Tumor Tissue-Specific Biomarkers of Colorectal Cancer by Anatomic Location and Stage


 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
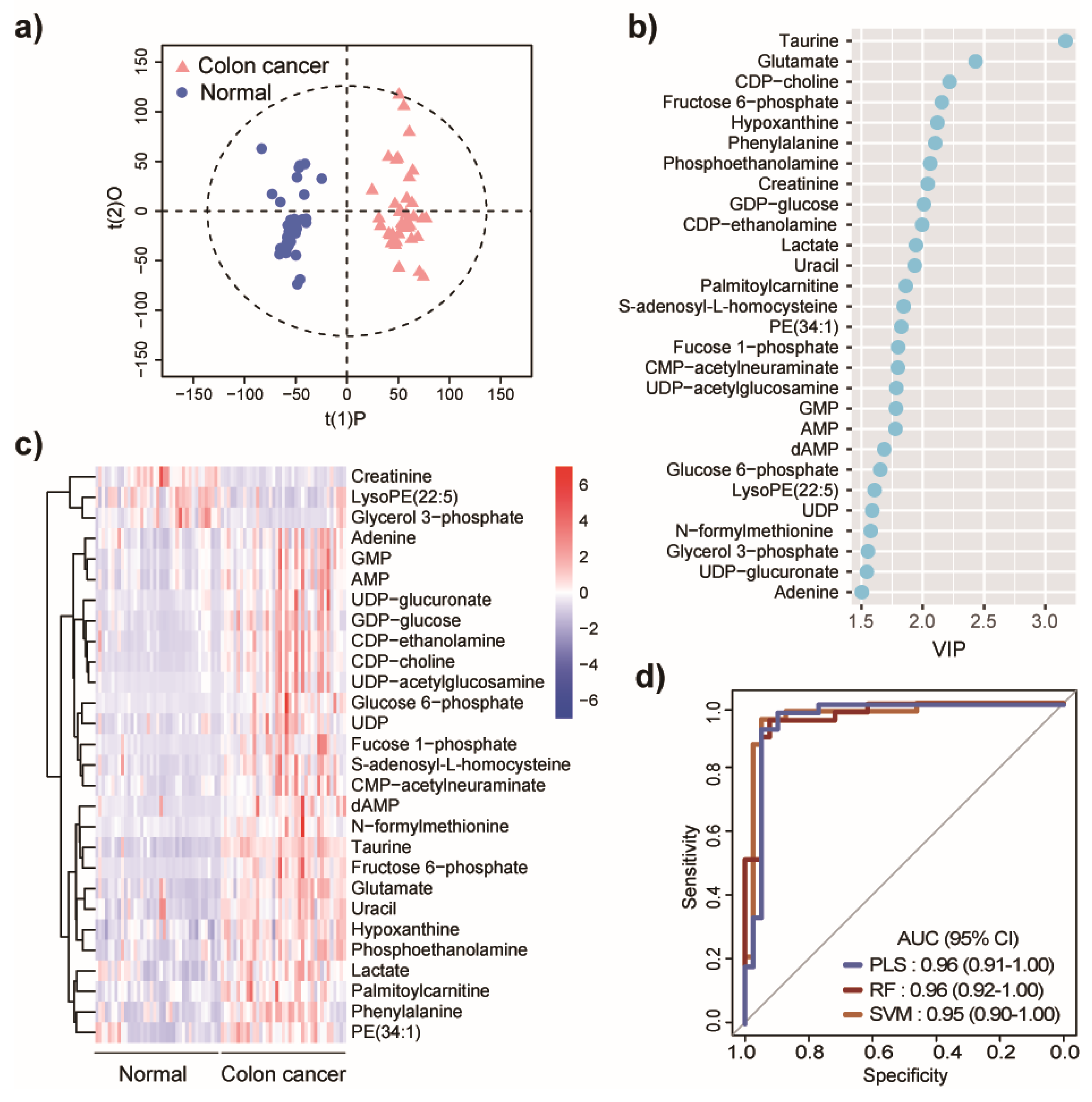
2.1. Metabolic Differences between Colon Cancer and Normal Controls
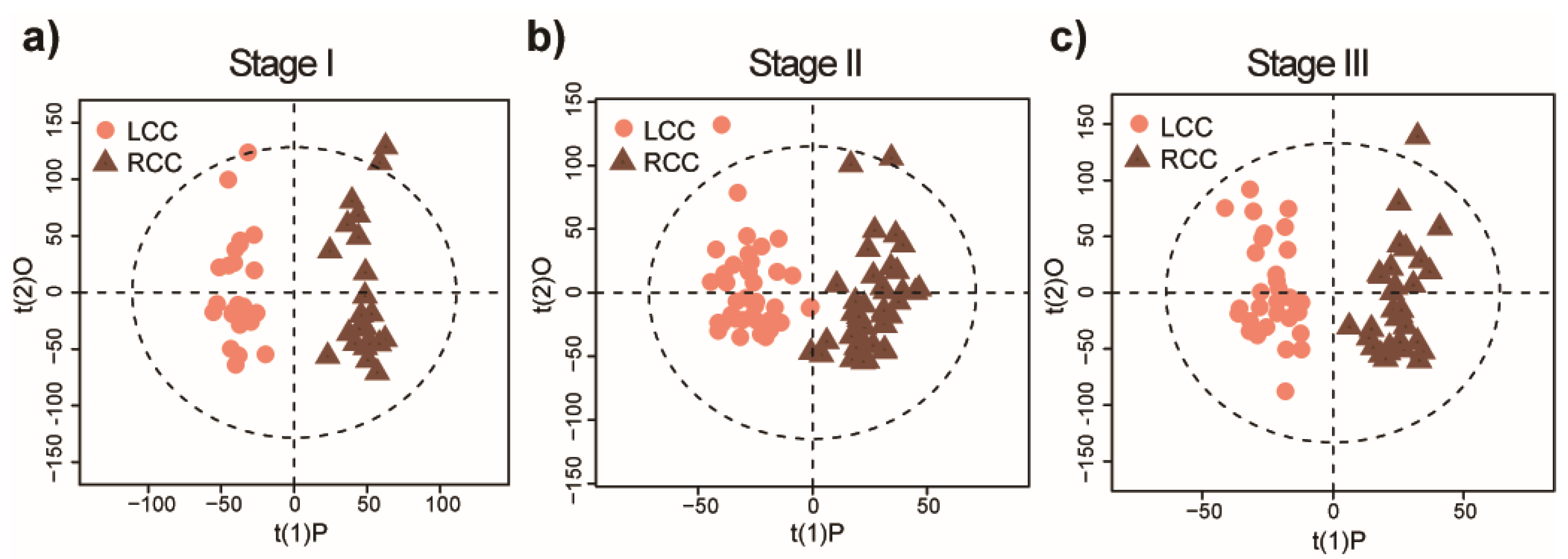
2.2. Distinct Metabolomic Signatures Associated with Anatomic Location
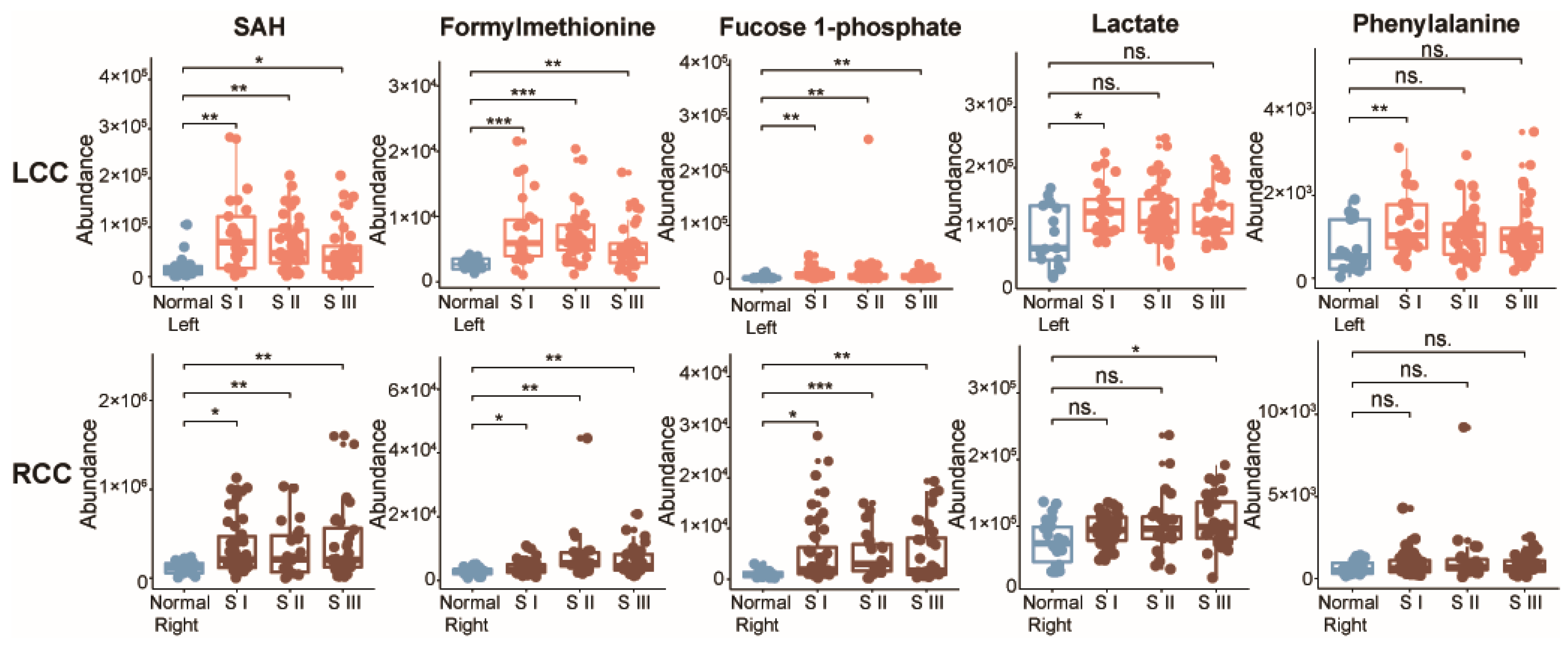
2.3. Potential Metabolic Biomarkers for Early-Stage LCC and RCC
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Sample Collection
4.3. Tissue Metabolite Extraction
4.4. UPLC-MS Analysis
4.5. UPLC-MS Data Processing
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Crooke, H.; Kobayashi, M.; Mitchell, B.; Nwokeji, E.; Laurie, M.; Kamble, S.; McKenna, M.; Masood, A.; Korytowsky, B. Estimating 1- and 5-year relative survival trends in colorectal cancer (CRC) in the United States: 2004 to 2014. J. Clin. Oncol. 2018, 36, 587. [Google Scholar] [CrossRef]
- Wolf, A.M.D.; Fontham, E.T.H.; Church, T.R.; Flowers, C.R.; Guerra, C.E.; LaMonte, S.J.; Etzioni, R.; McKenna, M.T.; Oeffinger, K.C.; Shih, Y.T.; et al. Colorectal cancer screening for average-risk adults: 2018 guideline update from the American Cancer Society. CA Cancer J. Clin. 2018, 68, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Muthukrishnan, M.; Arnold, L.D.; James, A.S. Patients’ self-reported barriers to colon cancer screening in federally qualified health center settings. Prev. Med. Rep. 2019, 15, 100896. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, Y.Y.; Zhao, W.W.; Deng, K.; Wang, Z.Z.; Yang, C.Y.; Ma, L.B.; Openkova, M.S.; Hou, Y.; Li, K. Metabolomics for biomarker discovery in the diagnosis, prognosis, survival and recurrence of colorectal cancer: A systematic review. Oncotarget 2017, 8, 35460–35472. [Google Scholar] [CrossRef] [Green Version]
