
Abstract
Every year there are > 33 million cases of Respiratory Syncytial Virus (RSV)-related respiratory infection in children under the age of five, making RSV the leading cause of lower respiratory tract infection (LRTI) in infants. RSV is a global infection, but 99% of related mortality is in low/middle-income countries. Unbelievably, 62 years after its identification, there remains no effective treatment nor vaccine for this deadly virus, leaving infants, elderly and immunocompromised patients at high risk. The success of all pathogens depends on their ability to evade and modulate the host immune response. RSV has a complex and intricate relationship with our immune systems, but a clearer understanding of these interactions is essential in the development of effective medicines. Therefore, in a bid to update and focus our research community’s understanding of RSV’s interaction with immune defences, this review aims to discuss how our current knowledgebase could be used to combat this global viral threat.
Similar content being viewed by others

Introduction
Respiratory syncytial virus (RSV) remains a significant burden to global health, with nearly all children thought to be exposed to this virus by the age of two, making it the most common cause of paediatric respiratory tract infection (RTI) and the biggest risk factor for severe infection in infants [1]. Elderly individuals are the second major group at risk of severe infection, with similar rates of intensive care admission and mortality to influenza. Indeed, RSV poses a particular threat to residents of long-term care facilities [2]. RSV outbrakes are observed worldwide and follow a seasonal pattern in most parts of the world, with significant occurrence during winter months [3, 4]. However, in some global regions, RSV infections occur throughout the year but still peak during the winter [5, 6].
Virus structure and epidemiology
RSV is a 15.2 kb, negative sense, RNA virus, with 10 genes producing 11 proteins. The viral envelope has three transmembrane proteins: the fusion glycoprotein (F), attachment glycoprotein (G) and the small hydrophobic (SH) protein. Other structural proteins are nucleoprotein (N), large RNA polymerase (L), phosphoprotein (P), matrix protein (M) and transcription factors (M2-1 & M2-2). In addition to these, RSV produces two non-structural (NS) proteins, NS1 and NS2 (Fig. 1) [7].

RSV structure. The RSV genome encodes for 10 genes, giving rise to 11 proteins. The non-structural proteins, NS1 and NS2, are not present in the viron, but are expressed in high levels on infection of the host cell
There are two major antigenic strains of RSV, RSV-A and RSV-B, which have distinct epitopes in the F and G proteins, as well as molecular differences in several genes [8, 9]. Both strains co-circulate, often alternating dominance annually [4]. There is some evidence that RSV-A is associated with higher morbidity, though other studies have shown no significant variation between strains [10,11,12,13,14,15]. The majority of RSV-infected patients recover after mild illness, however, a study in the UK reported a small percentage (6.9%), developed an acute respiratory infection (ARI) and required hospital admission, 2.7% needed intensive care and 1.5% required a ventilator [16]. It is estimated, that by the age of two, nearly all children have had at least one RSV infection, which could equate to 3.4 million children/year needing hospitalisation [17]. Large scale European epidemiological studies, showed that outbreak patterns vary between countries, with those further east seeing a later start to the RSV season, while more northerly countries have a longer RSV season [4]. Variation in disease burden is tied to metrological conditions; RSV fairs best in cool temperatures (6.3 °C) and high humidity (84%), meaning cool, dry winters are predicted to have less RSV-related illness, than warm, wet conditions [18].
RSV’s primary infection site is the respiratory tract epithelium. The virus is spread by droplet, contact and aerosol transmission [19, 20]. Infection can proceed to the lungs, causing serious disease; with the sloughing of dead cells enabling the virus to spread further into the respiratory airways [21]. There is also evidence that severe RSV infection in early life, increases the likelihood of asthma [22,23,24,25]. However, this phenomenon could also be a result of undiagnosed genetic predisposition to respiratory infection, thereby enhancing the observed associated with asthma development, highlighting the need for longer-term studies to fully elucidate this theory [26,27,28]. Once infected, infants are particularly susceptible to developing a severe infection; the immune system of an infant has fewer degenerative pathways than adults, relying heavily on their innate immune response and maternal antibodies to protect against infection [1, 29, 30]. The bronchial lumens of infants are underdeveloped, and are, therefore, narrower and more likely to be blocked by excessive mucus produced in response to infection, leading to reduced airflow, poor gas exchange in the alveoli and low blood oxygen levels [31]. Efficient clearance of a virus requires a strong Th1 response to activate IFN-γ producing cytotoxic T cells [32, 33]. However, multiple studies have shown that infants with RSV infection have a skewed response, producing a Th2 cytokine profile [34, 35]. RSV infection in young infants increases expression of Thymic Stromal Lymphopoietin (TLSP), which has been shown to be vital for immunopathology in mouse models and has been linked to later asthma development [25, 35]. TLSP alters T cell differentiation via dendritic cells (DCs), with TLSP primed DCs causing CD4 + T cells to express Th2 characteristic cytokines [36, 37]. TLSP is also able to induce type two innate lymphoid cells (ILC2) which play a significant role in allergy [25, 38]. RSV infection also increases expression of IL-33; this cytokine acts on both DCs and ILC2 to promote the differentiation of Th2 cells, increase mucus production and heighten airway sensitivity [39, 40]. The ability of RSV to increase levels of TLSP and IL-33 and thus promote a Th2 response, may indeed be responsible for negatively influencing the overall antiviral response.
With no vaccines currently available, limiting the spread of RSV infection remains under the control of good hygiene and hand washing; however, the close proximity of individuals at day-care centres and schools make these locations common epicentres of RSV outbreaks [19, 20, 41].
Treatments and vaccines
Despite affecting millions of people each year, there is still no fully effective, curative therapeutic available for RSV. Patients admitted to the hospital are generally given supportive treatments, including oxygen and airway clearance [42, 43]. Ribavirin is the only anti-viral drug on the market for use against RSV, though it is currently not recommended by the American Association of Paediatrics, guidelines vary between regions [42,43,44,45,46]. First developed in 1972, Ribavirin is a guanosine analogue, which limits the replication of several RNA and DNA viruses. While originally licenced for the treatment of RSV, its most significant use has been in the management of Hepatitis C Virus (HCV) [47]. In HCV treatment, Ribavirin was used to good effect in conjunction with other drugs, including the anti-viral cytokine, Interferon (IFN)-α [47,48,49]. While not widely prescribed, Ribavirin is still used in “extreme” RSV cases [42, 45, 50,51,52]. A prophylactic preventative anti-RSV antibody, Palivizumab, is also available. Produced by MedImmune, this monoclonal antibody is administered through monthly intramuscular injections during the RSV season [46, 53]. Because of its preventative nature, accurately measuring the effectiveness of Palivizumab has proved difficult. Evidence suggests that Palivizumab significantly reduces RSV-related hospitalisations [54,55,56]. However, updated guidelines from the American Academy of Paediatrics stated that Palivizumab “has limited effect on RSV hospitalizations on a population basis, no measurable effect on mortality, and a minimal effect on subsequent wheezing” [57]. Palivizumab’s high cost generally constrains its use to only “high risk” children, including infants born prematurely, those with congenital heart disease (CHD) or chronic lung disease (CLD) [58,59,60]. In addition, a randomized trial showed that Palivizumab should not be used as treatment; administering Palivizumab to infants with RSV bronchiolitis had no impact on outcomes [61]. Palivizumab’s effectiveness and cost are major limiting factors in its use against RSV; therefore, global research aims must remain focussed on the development of an effective treatment and preventative vaccine.
