
Abstract
Purpose
Hermansky–Pudlak syndrome (HPS) is characterized by oculocutaneous albinism, excessive bleeding, and often additional symptoms. Variants in ten different genes have been involved in HPS. However, some patients lack variants in these genes. We aimed to identify new genes involved in nonsyndromic or syndromic forms of albinism.
Methods
Two hundred thirty albinism patients lacking a molecular diagnosis of albinism were screened for pathogenic variants in candidate genes with known links to pigmentation or HPS pathophysiology.
Results
We identified two unrelated patients with distinct homozygous variants of the BLOC1S5 gene. Patients had mild oculocutaneous albinism, moderate bleeding diathesis, platelet aggregation deficit, and a dramatically decreased number of platelet dense granules, all signs compatible with HPS. Functional tests performed on platelets of one patient displayed an absence of the obligate multisubunit complex BLOC-1, showing that the variant disrupts BLOC1S5 function and impairs BLOC-1 assembly. Expression of the patient-derived BLOC1S5 deletion in nonpigmented murine Bloc1s5-/- melan-mu melanocytes failed to rescue pigmentation, the assembly of a functional BLOC-1 complex, and melanosome cargo trafficking, unlike the wild-type allele.
Conclusion
Mutation of BLOC1S5 is disease-causing, and we propose that BLOC1S5 is the gene for a new form of Hermansky–Pudlak syndrome, HPS-11.
Similar content being viewed by others

INTRODUCTION
Hermansky–Pudlak syndrome (HPS) comprises a group of rare recessive genetic diseases (estimated incidence ~1 in 106) in which patients suffer from oculocutaneous albinism (OCA), excessive bleeding, and in some cases lethal pulmonary fibrosis, immunodeficiency, and/or granulomatous colitis.1 These symptoms result primarily from defects of cell type–specific lysosome-related organelles (LROs) (melanosomes in pigment cells and dense granules in platelets).2,3 Pathogenic variants in ten different genes have been shown to cause distinct HPS isoforms. These genes encode subunits of four multisubunit protein complexes—adapter protein-3 (AP-3) and biogenesis of lysosome-related organelles complex (BLOC)-1, -2 and -3—that mediate intracellular membrane transport to maturing LROs.3 However, at least some patients lack variants in known HPS genes.4 Moreover, because of the multisystemic nature of the disorder and the variable severity of the symptoms, HPS is often undiagnosed. Thus, there likely exist additional forms of the disorder with variants in additional genes. Indeed, among the many mouse models of HPS are pathogenic variants in genes encoding BLOC-1 subunits not yet identified among HPS patients.
Albinism patients may have variable hypopigmentation of the skin, hair, and eyes, ranging from a total lack of melanin to residual or even near normal melanin production. However, all albinism patients suffer from ophthalmological anomalies including nystagmus, foveal hypoplasia, chiasmatic misrouting of the optic nerves, iris transillumination, retinal hypopigmentation, and reduced visual acuity. Pathogenic variants in any of 19 genes, including the 10 HPS genes, have been identified as causative for various forms of albinism or the closely related foveal hypoplasia and optic nerve misrouting without albinism (FHONDA)5,6,7,8 (Table S1). Because of the high phenotypic heterogeneity among albinism patients, establishment of genotype–phenotype correlations and clinical diagnoses can be difficult to achieve, leading to underdiagnosis of the disease.9
Despite a thorough search for both single nucleotide variants and copy-number variants of all 19 genes, our recent screening left 27% of patients without a molecular diagnosis.8 Missing variants may reside either in the introns or regulatory elements of known genes, which are not commonly analyzed in clinical laboratories, or in genes other than those identified so far.
To search for new albinism genes, we screened the genomes of 230 undiagnosed patients for variants in candidate genes. We focused on genes coding for subunits of AP-3 and BLOC-1, -2, and -3, in which variants have not been described so far, and on orthologues of additional genes known to control cargo trafficking to and from melanosomes and regulate melanosomal maturation2 (see Table S2). We describe the identification in two patients with oculocutaneous albinism and mild bleeding diathesis of homozygous pathogenic variants of the BLOC1S5 gene encoding a BLOC-1 subunit not previously identified as mutated in HPS patients.
MATERIALS AND METHODS
Ethics statement
This study was approved by the local authority (Comité de Protection des Personnes Sud Ouest et Outre Mer III). Informed consent was obtained from the patients and/or their parents before genetic analysis was performed. Authorization for publication and for the inclusion of photographs was obtained from the participants.
Platelet investigations
Platelet rich plasma (PRP) was obtained from blood collected into buffered 3.2% sodium citrate.
Light transmission aggregometry (LTA) was evaluated within 4 hours of blood collection using a SD-Soderel aggregometer or APACT-4004 aggregometer (Elitech, France) and different specific agonists that included 1.25 and 5 µg/ml collagen; 3, 5, and 10 µM adenosine diphosphate (ADP); 1 mM arachidonic acid; and 0.75 and 1.25 mg/l ristocetin or thrombin receptor agonist peptide (TRAP). Percentage maximal amplitude was evaluated using an internal reference interval.
