l-carnitine on carnitine levels, ambulatory activity, and oxidative stress biomarkers in the brain of old rats. Ann. N. Y. Acad. Sci. 2004, 1033, 117–131. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zhang, H.; Zhang, Z.; Wang, T.; Niu, J.; Cui, D.; Xu, S. Neuroprotective effects of pre-treatment with l-carnitine and acetyl-l-carnitine on ischemic injury in vivo and in vitro. Int. J. Mol. Sci. 2012, 13, 2078–2090. [Google Scholar] [CrossRef] [Green Version]
- Tyni, T.; Pourfarzam, M.; Turnbull, D.M. Analysis of mitochondrial fatty acid oxidation intermediates by tandem mass spectrometry from intact mitochondria prepared from homogenates of cultured fibroblasts, skeletal muscle cells, and fresh muscle. Pediatr. Res. 2002, 52, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Van Maldegem, B.T.; Wanders, R.J.; Wijburg, F.A. Clinical aspects of short-chain acyl-coa dehydrogenase deficiency. J. Inherit. Metab. Dis. 2010, 33, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhara, T.; Akiyama, T.; Ohse, M.; Koike, T.; Shibasaki, J.; Imai, K.; Cooper, A.J.L. Identification of new biomarkers of pyridoxine-dependent epilepsy by gc/ms-based urine metabolomics. Anal. Biochem. 2020, 113739. [Google Scholar] [CrossRef] [PubMed]
- Alpay Savasan, Z.; Yilmaz, A.; Ugur, Z.; Aydas, B.; Bahado-Singh, R.O.; Graham, S.F. Metabolomic profiling of cerebral palsy brain tissue reveals novel central biomarkers and biochemical pathways associated with the disease: A pilot study. Metabolites 2019, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercier, P.; Lewis, M.J.; Chang, D.; Baker, D.; Wishart, D.S. Towards automatic metabolomic profiling of high-resolution one-dimensional proton nmr spectra. J. Biomol. NMR 2011, 49, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.; Cloarec, O.; Holmes, E.; Nicholson, J.K.; Lindon, J.C. Scaling and normalization effects in nmr spectroscopic metabonomic data sets. Anal. Chem. 2006, 78, 2262–2267. [Google Scholar] [CrossRef] [PubMed]
- Bisgin, H.; Bera, T.; Ding, H.; Semey, H.G.; Wu, L.; Liu, Z.; Barnes, A.E.; Langley, D.A.; Pava-Ripoll, M.; Vyas, H.J.; et al. Comparing svm and ann based machine learning methods for species identification of food contaminating beetles. Sci. Rep. 2018, 8, 6532. [Google Scholar] [CrossRef]
- Graham, S.F.; Turkoglu, O.; Yilmaz, A.; Ustun, I.; Ugur, Z.; Bjorndhal, T.; Han, B.; Mandal, R.; Wishart, D.; Bahado-Singh, R.O. Targeted metabolomics highlights perturbed metabolism in the brain of autism spectrum disorder sufferers. Off. J. Metab. Soc. 2020, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Onan, A.; Korukoğlu, S. A feature selection model based on genetic rank aggregation for text sentiment classification. J. Inf. Sci. 2015, 43, 25–38. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The weka data mining software: An update. SIGKDD Explor. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Keerthi, S.S.; Lin, C.-J. Asymptotic behaviors of support vector machines with gaussian kernel. Neural Comput. 2003, 15, 1667–1689. [Google Scholar] [CrossRef]
- Abraham, A.; Pedregosa, F.; Eickenberg, M.; Gervais, P.; Mueller, A.; Kossaifi, J.; Gramfort, A.; Thirion, B.; Varoquaux, G. Machine learning for neuroimaging with scikit-learn. Front. Neuroinform. 2014, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. Metaboanalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; Wishart, D.S.; Xia, J. Using metaboanalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls | Epileptic | p-Value | |
|---|---|---|---|
| n | 15 | 15 | |
| Age, mean (SD) | 40.67(14.75) | 40.8(15.06273) | 0.98 a |
| Gender | |||
| Male | 9 | 9 | 1 b |
| Female | 6 | 6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lalwani, A.M.; Yilmaz, A.; Bisgin, H.; Ugur, Z.; Akyol, S.; Graham, S.F. The Biochemical Profile of Post-Mortem Brain from People Who Suffered from Epilepsy Reveals Novel Insights into the Etiopathogenesis of the Disease. Metabolites 2020, 10, 261. https://doi.org/10.3390/metabo10060261
Lalwani AM, Yilmaz A, Bisgin H, Ugur Z, Akyol S, Graham SF. The Biochemical Profile of Post-Mortem Brain from People Who Suffered from Epilepsy Reveals Novel Insights into the Etiopathogenesis of the Disease. Metabolites. 2020; 10(6):261. https://doi.org/10.3390/metabo10060261
Chicago/Turabian StyleLalwani, Ashna M., Ali Yilmaz, Halil Bisgin, Zafer Ugur, Sumeyya Akyol, and Stewart Francis Graham. 2020. "The Biochemical Profile of Post-Mortem Brain from People Who Suffered from Epilepsy Reveals Novel Insights into the Etiopathogenesis of the Disease" Metabolites 10, no. 6: 261. https://doi.org/10.3390/metabo10060261