RSV vaccine research began soon after the virus was first isolated in 1956, but has proven challenging. The 1966 trial of a formalin-inactivated RSV vaccine (FI-RSV), sensitised children to the virus, leading to enhanced disease in the immunised cohort. Unfortunately, this resulted in hospitalisations and the death of two children [62,63,64]. This failure of FI-RSV instilled caution over new RSV vaccines and halted the quest for a medicinal solution. However, this period of “reflection” sparked intensive research in the immune evasion and modulatory mechanisms of RSV; generating significant developments in our understanding of the virus, not least the discovery that RSV is attenuated upon deletion of the SH, M or NS1/2 genes, revealing these as prime targets for therapeutic intervention [65,66,67,68].
RSV presents a challenging, but essential global threat to harness. Several high-risk patient groups would particularly benefit from an effective RSV treatment. Indeed, RSV can be fatal for neonates and immunocompromised individuals, for whom any vaccines offer little protection. It is RSV’s multiple “anti-immune” effects, that cloud our current understanding. Therefore, increased knowledge of RSV’s cellular and molecular interactions and subversive mechanisms are fundamental in facilitating future drug and vaccine design.
Immune evasion mechanisms of RSV
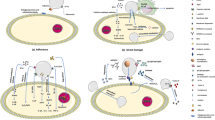
The immune response can be split into two branches: Innate and Adaptive. Together they provide comprehensive protection from pathogens, thus limiting damage to the host. The innate response is fast acting and non-specific, while the adaptive response provides targeted clearance of the pathogen and lasting immunological memory, that quickly eliminates the pathogen upon reinfection. However, the key to the success of all pathogens is their ability to evade and subvert the immune response. RSV has multiple mechanisms to limit host immunity, allowing it to replicate unhindered, ultimately leading to tissue damage and subsequent clinical symptoms of the disease. Viral evasion is often mediated via conserved mechanisms and limit the immune response at several stages of the viral life cycle [69, 70]. RSV’s interaction with the immune system at specific infection and replication points are key to its survival (Fig. 2). Having a genome that codes for 11 proteins provide RSV with multiple mechanisms to mask its replication and modulate the immune response [17]. While there is still much to elucidate, the effect of several specific RSV proteins upon immunity has been well characterised (Table 1).

The RSV Life Cycle. (1) The virion initially binds to the host cell through its G protein and membrane fusion is mediated by the F protein, which anchors into the membrane of the target cell and then folds on itself to bring the viral and host membranes into contact, resulting in membrane fusion. (2) The genome of the virus is used for protein synthesis, with large amounts of NS1/2 and sG protein produced shortly after infection. These proteins protect the replicating virus from the host immune defences. (3) The viral genome is replicated and structural proteins are produced. (4) The surface glycoproteins are synthesised in the Golgi body and deposited in the host membrane. (5) Assembly of the new virion takes place in the cytoplasm, before budding through the host cell membrane, picking up its surface glycoproteins as part of this process. sG protein is also released
Intracellular immunomodulation mechanisms
RSV, like many pathogens, influences cellular signalling pathways, thus disrupting the overall immune response, and limiting the speed and effectiveness of anti-viral clearance.
NS1 and NS2
The two non-structural (NS) proteins of RSV, NS1 and NS2 (which are made up of 139 and 124 amino acids, respectively), are the first proteins to be produced by the virus upon infection [7, 46]. These two proteins show little sequence homology, except for a short region at the C-terminus [71]. However, there is a high level of sequence identity between NS1 and NS2 of circulating RSV strains. Indeed, the conservation of the NS proteins between RSV strains suggests they hold an important role in viral replication.
As their name suggests, the NS proteins have no structural role, with NS1 and NS2 deficient RSV (∆NS1/2-RSV), still able to replicate, albeit with poor growth in immunocompetent cells [67]. The ∆NS1/2-RSV grows well in IFN receptor-deficient Vero cells [72, 73]. In immunocompetent cells Type I IFNs are released when a pathogen is detected; these IFNs act in an autocrine and paracrine manner, binding to their specific IFN receptors and triggering activation of the Janus kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathway. JAK/STAT signalling promotes the transcriptional upregulation of hundreds of IFN Sensitive Genes (ISGs) (often referred to as the “Interferome”), including cytokines, chemokines and anti-viral mediators [74]. ISGs are translated into effector proteins, which enhance the immune response and limit infection (Fig. 3). The Type I IFN pathway has been shown to be essential in clearing several viruses, including HCV and Influenza A [75, 76]. However, RSV infection only induces a weak anti-viral IFN response, that is insufficient to clear the virus [77,78,79].

TLR & IFN signalling. (1) Toll-like Receptor 3 & 8 detect intracellular pathogens by detecting dsRNA and ssRNA, respectively. Once initiated, signalling cascades activate transcription factors, (2) which upregulate anti-viral IFNs (Type I, II and III) and pro-inflammatory cytokines. (3) IFNs act on the infected and neighbouring cells by binding the Interferon receptors (e.g. IFNAR). (4) Change in receptor conformation allows the receptor-associated kinases, Tyk and Jak1, to trans-phosphorylate, which in turn phosphorylate receptor subunits, providing docking sites for STAT proteins. (5) Receptor-associated STATs become phosphorylated, dissociate from the receptor and form homo- or hetero-dimers. The IFN-α-activated STAT1:STAT2 dimer binds IRF9, forming a complex that translocates to the nucleus and stimulates the expression of Interferon Sensitive Genes (ISGs). RSV NS proteins have been shown to inhibit IFN signal transduction by impairing STAT activation
Several studies have investigated the ability of the RSV NS proteins to limit IFN signalling, with both NS1 and NS2 documented to suppress ISGs [80,81,82,83]. NS1 and NS2 are key to RSV’s regulation of the IFN response. NS1 harnesses a specific cellular E3 ligase, which selectively targets STAT2 for ubiquitination and proteasome-mediated degradation [81, 84]. In removing STAT2, RSV acts to limit anti-viral JAK/STAT signalling, thereby blocking the normal function of Type 1 IFNs and ultimately reducing ISG transcription [81, 82]. The presence of NS1 also modulates the T cell response, reducing the number of anti-viral CD8 + T cells and Th17 cells [85, 86], while also bolstering the activation of Th2 cells [34, 87], which, if uncontrolled, progresses the physiological symptoms of the disease. Additionally, the NS1 protein is associated with Mitochondrial Anti-viral Signalling Protein (MAVS), which may block its interaction with Retinoic Inducible Gene I (RIG-I), reducing IFN production in infected cells [88]. NS2 has also been implicated in targeting RIG-I, although this study was unable to show an interaction with MAVS [89].