Adenosine triphosphate (ATP) secretion from platelets was tested by a lumiaggregometer assay as recommended by the instrument manufacturer (Chrono-Log Corporation, USA) using PRP, firefly luciferase reagent, and 10 µmol/l TRAP, ADP, or collagen 14 as agonists.
High-performance liquid chromatography (HPLC) with electrochemical and amperometric detection was used to measure intraplatelet serotonin (5-HT) concentration in PRP samples.
Electron microscopy
For whole mount preparation, a few microliters of platelet rich plasma (PRP) were deposited on formvar-coated, carbon-stabilized grids for 1–5 minutes, rinsed in a drop of distilled water (10–15 seconds), dried from the edge with pieces of filter paper and air dried for 1 minute with gentle shaking without further chemical fixation or poststaining with contrasting agents. Samples were imaged and electron micrographs acquired with a transmission electron microscope (Tecnai Spirit G2; ThermoFisher Scientific, Eindhoven, The Netherlands) equipped with a 4k charge-coupled device (CCD) camera (Quemesa, EMSIS, Muenster, Germany) for patient 1, and JEOL 1400 (100 kev) equipped with a Gatan ORIUS SC600 camera for patient 2.
Sequencing of the candidate genes
DNA was extracted from peripheral blood leukocytes using an automated procedure as recommended by the manufacturer (Tecan EVO-ReliaPrep, Promega).
Next-generation sequencing (NGS) was performed using the ionTorrent technology on a S5XL instrument (Life Technologies, ThermoFisher Scientific, USA) (AmpliSeq panel) and bioinformatics analysis of variants were as previously described.8 See Supplementary Data for more details.
Amplification of BLOC1S5 exons 2 to 5 was performed as described in Supplementary Data.
Characterization of the BLOC1S5 gene deletion
The BLOC1S5 deletion was analyzed by high-resolution array comparative genomic hybridization (CGH) according to a procedure already described.8,10 One hundred forty-three probes were placed along BLOC1S5 with a median probe spacing of 173 bp.
Sequencing of the intron 2–4 deletion breakpoint was performed as described in Supplementary Data.
Cloning of control and mutant BLOC1S5 complementary DNA and generation of recombinant retroviral expression constructs
BLOC1S5 isoforms were amplified from RNA extracted from patient and control blood leukocytes by reverse-transcription polymerase chain reaction (RT-PCR) using primers located in exons 1 and 5 of the gene, based on the reference sequence NM_201280.3 (http://www.ensembl.org). RT-PCR products were first subcloned in the pCS2+ vector, and then inserted into pBMN-IRES-Hygro for retroviral transduction as described.11 See Supplementary Materials and Methods for detailed procedures.
Other procedures
A brief description of additional procedures is provided here. See Supplementary Materials and Methods for detailed procedures.
The melan-mu311 and wild-type melan-Ink4a were maintained in culture as described.12 Retrovirus production from transiently transfected Plat-E cells13 and retroviral transduction of melan-mu cells were carried out as described previously.11,14
Expression of the transgenes in melan-mu3 cells was evaluated by RT-PCR directed to the hygromycin resistance gene.
Immunofluorescence microscopy (IFM) analyses of fixed cells, image deconvolution with Microvolution software, and quantification of the area of marker overlap in double labeling experiments using ImageJ (http://fiji.sc/Fiji; National Institutes of Health) were done as described.15
Analysis of HA, Dysbindin, Pallidin, and Tubulin proteins in patients' platelets and melan-mu3 melanocytes before and after transduction with the recombinant retroviral expression constructs was performed by western blot analysis.
Spectrophotometric assay on melanocyte lysates was done as described.16
Statistical analyses
Statistical data are presented as mean ± SEM. Statistics were calculated in GraphPad Prism using ordinary one-way analysis of variance (ANOVA) or one-way ANOVA with nonparametric distribution (Kruskal–Wallis test by ranks) when specified. Significant differences between control or treated samples are indicated (****P < 0.0001; ***P < 0.001; **P < 0.01; P < 0.05). Only P < 0.05 was considered as statistically significant.
RESULTS
Identification of BLOC1S5 variants
Among our cohort of more than 1500 albinism patients, 25–30% remain without a molecular diagnosis despite a thorough analysis of all 19 albinism-related genes (Table S1).7,8 We hypothesized that some of these patients have pathogenic variants in candidate gene(s) selected for their potential involvement in the pathophysiology of albinism (Table S2). Therefore, 230 unsolved patients were screened for pathogenic variants of these genes using NGS.
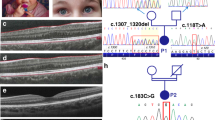
In two patients, we identified homozygous variants of the BLOC1S5 gene (NM_201280.2). BLOC1S5 is one of the eight subunits of the obligate multisubunit protein complex BLOC-1, which facilitates cargo transport to melanosomes and other lysosome-related organelles in specialized cell types.3 Pathogenic variants in the genes encoding three other BLOC-1 subunits (DTNBP1, BLOC1S3, and BLOC1S6) underlie Hermansky–Pudlak syndrome types 7, 8, and 9 respectively,17,18,19,20 and Bloc1s5 is mutated in the muted mouse model of HPS.21 Patient 1 harbored a BLOC1S5 deletion that was visualized in NGS data using an in-house bioinformatics plugin based on NGS read depth (Supplementary Materials and Methods). Further analysis showed that the deletion covered a 19,063-bp segment encompassing BLOC1S5 exons 3 and 4 (Fig. 1a, b). Detailed characterization of the deletion and of the molecular mechanism involved is provided in Supplementary Data. The deletion is referred to as NC_000006.11(BLOC1S5_v002):c.196–678_384+3483del and g.8023117_8042179del (Hg19) (ClinVar https://www.ncbi.nlm.nih.gov/clinvar/ submission number SUB6791269). The patient’s parents were both heterozygous for the deletion, as demonstrated by array-CGH (Figure S1A). Unaffected brother and sister were not tested.