- Erben, V.; Bhardwaj, M.; Schrotz-King, P.; Brenner, H. Metabolomics Biomarkers for Detection of Colorectal Neoplasms: A Systematic Review. Cancers 2018, 10, 246. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Xie, G.; Chen, T.; Qiu, Y.; Zou, X.; Zheng, M.; Tan, B.; Feng, B.; Dong, T.; He, P.; et al. Distinct urinary metabolic profile of human colorectal cancer. J. Proteome Res. 2012, 11, 1354–1363. [Google Scholar] [CrossRef]
- Zhu, J.J.; Djukovic, D.; Deng, L.L.; Gu, H.W.; Himmati, F.; Chiorean, E.G.; Raftery, D. Colorectal Cancer Detection Using Targeted Serum Metabolic Profiling. J. Proteome Res. 2014, 13, 4120–4130. [Google Scholar] [CrossRef]
- Qiu, Y.; Cai, G.; Zhou, B.; Li, D.; Zhao, A.; Xie, G.; Li, H.; Cai, S.; Xie, D.; Huang, C.; et al. A distinct metabolic signature of human colorectal cancer with prognostic potential. Clin. Cancer Res. 2014, 20, 2136–2146. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Mironova, V.; Chen, Y.; Lundh, E.P.F.; Zhang, Q.; Cai, Y.; Vasiliou, V.; Zhang, Y.; Garcia-Milian, R.; Khan, S.A.; et al. Molecular Pathway Analysis Indicates a Distinct Metabolic Phenotype in Women With Right-Sided Colon Cancer. Transl. Oncol. 2020, 13, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Rattray, N.J.W.; Zhang, Q.; Mironova, V.; Santos-Neto, A.; Hsu, K.S.; Rattray, Z.; Cross, J.R.; Zhang, Y.; Paty, P.B.; et al. Sex Differences in Colon Cancer Metabolism Reveal A Novel Subphenotype. Sci. Rep. 2020, 10, 4905. [Google Scholar] [CrossRef] [Green Version]
- Bufill, J.A. Colorectal-cancer-evidence for distinct genetic categories based on proximal or distal tumor location. Ann. Intern. Med. 1990, 113, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Yang, D.Y.; Yau, L.; Feng, S.B.; Cremolini, C.; Zhang, W.; Maus, M.K.H.; Antoniotti, C.; Langer, C.; Scherer, S.J.; et al. Primary tumor location as a prognostic factor in metastatic colorectal cancer. JNCI J. Nat. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Gervaz, P.; Usel, M.; Rapiti, E.; Chappuis, P.; Neyroud-Kaspar, I.; Bouchardy, C. Right colon cancer: Left behind. Eur. J. Surg. Oncol. 2016, 42, 1343–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrelli, F.; Tomasello, G.; Borgonovo, K.; Ghidini, M.; Turati, L.; Dallera, P.; Passalacqua, R.; Sgroi, G.; Barni, S. Prognostic survival associated with left-sided vs right-sided colon cancer a systematic review and meta-analysis. JAMA Oncol. 2017, 3, 211–219. [Google Scholar] [CrossRef]
- Schroll, M.M.; Ludwig, K.R.; Bauer, K.M.; Hummon, A.B. Calcitriol supplementation causes decreases in tumorigenic proteins and different proteomic and metabolomic signatures in right versus left-sided colon cancer. Metabolites 2018, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Xiao, Y.; Shu, D.; Liang, X.; Hu, X.; Xie, Y.; Lin, D.; Li, H. Metabolomics analysis in serum from patients with colorectal polyp and colorectal cancer by (1)H-NMR spectrometry. Dis. Mark. 2019, 2019, 3491852. [Google Scholar] [CrossRef] [Green Version]
- Zha, H.; Cai, Y.; Yin, Y.; Wang, Z.; Li, K.; Zhu, Z.J. SWATHtoMRM: Development of high-coverage targeted metabolomics method using SWATH technology for biomarker discovery. Anal. Chem. 2018, 90, 4062–4070. [Google Scholar] [CrossRef]
- Ikeda, A.; Nishiumi, S.; Shinohara, M.; Yoshie, T.; Hatano, N.; Okuno, T.; Bamba, T.; Fukusaki, E.; Takenawa, T.; Azuma, T.; et al. Serum metabolomics as a novel diagnostic approach for gastrointestinal cancer. Biomed. Chromatogr. 2012, 26, 548–558. [Google Scholar] [CrossRef]
- Farshidfar, F.; Weljie, A.M.; Kopciuk, K.A.; Hilsden, R.; McGregor, S.E.; Buie, W.D.; MacLean, A.; Vogel, H.J.; Bathe, O.F. A validated metabolomic signature for colorectal cancer: Exploration of the clinical value of metabolomics. Br. J. Cancer 2016, 115, 848–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efron, B.; Tibshirani, R. Improvements on cross-validation: The.632+ bootstrap method. J. Am. Stat. Assoc. 1997, 92, 548–560. [Google Scholar] [CrossRef]