Type 1 IFNs also sensitise infected cells to programmed cell death through the Fas-Associated protein with Death Domain (FADD) and Caspase-8 pathways. The death of infected cells effectively removes the virus’s “life-line”, limiting viral production and preventing infection of surrounding tissue [90]. In addition to limiting IFN signalling, NS2 also stimulates the phosphoinositide 3-kinase (PI3K) pathway, leading to delayed cell death and enhanced cell survival [71]. This prevents the action of NK and CD8 + T cells, allowing the virus to continue replicating in infected cells [71, 85]. RSV is also able to upregulate the expression of the Programmed Cell Death Ligand (PD-L)1, enabling infected cells to attenuate CD8 + T cell-mediated killing [91]; indeed, these discoveries reveal that the virus uses multiple molecular mechanisms to limit cell death. As well as the direct ubiquitination of STAT2, NS1 and NS2 have been linked to the reduction of several signalling molecules, including RIG-I, IRF3, TRAF3 and IKKε, further highlighting the broad spectrum of immune signalling components RSV targets to ensure the propagation of its infection and replication lifecycle [89, 92, 93].
Interestingly, type III IFNs have also been shown to be important in RSV infection. Much like type I IFNs, type III IFNs induce an anti-viral state, however, while type I IFNs are recognised by almost all cell types, type III IFNs are only detected by a subset of cells, typically those in the mucosal membrane, including macrophages, lymphocytes, Plasmacytoid DCs.
(pDCs) and epithelial cells [94]. This restricted expression of IFN III receptors allows enhanced response in specific cells without stimulating neighbouring cells. It is postulated that the restricted response to type III IFNs may have evolved to enable a selective anti-viral response in cells most likely to encounter viral infection [95, 96]. Although they bind different receptors, both type I and III IFNs activate the formation of Interferon Stimulated gamma factor 3 (ISGF3) (Fig. 3), leading to ISG production. Both type I and III IFNs are produced by and can act on airway epithelial cells, revealing type III IFN’s importance in the context of viral respiratory infections [94, 95]. Both type I and III IFNs are produced at low levels in response to RSV infection in the A549 cell line, however, the removal of the NS proteins causes an increase in all IFNs, highlighting the role of these RSV proteins in suppressing the innate immune signalling that induces these anti-viral cytokines [97, 98]. Additionally, investigating IFN production in primary nasal epithelial cells showed type III IFN production, but not type I, is induced by RSV [99]. This suggests that primary human nasal epithelial cells behave differently to cell lines, and that specific cell lines also mount differential responses; a factor that should be considered when reviewing the literature and designing physiologically relevant experiments [99,100,101].
While NS1 and NS2 together severely impair the IFN response in humans, it is thought that the scope of their interplay is still not fully understood. Indeed, how these proteins function in different species further confuses the issue, with bovine RSV (bRSV), NS proteins appearing to target IFN production by blocking IRF3 activation, rather than blocking the IFN-mediated JAK/STAT signalling [102].
Mechanisms to limit antigen recognition
The surface of each virion holds the F, G and SH proteins. The G protein enables attachment of the virus to the host cell and the F protein initiates fusion of the host and viral membranes [103]. To enable viral entry into the target cell, initial contact is made through the G protein which allows the engagement of a secondary receptor, causing the activation of the F protein and membrane fusion (Fig. 4). Which cell surface proteins are used by G and F are debated. Extensive studies using submerged cell lines have shown that heparan sulfate is needed for RSV entry [104, 105], while other studies have shown that CX3CR1 is sufficient for viral entry [106, 107]. Investigations using human airway epithelial (HAE) cultures have seen low levels of heparan sulfate expressed on the apical cell surface [108], suggesting that the use of heparan could be less clinically relevant [107,108,109,110].

The RSV F protein facilitates the fusion of the viral and host membranes. The stable form of the F protein anchors its hydrophobic N-terminus into the membrane of the target cell. The extended form of the F protein is not stable and the coils of the Heptad repeat A (blue) and Heptad repeat B (green) domains fold together. This overcomes the hydration force, to allow the viral capsid to fuse with the cell
Less is known about the role of the SH protein; while it is not needed for viral growth, the SH protein is conserved across all strains of RSV and is thought to influence virulence [46, 111, 112]. Their prominent external placement of the G and F proteins makes them key antigens for immune recognition, as a result, RSV has developed several mechanisms to limit host detection of both proteins.
G protein
The G protein is the major attachment protein of RSV and has been shown to bind heparan sulfate on the surface of immortalised cell lines and CX3CR1 on primary ciliated cells [107, 110, 113]; indeed, heparin reduces RSV infection of cell [114], and antibodies that prevent G protein–CX3CR1 interaction reduce RSV infection of mice [115]. While low levels of CX3CR1 are seen in both the upper and lower respiratory tract of infants, RSV has been shown to preferentially infect CX3CR1-expressing cells [116]. Another study found that removing RSV’s CX3C domain had no impact on the infective potential and replication of RSV [117]. To protect the G protein from detection it is heavily glycosylated. Glycosylation is a common viral strategy used to protect the antigenic protein from antibody recognition [118, 119]. This mechanism allows the viral glycan structure to change, frequently altering the macrostructure of the G protein and thus masking the protein backbone from antibodies, which effectively limits their affinity.
Large variation in oligosaccharide arrangement across RSV strains also generates antigenic variation, which, in turn, limits the efficiency of immune memory, as previously generated antibodies have a poor affinity to seasonal variants of the G protein [120]. In addition, G protein frameshifts, point mutations and premature stop codons are regularly observed between seasonal strains [121]. These “immune avoidance” strategies decrease the likelihood of neutralising antibodies (against the G protein), being protective between seasons, effectively rendering immunological memory redundant against RSV infection. To avoid the adaptive immune response further, the G protein contains two hypervariable regions within its ectodomain, allowing regular mutation of antigenic epitopes and preventing recognition by antibodies selected for during a previous infection, confounding immune memory further [11]. These variations in RSV are used as the basis for its strain classification; specifically into RSV-A and RSV-B genotypes [12, 122]. As well as suppressing an effective humoral response, antigen variability enhances virus pathogenicity; in 2010, a new genotype of RSV-A was discovered in Ontario, Canada, named RSV-A ON1. RSV-A ON1 contains a 72 nucleotide duplication at the C terminus of the G protein [123]. This strain is now observed worldwide; a study in Vietnam saw that as RSV-A ON1 spread, there was an increased risk of lower respiratory tract infections and pneumonia [124].