(a) Schematic representation of the deletion. Top: genomic representation of the BLOC1S5 gene (approximately to scale). The five exons are represented as black boxes. cen centromeric side, tel telomeric side. Middle: The region spanning exons 2–5 of the BLOC1S5 gene is expanded. The light gray boxes represent the 291-bp highly homologous segments (see Supplementary Data for details). The dark gray boxes represent the 57-bp perfectly homologous segment where nonallelic homologous recombination appears to have taken place in patient 1. Bottom: Deleted allele resulting from nonallelic homologous recombination, showing that the region between the two 291-bp highly homologous segments is missing. (b) Polymerase chain reaction (PCR) amplification of exons 2, 3, 4, and 5 of BLOC1S5 in genomic DNA from a control individual and from patient 1. A size marker (100 bp ladder) is shown on the left. Exons 3 and 4 are not amplified from the patient’s DNA. (c) Reverse-transcription PCR from leukocytes’ RNA of a control individual and patient 1. Reverse-transcription PCR (RT-PCR) primers where derived from BLOC1S5 exons 1 and 5, as shown in (d). RT- denotes without reverse transcriptase. RT+ denotes with reverse transcriptase. Bands at 763 bp (corresponding to complementary DNA [cDNA] BLOC1S5–202) and at 574 bp (corresponding to cDNA BLOC1S5-del1) are observed in the control and the patient, respectively. (d) Schematic representation of the messenger RNA (mRNA) isoforms identified in the control individual and in patient 1, as described in the main text. BLOC1S5–202 represents the theoretical wild-type isoform. Sizes of the cDNAs corresponding to the different isoforms are BLOC1S5–202 (763 bp) and BLOC1S5–205 (633 bp), BLOC1S5-del1 (574 bp), and BLOC1S5-del2 (542 bp).
A second patient (patient 2) harbored a homozygous 1-bp deletion in exon 4 of BLOC1S5, NM_201280.2:c.345del p.(Val116Serf19*), predicted to generate a reading-frameshift and protein truncation (ClinVar submission number SUB7267982). Biparental transmission of the variant could not be proven because the patient’s parents were unavailable for analysis. However, quantitative PCR analysis indicated the presence of two copies of exon 4 (data not shown), thus proving that the NM_201280.2:c.345del variant was homozygous.
American College of Medical Genetics and Genomics (ACMG) classification22 was in favor of patient 1's and patient 2's variants being pathogenic (patient 1: PVS1, PS3, PM2, PM3; patient 2: PVS1, PM2, PM3). Of note, these variants were not found in the gnomAD public database (https://gnomad.broadinstitute.org/) in either the homozygous or heterozygous state.
Clinical cases
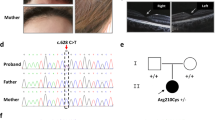
Patient 1, aged 2 months at the time of diagnosis and now 20 years old, was born to parents originating from French Flanders. She was referred to our laboratory with a clinical diagnosis of mild oculocutaneous albinism. Her creamy skin and blond hair (Fig. 2a, b) were lighter than observed in her mother, father, sister, and brother (Fig. 2d–f). Ophthalmological features included nystagmus, foveal hypoplasia (Fig. 2g–j), retinal hypopigmentation with increased visibility of choroid vessels (Fig. 2k–m, and Figure S2), iris transillumination, and visual acuity of 4/10 and 5/10 in the right and left eyes, respectively.

Upper left and right panel: cutaneous and ocular phenotype. Photographs show hypopigmentation of hair and skin of patient 1 (a, b), compared with her unaffected mother (c), father (d), brother (e), and sister (f), and of patient 2 (a’, b’). Bilateral foveal hypoplasia is seen in patient 1 (g: left eye; h: right eye), compared with a normal control (i) and a positive control (OCA1 patient) (j). Patient 2 does not have foveal hypoplasia (c’: left eye; d’: right eye). Retinal hypopigmentation is seen in patient 1 (k: left eye; l: right eye) compared with a normal control (m). Arrows point to hypoplasia of the macula. Patient 2 has low grade retinal hypopigmentation (e’: left eye; f’: right eye). Lower panel: whole mount electron microscopy of platelets. Platelets in healthy individuals displayed dense granules typically observed as round electron-dense structures (arrowheads) (a”–b”). Platelets from a HPS8 patient were used as a positive control for the absence of platelet dense granules (c”–d”). Platelets from the BLOC1S5 patient 1 harbored no round electron-dense structures (e”–f”). Most platelets from the BLOC1S5 patient 2 harbored no round electron-dense structures (g”–h”). Scale bar: 2 μm.