- Peng, D.X.; Lai, F.J. Using partial least squares in operations management research: A practical guideline and summary of past research. J. Oper. Manag. 2012, 30, 467–480. [Google Scholar] [CrossRef]
- Cross, A.J.; Moore, S.C.; Boca, S.; Huang, W.Y.; Xiong, X.; Stolzenberg-Solomon, R.; Sinha, R.; Sampson, J.N. A prospective study of serum metabolites and colorectal cancer risk. Cancer 2014, 120, 3049–3057. [Google Scholar] [CrossRef]
- Liesenfeld, D.B.; Grapov, D.; Fahrmann, J.F.; Salou, M.; Scherer, D.; Toth, R.; Habermann, N.; Bohm, J.; Schrotz-King, P.; Gigic, B.; et al. Metabolomics and transcriptomics identify pathway differences between visceral and subcutaneous adipose tissue in colorectal cancer patients: The ColoCare study. Am. J. Clin. Nutr. 2015, 102, 433–443. [Google Scholar] [CrossRef]
- Rattray, N.J.W.; Charkoftaki, G.; Rattray, Z.; Hansen, J.E.; Vasiliou, V.; Johnson, C.H. Environmental influences in the etiology of colorectal cancer: The premise of metabolomics. Curr. Pharmacol. Rep. 2017, 3, 114–125. [Google Scholar] [CrossRef]
- Chan, E.C.Y.; Koh, P.K.; Mal, M.; Cheah, P.Y.; Eu, K.W.; Backshall, A.; Cavill, R.; Nicholson, J.K.; Keun, H.C. Metabolic profiling of human colorectal cancer using high-resolution magic angle spinning nuclear magnetic resonance (HR-MAS NMR) spectroscopy and gas chromatography mass spectrometry (GC/MS). J. Proteome Res. 2009, 8, 352–361. [Google Scholar] [CrossRef]
- Jimenez, B.; Mirnezami, R.; Kinross, J.; Cloarec, O.; Keun, H.C.; Holmes, E.; Goldin, R.D.; Ziprin, P.; Darzi, A.; Nicholson, J.K. 1H HR-MAS NMR spectroscopy of tumor-induced local metabolic “field-effects” enables colorectal cancer staging and prognostication. J. Proteome Res. 2013, 12, 959–968. [Google Scholar] [CrossRef]