As well as limiting the antibody response, heavy glycosylation of the G protein also hampers antigen presentation to T cells. To activate T cells viral antigen are presented by antigen-presenting cells (APC), such as DCs, through the major histocompatibility complex (MHC) and costimulatory molecules. The resulting structure forms an immunological synapse [125]. This stabilises the interaction between the MHC and T cell receptor, allowing correct alignment and activation of cognate T cells. If the presented antigen has sufficient affinity to the three complementary-determining regions of the T cell receptor, the T cell is activated and begins affinity maturation. Heavy glycosylation of proteins can disrupt this process, curtailing T cell activation [118]. During processing for presentation, antigens have some or all of the oligosaccharide groups removed by N-glycanase, before proteolytic cleavage within lysosomes. Any remaining large oligosaccharides can affect the proteolytic cleavage, impacting the T cell repertoire produced. These short antigens bind to vacant MHC molecules in the lysosome, before being trafficked to the surface of the cell [126]. As a result, RSV infection generates a limited T cell response against the G protein. As CD4 + T cell help is critical in the affinity maturation of B cells, this process further hinders the generation of high affinity neutralising antibodies that can target the G protein.
The RSV G protein can also be produced in a soluble form (sG), which acts as an immune “decoy” (Fig. 2). The sG protein is produced in large quantities at the beginning of the viral life cycle, flooding the surrounding area and limiting the effectiveness of G-specific antibodies upon the actual RSV virion [127]. The creation of sG proteins is achieved by initiating transcription at the second AUG codon; this removes 65 amino acids from the N-terminal, the transmembrane region that normally anchors the G protein in the viral capsid. This shorter, truncated version has a hydrophobic amino terminus and is trafficked out of the infected cell and can be detected in the culture medium [128]. The combination of highly variable glycosylation and sG protein release, could limit the effectiveness of any G-specific antibodies and hinder effective immune memory. When studied in vivo it was found that sG protects RSV neutralisation from both G- and F-specific antibodies [127]; while the antibody decoy model suggests that sG would result in protection from G-specific antibodies, sG also reduced levels of F-specific antibodies, suggesting that sG uses multiple mechanisms to reduce the antibody response. Indeed, these modulatory effects of sG protein could also alter cytokine responses, which, in turn, limit the Th1 response [129, 130]. Interestingly, RSV lacking sG increases pro-inflammatory modulators, such as the chemokines CCL5 and IL-8; these findings suggest a role for the sG in blocking the recruitment of immune cells (specifically T cell and neutrophils), to the site of infection [130]. Furthermore, sG protein also limits the impact of antibody-dependent cell-mediated cytotoxicity and clearance of virus particles through the complement system [131], revealing yet another immune-modulatory effect of this soluble viral protein. Importantly, while these discoveries reveal how the RSV sG protein launches an effective immune evasion strategy, which affects both the innate and adaptive responses, they also highlight that it is not the only mechanism by which RSV suppresses and avoids immunity.
The G protein contains a CX3C motif (in both in its soluble and membrane-bound form), which acts to limit immune cell recruitment and thus modify the immune response [106, 132]. Through its CXC3 motif, RSV’s G protein acts as a mimic of the chemokine CX3CL1 (Fractalkine). As well as blocking the action of CX3CL1, the RSV’s CX3C motif allows the attachment of viral particles through the CX3CR receptor, aiding RSV infectivity [107, 117]. In addition, the CX3CR region reduces IFN production, with reduced levels of IFN-α2, IFN-λ1 and IFN-λ2 observed from A549 cells infected with WT RSV, compared to a CX3CR motif mutant version of RSV. This reduced IFN response limits the antiviral activity of the cell, increasing opportunity for RSV replication [132]. Co-culturing PBMCs with A549 epithelial cells infected with WT or CX3C-mutated RSV strains influenced the expression of anti-viral cytokines in several immune cell types. A greater proportion of monocytes and pDCs produced IFN-α and TNF-α, and more CD8 + and CD4 + T cells produced IFN-γ, when co-cultured with A549 cells infected the CX3C-mutated RSV, compared to the WT strain [132]. As pDCs are a major source of Type I IFNs and hold an important role in shaping the overall immune response, RSV’s influence over this cell type, through its G protein, is of major importance to our understanding of RSV’s immune evasion strategy. Furthermore, the presence of the CX3CR motif is also associated with reduced T cell trafficking, CD8 + T cell function and IFN-γ expression, revealing yet another key function for this region in blocking effective immunity [106]. Collectively, these discoveries show the RSV G protein to have a broad role in immunomodulation, allowing the virus to limit the impact of G-specific antibodies, reduce immune cell migration to the site of infection and alter key cytokine production, thus impacting the normal function of several immune cell types.
F protein
The activity of the F protein is thought to be essential for viral replication and is highly conserved between RSV-A and RSV-B strains [122, 133]. Its positioning on RSV’s surface and high level of sequence homology between strains makes the F protein a prime target for vaccine development. The monoclonal antibody therapeutic, Palivizumab, targets the F protein and can neutralise RSV replication, offering some protection to high-risk patients. While this indicates that targeting the F protein provides protection against the majority of RSV strains, emerging RSV strains that contain a N276S mutation within the F protein, are resistant to Palivizumab [134, 135]. When the virion comes into contact with the host cell, the F protein undergoes a conformational shift, with the hydrophobic N-terminus anchoring into the target membrane (Fig. 4). The F protein then folds back in on itself to bring the viral and target membranes together causing membrane fusion [136,137,138,139,140]. Initial work on designing antibodies and small molecules against the F protein were hampered by the multiple confirmations the protein forms; indeed, targeting the post-fusion F protein form had little clinical benefit. The crystal structures of both the pre- and post-fusion F protein have now been solved which, it is hoped, will lead to the development of new therapeutics which can inhibit its function [133, 141]. Previous small peptides have been developed for therapeutic use against the RSV F protein, but high costs and the requirement for frequent injections limits their appeal [113].
Though the G protein is the main attachment protein of RSV, the F protein is also able to bind host cells; this degenerate function protects against potential loss of function mutations in the G protein, which is much less stable than the F protein [137].
The F protein has been shown to allow preferential infection of neonatal B regulatory cells (nBreg). F protein binds to nBregs through the B cell receptor (BCR), causing cellular upregulation of CX3CR1, which is bound by the G protein, through its CX3C domain, allowing infection of the cell. RSV infection of nBregs causes an increase in the production of anti-inflammatory IL-10, thus supressing Th1 activity [142]. This activity was observed to be specific to nBreg cells found in both cord and infant blood immediately after birth but significantly decreased with age, with less than 2% of CD19 + B cells being nBregs by the age of 12 months. Indeed, this observation may explain why younger infants have such a significant risk factor for severe RSV infection.
SH protein
The final protein on the surface of the RSV capsid is a type II transmembrane protein, the small hydrophobic (SH) protein. Structural studies suggest that SH functions as a viroporin and may be able to form pores in cell membranes, altering membrane permeability [143, 144]. SH deletion mutants (ΔSH-RSV) are still able to enter cells and replicate, however, murine experiments have shown that the ΔSH-RSV is less virulent than the WT strain [111, 112, 143, 145]. Although the SH protein mechanism is not yet fully understood, research suggests that it has a role in prolonging the life of infected cells; this insensitivity to apoptosis permits increased viral replication [111]. The presence of the SH also influences cytokine production, with the ΔSH-RSV inducing increased IL-1β and TNF expression [111, 146]. As a result, SH deletion mutants have been explored as live attenuated vaccine candidates, though, to date, none have been brought to market [147].