Mild signs of bleeding diathesis were not reported initially by the patient. After the BLOC1S5 homozygous deletion and its potential link to HPS were identified, the patient’s history was found to include the following: bruises appeared easily in infancy but were not large and did not spread, episodes of epistaxis occurred once or twice a year and lasted for about a week, gingival bleeding was observed but there was no history of postsurgical hemorrhage, menorrhagia was absent. Platelet aggregation in response to a low concentration of collagen (1.25 µg/ml) was reduced and delayed in the patient relative to controls, with a latency of 54 seconds (33 seconds for the control), velocity of 22%/30 seconds (65%/30 seconds for the control) and maximal amplitude of 30% (83% for the control) (Figure S3A). Platelet aggregation was normal in response to a high concentration of collagen (5 µg/ml) and to other agonists, including ADP (3, 7, or 10 µM), arachidonic acid (1 mM), and ristocetin (0.75 mg/ml or 1.5 mg/ml; Figure S3A). The weak aggregation in response to low levels of collagen but normal responses to ADP (which may complement reduced ADP release from dense granules) and ristocetin (which bypasses activation pathways) are consistent with a mild defect in dense granule secretion.23 Consistently, lumiaggregometry assay indicated that patient’s platelets had low ATP release after TRAP activation (0.19 nM/108 platelets; normal values > 0.30 nM/108 platelets), and platelet serotonin storage, quantified by HPLC, was very low (9 ng/109 platelets; normal values, 350–980 ng/109 platelets) (not shown).
Platelets from patient 1, as well as from a healthy donor and a well-characterized HPS patient as controls, were analyzed by whole mount electron microscopy for dense granule content. In the healthy control, platelet dense granules appeared as 3–8 electron-dense round structures (arrowheads in Fig. 2a”, b”). By contrast, platelets from a HPS-8 patient (Fig. 2c”, d”) and from patient 1 (Fig. 2e”, f”) contained almost no dense granules (<0.1 granule per platelet). These data are consistent with the mild platelet aggregation deficiency and serotonin storage pool defect observed in the patient. The data suggest that Patient 1 has a mild form of Hermansky–Pudlak syndrome, consistent with a BLOC-1 deficiency.
Patient 2 was born to nonconsanguineous parents originating from the same village of Slovenia, both with black hair. She is the only one affected of a sibship of five. She presented at birth with yellow skin and white platinum hair. At the age of 39 years she had pigmented skin, blond hair, brown iris, and numerous pigmented naevi. Mild ocular albinism included nystagmus, grade 1 retinal hypopigmentation, iris transillumination, optic nerve decussation anomalies on visual evoked potentials, strabismus, photophobia, visual acuity of 6/10 on both eyes, and normal fovea (Fig. 2a’–f’). Clinical report indicated important epistaxis, mostly in childhood, easy or unexplained bruising, menorrhagia improved by contraception, excessive blood loss after deliveries, surgery and dental extraction, as well as abdominal pain, dyspnea, and recurrent infections (pneumonia, herpes, conjunctivitis). These features suggested a syndromic form of albinism.
Further investigations showed that patient 2 had normal platelet counts (139–197 × 109 platelets/L) and von Willebrand factor (VWF: RCo = 60% [50–150]). Platelet aggregation tested using different agonists showed abnormal profiles upon collagen, arachidonic acid, and TRAP stimulation (Figure S3B). ATP release was also recorded by measuring luminescence from firefly luciferin-luciferase reaction using 10 µM ADP, 2 µg/mL collagen or 50 µM TRAP-6, only revealing a slight decrease after stimulation with collagen (0.34 nmol; normal value >0.49 nmol). Electron microscopy of whole mount platelets showed a strong reduction of δ-granules numbers (mean ± SD = 0.2 ± 0.5 granule per platelet; normal values = 4.14–7.74) (Fig. 2g”–h”). Altogether these data were compatible with a quantitative defect of dense bodies.
Impact of the BLOC1S5 deletion on BLOC-1 assembly and function
BLOC-1 is an obligate multisubunit complex that is unstable in the absence of any of its subunits. Hence, expression of a subunit that is incapable of assembling into the complex results in the degradation of the other BLOC-1 subunits.24,25,26 To test whether the BLOC1S5 deletion impacted BLOC-1 stability, western blot analysis was performed on lysates of patient 1 platelets for the BLOC-1 subunits Pallidin (BLOC1S6) and Dysbindin (DTNBP1). The results showed that relative to lysates from controls, levels of both Pallidin and Dysbindin are reduced or absent in the patient’s platelets (Fig. 3). We were unable to detect a specific band in control platelets with available antibodies to Muted/BLOC1S5 (not shown).

Platelet lysates were analyzed with anti-Dysbindin (left), anti-Pallidin (right) antibodies. An anti-GPIIb antibody was used as a reference. Sizes of detected proteins are indicated in kDa.