- Mirnezami, R.; Jimenez, B.; Li, J.V.; Kinross, J.M.; Veselkov, K.; Goldin, R.D.; Holmes, E.; Nicholson, J.K.; Darzi, A. Rapid diagnosis and staging of colorectal cancer via high-resolution magic angle spinning nuclear magnetic resonance (HR-MAS NMR) spectroscopy of intact tissue biopsies. Ann. Surg. 2014, 259, 1138–1149. [Google Scholar] [CrossRef]
- Qiu, Y.; Cai, G.; Su, M.; Chen, T.; Liu, Y.; Xu, Y.; Ni, Y.; Zhao, A.; Cai, S.; Xu, L.X.; et al. Urinary metabonomic study on colorectal cancer. J. Proteome Res. 2010, 9, 1627–1634. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | m/z | RT | FC a | VIP b | pc | pd | pe | pf |
|---|---|---|---|---|---|---|---|---|
| SAH | 385.1282 | 363.2 | 0.29 | 1.8 | 0.028 | 0.001 | 0.026 | N.S. |
| Formylmethionine | 176.0393 | 188.9 | 0.64 | 1.9 | 0.010 | <0.001 | 0.033 | N.S. |
| Fucose 1-phosphate | 243.0285 | 438.0 | 0.38 | 1.3 | 0.026 | 0.001 | 0.010 | N.S. |
| Lactate | 89.0233 | 218.2 | 0.75 | 2.1 | 0.008 | 0.046 | N.S. | N.S. |
| Phenylalanine | 207.1130 | 252.0 | 0.53 | 2.3 | 0.003 | 0.019 | N.S. | N.S. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, Y.; Rattray, N.J.W.; Zhang, Q.; Mironova, V.; Santos-Neto, A.; Muca, E.; Vollmar, A.K.R.; Hsu, K.-S.; Rattray, Z.; Cross, J.R.; et al. Tumor Tissue-Specific Biomarkers of Colorectal Cancer by Anatomic Location and Stage. Metabolites 2020, 10, 257. https://doi.org/10.3390/metabo10060257
Cai Y, Rattray NJW, Zhang Q, Mironova V, Santos-Neto A, Muca E, Vollmar AKR, Hsu K-S, Rattray Z, Cross JR, et al. Tumor Tissue-Specific Biomarkers of Colorectal Cancer by Anatomic Location and Stage. Metabolites. 2020; 10(6):257. https://doi.org/10.3390/metabo10060257
Chicago/Turabian StyleCai, Yuping, Nicholas J. W. Rattray, Qian Zhang, Varvara Mironova, Alvaro Santos-Neto, Engjel Muca, Ana K. Rosen Vollmar, Kuo-Shun Hsu, Zahra Rattray, Justin R. Cross, and et al. 2020. "Tumor Tissue-Specific Biomarkers of Colorectal Cancer by Anatomic Location and Stage" Metabolites 10, no. 6: 257. https://doi.org/10.3390/metabo10060257