N protein
The nucleoprotein, together with the phosphoprotein (P), coats the RSV RNA genome in a nucleocapsid to protect it from degradation [7]. Structural analyses have shown that it forms a left-handed helix around the viral genome [148]. As well as this key structural role, the N protein has also been shown to have an immune evasion function. Despite being a nucleoprotein, N protein is also present on the surface of infected epithelial cells and DCs early during infection [149]. Additionally, the presence of N protein in a bi-lipid membrane prevents the formation of mature immunological synapses, leading to a reduction in T cell activation [149]. The ability of RSV to modulate T cell activation also affects antibody production; with fewer naïve T cells activated, there are fewer T follicular helper cells and consequently a reduction in B cell activation. Lifland et al., found that after 6 h RSV infection, the N protein co-localised with RIG-1 and Melanoma Differentiation Associated Gene 5 (MDA5), and later during infection, viral inclusion bodies were observed containing MAVS and MDA5 [150]. This interaction of the N protein with the RIG pathway components, limits the subsequent anti-viral IFN response, thereby enabling more efficient, undisturbed, RSV replication.
Future vaccine and treatment development
With an estimated 3.2 million RSV-related hospital admissions each year, the development of vaccines is key to protection and control of this virus [151]. The nature of RSV’s infectivity and immune evasion strategies has hindered the development of a vaccine that balances safety and efficacy. The spectrum of different immune profiles of the high-risk groups (infants, children, pregnant women and the elderly), adds a layer of complexity, that has stunted successful vaccine development [134]. Where available, the standard treatment of care for children hospitalised with RSV-related LRTI is to mitigate the symptoms of bronchiolitis with fluids and oxygen. Ribavirin is administered in some countries, though its often morbid side effects (such as potential teratogenicity), high cost and poor effectiveness has limited its widespread use against RSV [44, 45, 50,51,52].
The primary purpose of any vaccine is to generate a significant long-lasting antibody and memory B and T cell response, thus protecting against future infection. However, “natural” RSV infection only generates short-lived antibody responses, with two RSV infections thought to be required to generate protective antibodies, which even then can rapidly wane [103, 152]. Maternal antibodies provide protection for new-born infants, but the rate of infant RSV infections peak around 2–3 months of age, with maternal antibodies declining to seronegative levels at ~ 2.5 months. This process is thought to rely on the mother having had an RSV infection close to birth, thus enabling antibody transfer [153, 154]. However, one study showed that high titres of RSV antibodies in cord blood did not reduce the infant’s number of RSV infections, although it did reduce the severity, indicating that maternal anti-RSV antibodies have some protective effect. Interestingly, the antibody transfer rate was lower in male infants than females, leaving the males at higher risk of RSV-related hospital admittance [30].
The first clinically trialled vaccine was formalin-inactivated RSV (FI-RSV) in 1966 [62]. While several other successful formalin-inactivated vaccines exist, including those against polio, hepatitis A and Rabies [155,156,157], the formalin-inactivated RSV vaccine not only provided no protection in RSV naïve infants but increased the risk of significant infection [134, 158]. The cause of this vaccine-enhanced disease is still not fully understood. This initial clinical trial failure resulted in subsequent caution, which has most certainly contributed to the current absence of a licenced RSV vaccine. Advances in crystallography have given insights into the native form of key RSV proteins, including the structure of the F protein [133, 138, 141], which will likely guide and direct future vaccine development. Indeed, the fact that Palivizumab offers some protection indicates that a targeted antibody response can protect against RSV infection [54, 134, 138], though the emergence of resistant Palivizumab strains of RSV suggests any vaccine will require continued maintenance, similar to that against Influenza.
There are now several dozen RSV vaccines under clinical trial, including some maternal vaccines, which could offer protection for neonates in the future [159, 160]. Use of viral proteins, particularly G and F, should theoretically, generate a strong antibody response. While the design of an effective vaccine is a priority, in its absence, the development of a curative treatment remains key. Indeed, when or if, an effective vaccine is found, treatment will always be essential for those do not have sufficient vaccine protection.
An ideal curative treatment will be a direct-acting anti-viral, or a therapeutic that targets a conserved immune evasion mechanism of RSV, thus restoring natural immunity against RSV and limiting viral replication. To better inform the design of these new therapeutics and vaccines the research community must work towards defining the role of all 11 RSV proteins and determine all the processes by which RSV suppresses immunity, while also monitoring the genetic variation of the virus over time. Armed with this knowledge, we will be better placed to develop vaccines and therapeutics that protect our global populations from this ongoing viral threat.
References
Nair H et al (2010) Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375:1545–1555
Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE (2005) Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 352:1749–1759
Terletskaia-Ladwig E, Enders G, Schalasta G, Enders M (2005) Defining the timing of respiratory syncytial virus (RSV) outbreaks: an epidemiological study. BMC Infect Dis 5:20
Broberg EK, Waris M, Johansen K, Snacken R, Penttinen P, European Influenza Surveillance N (2018) Seasonality and geographical spread of respiratory syncytial virus epidemics in 15 European countries, 2010 to 2016. Euro Surveill 23:17–284
Ali S et al (2013) Functional genetic variation in NFKBIA and susceptibility to childhood asthma, bronchiolitis, and bronchopulmonary dysplasia. J Immunol 190:3949–3958
Moyes J et al (2017) Respiratory syncytial virus in adults with severe acute respiratory illness in a high HIV prevalence setting. J Infect 75:346–355
Collins PL, Fearns R, Graham BS (2013) Respiratory syncytial virus: virology, reverse genetics, and pathogenesis of disease. Curr Top Microbiol Immunol 372:3–38