The instability of BLOC-1 in patient 1 platelets suggested that BLOC-1 function might be impaired. To definitively test whether the patient 1–derived BLOC1S5 intragenic homozygous deletion impairs BLOC-1 function and might be causal for the HPS phenotype, we tested whether this allele was capable of rescuing the pigmentation and trafficking defects in murine Bloc1s5mu/mu melanocytes that lack a functional Muted/BLOC1S5 protein because of a retrotransposon insertion in the Bloc1s5 locus.21 Immortalized melan-mu melanocytes derived from these mice lack melanin and misaccumulate BLOC-1-dependent melanosome proteins, such as TYRP1, in early endosomes.11 Stable “rescue” of these cells by expression of the epitope-tagged wild-type human BLOC1S5 protein restores pigmentation and normal cargo localization.11 Thus, to evaluate the activity of patient 1–derived isoforms, we expressed them or control alleles as HA.11 epitope-tagged proteins by retroviral transduction in melan-mu cells, and assessed both pigmentation and TYRP1 localization.
The gene rearrangement identified in patient 1 was predicted to result in the in-frame deletion of exons 3 and 4 of the BLOC1S5 gene (NM_201280.2 transcript). Accordingly, RT-PCR of full-length BLOC1S5 complementary DNA (cDNA) from the patient amplified a product containing exons 1, 2, and 5, but not exons 3 and 4 (574-bp band in Fig. 1c, corresponding to BLOC1S5-del1 represented in Fig. 1d). The patient’s cDNA also contained a shorter fragment lacking 32 bp of exon 2 (BLOC1S5-del2, Fig. 1d) that did not correspond to any alternative transcript reported in Ensembl (http://www.ensembl.org) and that was expected to induce a translation frameshift. In contrast, RT-PCR of control leukocytes' messenger RNA (mRNA) produced two cDNA species, one comprising exons 1–5 of BLOC1S5 (763 band in Fig. 1c) corresponding to the full length NM_201280.3 (BLOC1S5–202) (http://www.ensembl.org) mRNA isoform, and the other comprising exons 1, 2, 4, and 5 of BLOC1S5, corresponding to isoform NM_001199323 (BLOC1S5–205) and predicted to be out of frame (see Fig. 1d) (the band at 633 bp is indistinguishable from the smear observed in Fig. 1c). All BLOC1S5 cDNA species, control or patient-derived, are schematically presented in Fig. 1d.
Whereas immortalized wild-type melan-Ink4a melanocytes from C57BL/6J-Ink4a-Arf-/- mice contain pigmented melanosomes detected by bright field microscopy (Fig. 4a), melan-mu melanocytes are unpigmented (Fig. 4b). Pigmentation of melan-mu was rescued by stable expression of wild-type HA-tagged human BLOC1S525 (Fig. 4c) or of HA-BLOC1S5–202 (full-length BLOC1S5 isoform isolated from a control donor; Fig. 4h) but not of HA-tagged BLOC1S6 (Fig. 4d), a distinct human BLOC-1 subunit. By contrast, the expression of any of the truncated BLOC1S5 isoforms—HA—BLOC1S5-del1 and HA-BLOC1S5-del2 from the patient or HA-BLOC1S5–205 from the control—was unable to rescue pigmentation (Fig. 4e–g). These results were confirmed by a quantitative melanin content assay of cell lysates (Fig. 4i). Expression of the hygromycin resistance gene from the internal ribosome entry site of the retroviral backbone confirmed stable mRNA expression of all constructs (Fig. 4j). These data indicate that neither the patient-derived BLOC1S5 mRNA species (HA-BLOC1S5-del1 and HA-BLOC1S5-del2) nor the truncated alternative physiological allele (HA-BLOC1S5–205) are functional.

(a–h) Melanin content of fixed mouse melanocyte cell lines melan-Ink4a (wild-type), melan-mu (from Bloc1s5mu/mu mice) or melan-mu stably expressing the indicated HA.11-tagged Muted (BLOC1S5) or Pallidin (BLOC1S6) transgenes was assessed by bright field microscopy. Unpigmented cells are outlined in white. Scale: 10 µm. (i) Melanin content in lysates from the indicated cell lines was assayed by spectrophotometry. Plat-E is a nonpigmented cell line used as a negative control. Data are normalized to values from melan-mu and represent mean ± SEM from at least three experiments. Statistical analysis was performed using the Kruskal–Wallis test by ranks. *P < 0.05; ****P < 0.0001. (j) Reverse-transcription polymerase chain reaction (RT-PCR) analysis to confirm transgene expression. Complementary DNA (cDNA) was amplified from each of the indicated cell lines using primers for the hygromycin resistance gene expressed from the internal ribosome entry site of the pBMN-Hygro-IRES retroviral backbone, and products were fractionated by agarose gel electrophoresis. Shown is a representative of two experiments. Positions of DNA markers (bp) are shown at left. (k) Whole-cell lysates of indicated cell lines fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) were immunoblotted for the HA.11 epitope tag, BLOC-1 subunits Dysbindin and Pallidin, or γ-tubulin as a control. Shown is a representative of three experiments. Relevant bands (right) and positions of molecular weight markers (kDa, left) are indicated. Note that the higher molecular weight band in the Pallidin blot is HA-tagged Pallidin, whereas the lower molecular weight band represents endogenous Pallidin.
Consistent with the BLOC-1 instability observed in patient 1 platelets relative to controls, the levels of endogenous Dysbindin and Pallidin subunits were dramatically reduced in melan-mu cells relative to their levels in lysates of wild-type melan-Ink4a melanocytes (Fig. 5k, lanes 1 and 2). Expression of either HA-tagged wild type24 or HA-tagged full-length isoform HA-BLOC1S5–202 isolated from a control donor rescued Dysbindin and Pallidin expression to levels similar to those in melan-Ink4a cells (Fig. 4k, lanes 1, 3, and 8), indicating BLOC-1 stabilization. In contrast, neither HA-tagged Pallidin nor the HA-tagged truncated patient or control isoforms rescued Dysbindin and Pallidin expression (Fig. 4k, lanes 4–7). Moreover, the HA-tagged truncated isoforms of BLOC1S5 were not detected by immunoblotting, despite expression of the hygromycin resistance marker from the same mRNA (Fig. 4j). These data indicate that the patient’s truncated BLOC1S5 isoforms and the alternative truncated transcript from controls are unstable and unable to assemble with other BLOC-1 subunits.

(a–h) Mouse melanocyte cell lines melan-Ink4a, melan-mu, or melan-mu stably expressing the indicated HA.11-tagged Muted or Pallidin transgenes were fixed, immunolabeled for TYRP1 (magenta) and transferrin receptor (TfR, green), and analyzed by immunofluorescence microscopy and image deconvolution (dIFM) and by bright field microscopy to detect melanin. Scale: 10 µm. Insets of boxed regions are magnified ten times. TYRP1 localized on TfR-positive compartments (yellow arrowheads) or TYRP1 (white arrow) and TfR (white arrowhead) localized on discrete compartments are indicated. (i) Percent area of overlap between TYRP1 and TfR in melanocyte cell lines. Data are shown as dot plots with mean ± SEM from at least 14 cells representing three independent experiments. Statistical analysis was performed using a one-way analysis of variance (ANOVA). ****P < 0.0001.
To test whether patient 1–derived BLOC1S5 isoforms could partially rescue BLOC-1 function in protein transport in melan-mu cells, we used immunofluorescence microscopy and image deconvolution (dIFM) to visualize TYRP1 localization relative to pigment granules visualized by bright field microscopy and to an early endosome marker, Transferrin receptor (TfR). In melan-Ink4a melanocytes, most TYRP1 localized to pigment granules at steady state and only 7% of the total TYRP1-containing structures (by area) colocalized with TfR (Fig. 5a, i). In contrast, in melan-mu cells, 27% of TYRP1 colocalized with TfR (Fig. 5b, i). Stable expression of full-length BLOC1S5 mRNA (HA-BLOC1S5 or HA-BLOC1S5–202) rescued TYRP1 localization, significantly decreasing TYRP1 localization to TfR-containing compartments to 14% or 10%, respectively (Fig. 5c, h, i). In contrast, stable expression of BLOC1S6 or of truncated isoforms BLOC1S5-del1, HA-BLOC1S5-del2, or HA-BLOC1S5–205 did not significantly decrease the percentage of TYRP1 that colocalized with TfR (Fig. 5d–g, i). These data indicate that the patient-derived isoforms (HA-BLOC1S5-del1 and HA-BLOC1S5-del2) and the normal truncated form (HA-BLOC1S5–205) of BLOC1S5 are not functional for melanosome cargo trafficking.
Similar investigations were not conducted on the patient 2–derived allele, because this patient was identified at a later stage, and the nature of the reading frameshift 1-bp deletion suggested it was very likely pathogenic. It was therefore expected that this allele would display the same behavior as the exon 3–4 deletion in the melan-mu cells complementation assays.
DISCUSSION
We identified two patients with variants of the BLOC1S5 gene (NM_201280.2). Patient 1 (20 years old) was initially diagnosed only with oculocutaneous albinism, but secondarily reported mild bleeding diathesis characterized by easy bruising and infrequent episodes of epistaxis and gingival bleeding. Patient 2 (39 years old) reported menorrhagia and excessive blood loss after deliveries and surgery. Further analyses documented in both patients a lack of detectable platelet dense granules, concomitant with mildly impaired activation-induced ATP release and platelet aggregation in vitro. These features are consistent with a diagnosis of HPS20 (see Table S3 for a comparison of the main clinical features observed in different HPS forms).
BLOC1S5 is a subunit of BLOC-1, and pathogenic variants in the genes encoding three other BLOC-1 subunits cause HPS types 7–9. BLOC-1 was severely destabilized in lysates from patient 1–derived platelets, as demonstrated by the absence of the Dysbindin and Pallidin subunits by immunoblotting. Moreover, the patient’s BLOC1S5 allele was unable to restore BLOC-1 stability or function in melanogenesis and protein sorting in murine melanocytes lacking the orthologous BLOC1S5/Muted protein. These data strongly suggest this represents a loss of function variant.
NGS analysis of patients 1 and 2 DNA did not detect pathogenic variants in any of the 19 known albinism genes in either homozygous or heterozygous state. Since it is unlikely that our analyses would have missed two variants in the same gene, we reject the possibility that one of these genes is involved in the disease in these patients.