Mufson MA, Orvell C, Rafnar B, Norrby E (1985) Two distinct subtypes of human respiratory syncytial virus. J Gen Virol 66(Pt 10):2111–2124
Johnson PR Jr, Olmsted RA, Prince GA, Murphy BR, Alling DW, Walsh EE, Collins PL (1987) Antigenic relatedness between glycoproteins of human respiratory syncytial virus subgroups A and B: evaluation of the contributions of F and G glycoproteins to immunity. J Virol 61:3163–3166
Melero JA, Moore ML (2013) Influence of respiratory syncytial virus strain differences on pathogenesis and immunity. Curr Top Microbiol Immunol 372:59–82
Tabatabai J, Prifert C, Pfeil J, Grulich-Henn J, Schnitzler P (2014) Novel respiratory syncytial virus (RSV) genotype ON1 predominates in Germany during winter season 2012–13. PLoS ONE 9:e109191
Tan L, Coenjaerts FE, Houspie L, Viveen MC, van Bleek GM, Wiertz EJ, Martin DP, Lemey P (2013) The comparative genomics of human respiratory syncytial virus subgroups A and B: genetic variability and molecular evolutionary dynamics. J Virol 87:8213–8226
Pandya CM, Callahan MS, Savchenko GK, Stobart CC (2019) A Contemporary view of respiratory syncytial virus (RSV) biology and strain-specific differences. Pathogens 8:67
Gilca R, De Serres G, Tremblay M, Vachon ML, Leblanc E, Bergeron MG, Dery P, Boivin G (2006) Distribution and clinical impact of human respiratory syncytial virus genotypes in hospitalized children over 2 winter seasons. J Infect Dis 193:54–58
Martinello RA, Chen MD, Weibel C, Kahn JS (2002) Correlation between respiratory syncytial virus genotype and severity of illness. J Infect Dis 186:839–842
Deshpande SA, Northern V (2003) The clinical and health economic burden of respiratory syncytial virus disease among children under 2 years of age in a defined geographical area. Arch Dis Child 88:1065–1069
Lambert L, Sagfors AM, Openshaw PJ, Culley FJ (2014) Immunity to RSV in early-life. Front Immunol 5:466
Price RHM, Graham C, Ramalingam S (2019) Association between viral seasonality and meteorological factors. Sci Rep 9:929
Kulkarni H, Smith CM, Lee DOH, Hirst RA, Easton AJ, O’Callaghan C (2016) Evidence of respiratory syncytial virus spread by aerosol. Time to revisit infection control strategies? Am J Respir Crit Care Med 194:308–316
Public Health England (2016) Infection control precautions to minimise transmission of acute respiratory tract infections in healthcare settingsed. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/585584/RTI_infection_control_guidance.pdf. Accessed 06 Jan 2018
Griffiths C, Drews SJ, Marchant DJ (2017) Respiratory syncytial virus: infection, detection, and new options for prevention and treatment. Clin Microbiol Rev 30:277–319
Henderson J, Hilliard TN, Sherriff A, Stalker D, Al Shammari N, Thomas HM (2005) Hospitalization for RSV bronchiolitis before 12 months of age and subsequent asthma, atopy and wheeze: a longitudinal birth cohort study. Pediatr Allergy Immunol 16:386–392
Sigurs N, Aljassim F, Kjellman B, Robinson PD, Sigurbergsson F, Bjarnason R, Gustafsson PM (2010) Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax 65:1045–1052
Fauroux B, Simoes EAF, Checchia PA, Paes B, Figueras-Aloy J, Manzoni P, Bont L, Carbonell-Estrany X (2017) The burden and long-term respiratory morbidity associated with respiratory syncytial virus infection in early childhood. Infect Dis Ther 6:173–197
Malinczak CA, Fonseca W, Rasky AJ, Ptaschinski C, Morris S, Ziegler SF, Lukacs NW (2019) Sex-associated TSLP-induced immune alterations following early-life RSV infection leads to enhanced allergic disease. Mucosal Immunol 12:969–979
Scheltema NM et al (2018) Respiratory syncytial virus prevention and asthma in healthy preterm infants: a randomised controlled trial. Lancet Respir Med 6:257–264
Wu P, Hartert TV (2011) Evidence for a causal relationship between respiratory syncytial virus infection and asthma. Expert Rev Anti-infective Therapy 9:731–745
Hunderi JOG, Rolfsjord LB, Carlsen KCL, Holst R, Bakkeheim E, Berents TL, Carlsen KH, Skjerven HO (2020) Virus, allergic sensitisation and cortisol in infant bronchiolitis and risk of early asthma. ERJ Open Res. https://doi.org/10.1183/23120541.00268-2019
Strunk T, Currie A, Richmond P, Simmer K, Burgner D (2011) Innate immunity in human newborn infants: prematurity means more than immaturity. J Matern Fetal Neonatal Med 24:25–31
Chu HY et al (2017) Transplacental transfer of maternal respiratory syncytial virus (RSV) antibody and protection against RSV disease in infants in rural Nepal. J Clin Virol 95:90–95
Smith LJ, McKay KO, van Asperen PP, Selvadurai H, Fitzgerald DA (2010) Normal development of the lung and premature birth. Paediatr Respir Rev 11:135–142
Muraro E, Merlo A, Martorelli D, Cangemi M, Dalla Santa S, Dolcetti R, Rosato A (2017) Fighting viral infections and virus-driven tumors with cytotoxic CD4(+) T Cells. Front Immunol 8:197
Farrar JD, Asnagli H, Murphy KM (2002) T helper subset development: roles of instruction, selection, and transcription. J Clin Invest 109:431–435
Becker Y (2006) Respiratory syncytial virus (RSV) evades the human adaptive immune system by skewing the Th1/Th2 cytokine balance toward increased levels of Th2 cytokines and IgE, markers of allergy—a review. Virus Genes 33:235–252
Lee HC, Headley MB, Loo YM, Berlin A, Gale M Jr, Debley JS, Lukacs NW, Ziegler SF (2012) Thymic stromal lymphopoietin is induced by respiratory syncytial virus-infected airway epithelial cells and promotes a type 2 response to infection. J Allergy Clin Immunol 130(1187–1196):e5
Soumelis V et al (2002) Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol 3:673–680
Ziegler SF, Roan F, Bell BD, Stoklasek TA, Kitajima M, Han H (2013) The biology of thymic stromal lymphopoietin (TSLP). Adv Pharmacol 66:129–155
McKenzie AN (2014) Type-2 innate lymphoid cells in asthma and allergy. Ann Am Thorac Soc 11(Suppl 5):S263–S270
Saravia J et al (2015) Respiratory syncytial virus disease is mediated by age-variable IL-33. PLoS Pathog 11:e1005217
Besnard AG, Togbe D, Guillou N, Erard F, Quesniaux V, Ryffel B (2011) IL-33-activated dendritic cells are critical for allergic airway inflammation. Eur J Immunol 41:1675–1686
Rodriguez-Fernandez R et al (2017) Respiratory syncytial virus genotypes, host immune profiles, and disease severity in young children hospitalized with bronchiolitis. J Infect Dis 217:24–34
Eiland LS (2009) Respiratory syncytial virus: diagnosis, treatment and prevention. J Pediatr Pharmacol Ther 14:75–85
Ralston SL et al (2014) Clinical practice guideline: the diagnosis, management, and prevention of bronchiolitis. Pediatrics 134:e1474–e1502
Academy Academy of Pediatrics Subcommittee on, D. and Management of, B. (2006) Diagnosis and management of bronchiolitis. Pediatrics 118:1774–1793
HSPC (2019) RSV fact sheet. https://www.hpsc.ie/a-z/respiratory/respiratorysyncytialvirus/factsheet/ Accessed 12 Dec 2019
Schmidt ME, Varga SM (2017) Modulation of the host immune response by respiratory syncytial virus proteins. J Microbiol 55:161–171
Te HS, Randall G, Jensen DM (2007) Mechanism of action of ribavirin in the treatment of chronic hepatitis C. Gastroenterol Hepatol (N Y) 3:218–225
González PA, Bueno SM, Carreño LJ, Riedel CA, Kalergis AM (2012) Respiratory syncytial virus infection and immunity. Rev Med Virol 22:230–244
Thomas E, Ghany MG, Liang TJ (2012) The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir Chem Chemother 23:1–12
Conrad DA, Christenson JC, Waner JL, Marks MI (1987) Aerosolized ribavirin treatment of respiratory syncytial virus infection in infants hospitalized during an epidemic. Pediatr Infect Dis J 6:152–158
Martin JT, Kugler JD, Gumbiner CH, Brown JC, Murphy KR, Colombo JL, McManus BM (1990) Refractory congestive heart failure after ribavirin in infants with heart disease and respiratory syncytial virus. Nebr Med J 75:23–26
Foolad F, Prayag A, Ghantoji SS, Ariza-Heredia EJ, Chemaly RF (2017) Use of oral ribavirin for the treatment of RSV infections in hematopoietic cell transplant (HCT) recipients. Biol Blood Marrow Transplant 23:S189
Medimmune (1999) SYNAGIS® (PALIVIZUMAB) https://www.accessdata.fda.gov/drugsatfda_docs/label/2002/palimed102302LB.pdf Accessed 04 May 2019
Anderson EJ, Carosone-Link P, Yogev R, Yi J, Simões EAF (2017) Effectiveness of palivizumab in high-risk infants and children: a propensity score weighted regression analysis. Pediatr Infect Dis J 36:699–704
Morris SK, Dzolganovski B, Beyene J, Sung L (2009) A meta-analysis of the effect of antibody therapy for the prevention of severe respiratory syncytial virus infection. BMC Infect Dis 9:106
Wegzyn C, Toh LK, Notario G, Biguenet S, Unnebrink K, Park C, Makari D, Norton M (2014) Safety and effectiveness of palivizumab in children at high risk of serious disease due to respiratory syncytial virus infection: a systematic review. Infect Dis Ther 3:133–158
Academy Academy of Pediatrics Committee on Infectious, D. and American Academy of Pediatrics Bronchiolitis Guidelines, C. (2014) Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics 134:415–420
Welliver RC, Checchia PA, Bauman JH, Fernandes AW, Mahadevia PJ, Hall CB (2010) Fatality rates in published reports of RSV hospitalizations among high-risk and otherwise healthy children. Curr Med Res Opin 26:2175–2181
Nuijten M, Lebmeier M, Wittenberg W (2009) Cost effectiveness of palivizumab for RSV prevention in high-risk children in the Netherlands. J Med Econ 12:291–300
Whelan B et al (2016) Review of the home care programmes for respiratory syncytial virus (RSV) prophylaxis in Ireland and The Netherlands. Drugs Ther Perspect 32:119–130
Alansari K, Toaimah FH, Almatar DH, El Tatawy LA, Davidson BL, Qusad MIM (2019) Monoclonal antibody treatment of RSV bronchiolitis in young infants: a randomized trial. Pediatrics. https://doi.org/10.1542/peds.2018-2308
Kim HW, Canchola JG, Brandt CD, Pyles G, Chanock RM, Jensen K, Parrott RH (1969) Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol 89:422–434
Openshaw PJ, Culley FJ, Olszewska W (2001) Immunopathogenesis of vaccine-enhanced RSV disease. Vaccine 20(Suppl 1):S27–31
Acosta PL, Caballero MT, Polack FP (2015) Brief history and characterization of enhanced respiratory syncytial virus disease. Clin Vaccine Immunol 23:189–195
Karron RA, Buchholz UJ, Collins PL (2013) Live-attenuated respiratory syncytial virus vaccines. Curr Top Microbiol Immunol 372:259–284
McFarland EJ et al (2018) Live-attenuated respiratory syncytial virus vaccine candidate with deletion of RNA synthesis regulatory protein M2–2 is highly immunogenic in children. J Infect Dis 217:1347–1355
Jin H, Zhou H, Cheng X, Tang R, Munoz M, Nguyen N (2000) Recombinant respiratory syncytial viruses with deletions in the NS1, NS2, SH, and M2–2 genes are attenuated in vitro and in vivo. Virology 273:210–218
Teng MN, Whitehead SS, Bermingham A, St Claire M, Elkins WR, Murphy BR, Collins PL (2000) Recombinant respiratory syncytial virus that does not express the NS1 or M2–2 protein is highly attenuated and immunogenic in chimpanzees. J Virol 74:9317–9321
Beachboard DC, Horner SM (2016) Innate immune evasion strategies of DNA and RNA viruses. Curr Opin Microbiol 32:113–119
Bowie AG, Unterholzner L (2008) Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol 8:911–922
Bitko V, Shulyayeva O, Mazumder B, Musiyenko A, Ramaswamy M, Look DC, Barik S (2007) Nonstructural proteins of respiratory syncytial virus suppress premature apoptosis by an NF-kappaB-dependent, interferon-independent mechanism and facilitate virus growth. J Virol 81:1786–1795
Diaz MO, Ziemin S, Le Beau MM, Pitha P, Smith SD, Chilcote RR, Rowley JD (1988) Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell lines. Proc Natl Acad Sci U S A 85:5259–5263
Osada N, Kohara A, Yamaji T, Hirayama N, Kasai F, Sekizuka T, Kuroda M, Hanada K (2014) The genome landscape of the african green monkey kidney-derived vero cell line. DNA Res 21:673–683
Schneider WM, Chevillotte MD, Rice CM (2014) Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545
Killip MJ, Fodor E, Randall RE (2015) Influenza virus activation of the interferon system. Virus Res 209:11–22
Huang M, Jiang JD, Peng Z (2014) Recent advances in the anti-HCV mechanisms of interferon. Acta Pharm Sin B 4:241–247
Randall RE, Goodbourn S (2008) Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89:1–47
Taylor KE, Mossman KL (2013) Recent advances in understanding viral evasion of type I interferon. Immunology 138:190–197
Durbin JE, Johnson TR, Durbin RK, Mertz SE, Morotti RA, Peebles RS, Graham BS (2002) The role of IFN in respiratory syncytial virus pathogenesis. J Immunol 168:2944–2952
Zhang W et al (2005) Inhibition of respiratory syncytial virus infection with intranasal siRNA nanoparticles targeting the viral NS1 gene. Nat Med 11:56–62
Elliott J et al (2007) Respiratory syncytial virus NS1 protein degrades STAT2 by using the Elongin-Cullin E3 ligase. J Virol 81:3428–3436
Lo MS, Brazas RM, Holtzman MJ (2005) Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and alpha/beta interferon responsiveness. J Virol 79:9315–9319
Ramaswamy M, Shi L, Varga SM, Barik S, Behlke MA, Look DC (2006) Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology 344:328–339
Spann KM, Tran KC, Collins PL (2005) Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J Virol 79:5353–5362