Laboratory analyses of platelet aggregation and delta storage disease were not as severe as in the more common HPS-1 patients. This is typical of HPS-7, -8, and -9 patients with BLOC-1 subunit variants,18,20,27 in contrast to the more dramatic platelet disorder observed in corresponding mouse HPS models.28,29,30 However, it should be noted that these are rare forms of HPS, since only a few patients have been reported so far (8 for HPS-7, 9 for HPS-8, 4 for HPS-9).20 Bleeding diathesis and mild albinism were described in all of these patients. Pulmonary disease, associated with all BLOC-3- and AP-3-deficient patients, and granulomatous colitis, associated with some BLOC-3 and BLOC-2-deficient patients, were reported in only one patient with HPS-7, whereas one patient with HPS-9 had recurrent cutaneous infections and a prolonged episode of fever with seizures (similar to HPS-2 and -10 patients with AP-3 deficiency). Our patients’ symptoms are thus consistent with the majority of BLOC-1-deficient patients with a mild form of HPS (Table S3). Of note, HPS patients may show either an absence or a reduced number of delta granules and mild platelet storage pool disease. Screening of all HPS genes in individuals with a bleeding diathesis with or without recognized oculocutaneous albinism may therefore yield additional HPS patients.
It was expected that alterations in genes encoding other BLOC-1 subunits might cause additional forms of HPS. Indeed, mouse mutants in genes encoding five BLOC-1 subunits are associated with hypopigmentation phenotypes and features of syndromic forms of albinism. These are cappuccino (Bloc1s4), reduced pigmentation (Bloc1s3), muted (Bloc1s5), pallid (Bloc1s6), and sandy (Dtnbp1/Bloc1s8). Note that the three additional BLOC-1 subunits, BLOC1S1, BLOC1S2, and BLOC1S7, are shared with a separate protein complex called BORC that is essential in mice and possibly also in humans.31,32 Of the BLOC-1-specific subunits, variants in the human orthologs of pallid/ Bloc1s6, reduced pigmentation/Bloc1s3, and sandy/Bloc1s8 underlie HPS-7, -8, and -9, respectively. We now demonstrate that pathogenic variants in the human ortholog of muted/Bloc1s5 underlie a novel form of HPS. Based on our observations in two patients, we recommend that BLOC1S5 should now be included among the panel of genes tested for variants as a molecular diagnosis for all patients with albinism or a HPS-like phenotype. We also predict that variants in BLOC1S4 may underlie another form of HPS. Because of the additional complications of HPS relative to nonsyndromic albinism and the difficulty in their diagnosis, all HPS genes should be included in screens of albinism patients.
Our data demonstrate the pathogenicity of BLOC1S5 homozygous variants identified in two patients with syndromic oculocutaneous albinism corresponding to HPS. We propose that this form of HPS should be considered as HPS-11.
References
Seward SL Jr, Gahl WA. Hermansky–Pudlak syndrome: health care throughout life. Pediatrics. 2013;132:153–160.
Delevoye C, Marks MS, Raposo G. Lysosome-related organelles as functional adaptations of the endolysosomal system. Curr Opin Cell Biol. 2019;59:147–158.
Bowman SL, Bi-Karchin J, Le L, Marks MS. The road to LROs: insights into lysosome-related organelles from Hermansky–Pudlak syndrome and other rare diseases. Traffic. 2019;20:404–435.
Nazarian R, Huizing M, Helip-Wooley A, Starcevic M, Gahl WA, Dell’Angelica EC. An immunoblotting assay to facilitate the molecular diagnosis of Hermansky–Pudlak syndrome. Mol Genet Metab. 2008;93:134–144.
Montoliu L, Marks MS. A new type of syndromic albinism associated with mutations in AP3D1. Pigment Cell Melanoma Res. 2017;30:5–7.
Montoliu L, Grønskov K, Wei AH, et al. Increasing the complexity: new genes and new types of albinism. Pigment Cell Melanoma Res. 2014;27:11–18.
Arveiler B, Lasseaux E, Morice-Picard F. Clinical and genetic aspects of albinism. Presse Med. 2017;46:648–654.
Lasseaux E, Plaisant C, Michaud V, et al. Molecular characterization of a series of 990 index patients with albinism. Pigment Cell Melanoma Res. 2018;31:466–474.
Arveiler B, Michaud V, Lasseaux E. Albinism: an underdiagnosed condition. J Dermatol Invest. 2019. https://doi.org/10.1016/j.jid.2019.12.010 [Epub ahead of print].
Morice-Picard F, Lasseaux E, Cailley D, et al. High-resolution array-CGH in patients with oculocutaneous albinism identifies new deletions of the TYR, OCA2, and SLC45A2 genes and a complex rearrangement of the OCA2 gene. Pigment Cell Melanoma Res. 2014;27:59–71.
Setty SRG, Tenza D, Truschel ST, et al. BLOC-1 is required for cargo-specific sorting from vacuolar early endosomes toward lysosome-related organelles. Mol Biol Cell. 2007;18:768–780.
Sviderskaya EV, Hill SP, Evans-Whipp TJ, et al. p16(Ink4a) in melanocyte senescence and differentiation. J Natl Cancer Inst. 2002;94:446–454.
Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–1066.
Meng R, Wang Y, Yao Y, et al. SLC35D3 delivery from megakaryocyte early endosomes is required for platelet dense granule biogenesis and is differentially defective in Hermansky–Pudlak syndrome models. Blood. 2012;120:404–414.
Dennis MK, Delevoye C, Acosta-Ruiz A, et al. BLOC-1 and BLOC-3 regulate VAMP7 cycling to and from melanosomes via distinct tubular transport carriers. J Cell Biol. 2016;214:293–308.
Delevoye C, Hurbain I, Tenza D, et al. AP-1 and KIF13A coordinate endosomal sorting and positioning during melanosome biogenesis. J Cell Biol. 2009;187:247–264.
Li W, Zhang Q, Oiso N, et al. Hermansky–Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat Genet. 2003;35:84–89.
Morgan NV, Pasha S, Johnson CA, et al. A germline mutation in BLOC1S3/reduced pigmentation causes a novel variant of Hermansky–Pudlak syndrome (HPS8). Am J Hum Genet. 2006;78:160–166.
Badolato R, Prandini A, Caracciolo S, et al. Exome sequencing reveals a pallidin mutation in a Hermansky-Pudlak-like primary immunodeficiency syndrome. Blood. 2012;119:3185–3187.
Huizing M, Malicdan MCV, Wang JA, et al. Hermansky–Pudlak syndrome: mutation update. Hum Mutat. 2020;41:543–580.
Zhang Q, Li W, Novak EK, et al. The gene for the muted (mu) mouse, a model for Hermansky–Pudlak syndrome, defines a novel protein which regulates vesicle trafficking. Hum Mol Genet. 2002;11:697–706.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Dawood BB, Lowe GC, Lordkipanidze M, et al. Evaluation of participants with suspected heritable platelet function disorders including recommendation and validation of a streamlined agonist panel. Blood. 2012;120:5041–5049.
Falcon-Perez JM, Starcevic M, Gautam R, Dell’Angelica EC. BLOC-1, a novel complex containing the pallidin and muted proteins involved in the biogenesis of melanosomes and platelet dense granules. J Biol Chem. 2002;277:28191–28199.
Moriyama K, Bonifacino JS. Pallidin is a component of a multi-protein complex involved in the biogenesis of lysosome-related organelles. Traffic. 2002;3:666–677.
Starcevic M, Dell’Angelica EC. Identification of snapin and three novel proteins (BLOS1, BLOS2 and BLOS3/reduced pigmentation) as subunits of biogenesis of lysosome-related organelles complex-1 (BLOC-1). J Biol Chem. 2004;279:28393–28401.
Bryan MM, Tolman NJ, Simon KL, et al. Clinical and molecular phenotyping of a child with Hermansky–Pudlak syndrome-7, an uncommon genetic type of HPS. Mol Genet Metab. 2017;120:378–383.
Meng R, Wu J, Harper DC, et al. Defective release of a granule and lysosome contents from platelets in mouse Hermansky–Pudlak syndrome models. Blood. 2015;125:1623–1632.
Novak EK, Hui SW, Swank RT. Platelet storage pool deficiency in mouse pigment mutations associated with seven distinct genetic loci. Blood. 1984;63:536–544.
Reddington M, Novak EK, Hurley E, Medda C, McGarry MP, Swank RT. Immature dense granules in platelets from mice with platelet storage pool disease. Blood. 1987;69:1300–1306.
Pu J, Schindler C, Jia R, Jarnik M, Backlund P, Bonifacino JS. BORC, a multisubunit complex that regulates lysosome positioning. Dev Cell. 2015;33:176–188.
Snouwaert JN, Church RJ, Jania L, et al. A mutation in the Borcs7 subunit of the lysosome regulatory BORC complex results in motor deficits and dystrophic axonopathy in mice. Cell Rep. 2018;24:1254–1265.
Acknowledgements
The authors are grateful to the patients, and to the parents and siblings of patient 1 for participating in this study, and to the French Albinism Association Genespoir, French Agence Nationale de la Recherche (ANR-10-LBX-0038, ANR-10-IDEX-0001-02 PSL, PICT-IBiSA ANR10-INBS-04), and US National Institutes of Health grant R01 EY015625 from the National Eye Institute, for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Pennamen, P., Le, L., Tingaud-Sequeira, A. et al. BLOC1S5 pathogenic variants cause a new type of Hermansky–Pudlak syndrome. Genet Med 22, 1613–1622 (2020). https://doi.org/10.1038/s41436-020-0867-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0867-5
Keywords
This article is cited by
-
The experience of albinism in France: a qualitative study on dyads of parents and their adult child with albinism
BMC Medicine (2024)
-
ZEB1 controls a lineage-specific transcriptional program essential for melanoma cell state transitions
Oncogene (2024)
-
Progressive pulmonary fibrosis in a murine model of Hermansky-Pudlak syndrome
Respiratory Research (2022)
-
Dysregulated myosin in Hermansky-Pudlak syndrome lung fibroblasts is associated with increased cell motility
Respiratory Research (2022)
-
Hermansky-Pudlak syndrome type 1 causes impaired anti-microbial immunity and inflammation due to dysregulated immunometabolism
Mucosal Immunology (2022)