Kotelkin A, Belyakov IM, Yang L, Berzofsky JA, Collins PL, Bukreyev A (2006) The NS2 protein of human respiratory syncytial virus suppresses the cytotoxic T-cell response as a consequence of suppressing the type I interferon response. J Virol 80:5958–5967
Munir S, Hillyer P, Le Nouën C, Buchholz UJ, Rabin RL, Collins PL, Bukreyev A (2011) Respiratory syncytial virus interferon antagonist NS1 protein suppresses and skews the human T lymphocyte response. PLoS Pathog 7:e1001336
Tripp RA, Hou S, Etchart N, Prinz A, Moore D, Winter J, Anderson LJ (2001) CD4(+) T cell frequencies and Th1/Th2 cytokine patterns expressed in the acute and memory response to respiratory syncytial virus I-E(d)-restricted peptides. Cell Immunol 207:59–71
Boyapalle S, Wong T, Garay J, Teng M, San Juan-Vergara H, Mohapatra S, Mohapatra S (2012) Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS ONE 7:e29386
Ling Z, Tran KC, Teng MN (2009) Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J Virol 83:3734–3742
Malireddi RK, Kanneganti TD (2013) Role of type I interferons in inflammasome activation, cell death, and disease during microbial infection. Front Cell Infect Microbiol 3:77
Telcian AG et al (2011) RSV-induced bronchial epithelial cell PD-L1 expression inhibits CD8+ T cell nonspecific antiviral activity. J Infect Dis 203:85–94
Swedan S, Musiyenko A, Barik S (2009) Respiratory syncytial virus nonstructural proteins decrease levels of multiple members of the cellular interferon pathways. J Virol 83:9682–9693
Ren J, Liu T, Pang L, Li K, Garofalo RP, Casola A, Bao X (2011) A novel mechanism for the inhibition of interferon regulatory factor-3-dependent gene expression by human respiratory syncytial virus NS1 protein. J Gen Virol 92:2153–2159
Zhou JH, Wang YN, Chang QY, Ma P, Hu Y, Cao X (2018) Type III interferons in viral infection and antiviral immunity. Cell Physiol Biochem 51:173–185
Wack A, Terczyńska-Dyla E, Hartmann R (2015) Guarding the frontiers: the biology of type III interferons. Nat Immunol 16:802–809
Broggi A, Granucci F, Zanoni I (2020) Type III interferons: balancing tissue tolerance and resistance to pathogen invasion. J Exp Med. https://doi.org/10.1084/jem.20190295
Spann KM, Tran KC, Chi B, Rabin RL, Collins PL (2004) Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages [corrected]. J Virol 78:4363–4369
Hastie ML et al (2012) The human respiratory syncytial virus nonstructural protein 1 regulates type I and type II interferon pathways. Mol Cell Proteomics 11:108–127
Okabayashi T et al (2011) Type-III interferon, not type-I, is the predominant interferon induced by respiratory viruses in nasal epithelial cells. Virus Res 160:360–366
Glaser L, Coulter PJ, Shields M, Touzelet O, Power UF, Broadbent L (2019) Airway epithelial derived cytokines and chemokines and their role in the immune response to respiratory syncytial virus infection. Pathogens 8:106
Hillyer P et al (2018) Differential responses by human respiratory epithelial cell lines to respiratory syncytial virus reflect distinct patterns of infection control. J Virol. https://doi.org/10.1128/JVI.02202-17
Bossert B, Marozin S, Conzelmann KK (2003) Nonstructural proteins NS1 and NS2 of bovine respiratory syncytial virus block activation of interferon regulatory factor 3. J Virol 77:8661–8668
Piedimonte G, Perez MK (2014) Respiratory syncytial virus infection and bronchiolitis. Pediatr Rev 35:519–530
Crim RL, Audet SA, Feldman SA, Mostowski HS, Beeler JA (2007) Identification of linear heparin-binding peptides derived from human respiratory syncytial virus fusion glycoprotein that inhibit infectivity. J Virol 81:261–271
Feldman SA, Audet S, Beeler JA (2000) The fusion glycoprotein of human respiratory syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J Virol 74:6442–6447
Harcourt J, Alvarez R, Jones LP, Henderson C, Anderson LJ, Tripp RA (2006) Respiratory syncytial virus G protein and G protein CX3C motif adversely affect CX3CR1+ T cell responses. J Immunol 176:1600–1608
Johnson SM, McNally BA, Ioannidis I, Flano E, Teng MN, Oomens AG, Walsh EE, Peeples ME (2015) Respiratory syncytial virus uses CX3CR1 as a receptor on primary human airway epithelial cultures. PLoS Pathog 11:e1005318
Zhang L, Bukreyev A, Thompson CI, Watson B, Peeples ME, Collins PL, Pickles RJ (2005) Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium. J Virol 79:1113–1124
Chirkova T et al (2015) CX3CR1 is an important surface molecule for respiratory syncytial virus infection in human airway epithelial cells. J Gen Virol 96:2543–2556
Cagno V, Tseligka ED, Jones ST, Tapparel C (2019) Heparan sulfate proteoglycans and viral attachment: true receptors or adaptation bias? Viruses. https://doi.org/10.3390/v11070596
Fuentes S, Tran KC, Luthra P, Teng MN, He B (2007) Function of the respiratory syncytial virus small hydrophobic protein. J Virol 81:8361–8366
Bukreyev A, Whitehead SS, Murphy BR, Collins PL (1997) Recombinant respiratory syncytial virus from which the entire SH gene has been deleted grows efficiently in cell culture and exhibits site-specific attenuation in the respiratory tract of the mouse. J Virol 71:8973–8982
McLellan JS, Ray WC, Peeples ME (2013) Structure and function of respiratory syncytial virus surface glycoproteins. Curr Top Microbiol Immunol 372:83–104
Krusat T, Streckert HJ (1997) Heparin-dependent attachment of respiratory syncytial virus (RSV) to host cells. Arch Virol 142:1247–1254
Choi Y, Mason CS, Jones LP, Crabtree J, Jorquera PA, Tripp RA (2012) Antibodies to the central conserved region of respiratory syncytial virus (RSV) G protein block RSV G protein CX3C-CX3CR1 binding and cross-neutralize RSV A and B strains. Viral Immunol 25:193–203
Anderson CS et al (2019) CX3CR1 as a respiratory syncytial virus receptor in pediatric human lung. Pediatr Res. https://doi.org/10.1038/s41390-019-0677-0
Teng MN, Collins PL (2002) The central conserved cystine noose of the attachment G protein of human respiratory syncytial virus is not required for efficient viral infection in vitro or in vivo. J Virol 76:6164–6171
Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA (2001) Glycosylation and the immune system. Science 291:2370–2376
Vigerust DJ, Shepherd VL (2007) Virus glycosylation: role in virulence and immune interactions. Trends Microbiol 15:211–218
Collins PL, Graham BS (2008) Viral and host factors in human respiratory syncytial virus pathogenesis. J Virol 82:2040–2055
Melero JA, García-Barreno B, Martínez I, Pringle CR, Cane PA (1997) Antigenic structure, evolution and immunobiology of human respiratory syncytial virus attachment (G) protein. J Gen Virol 78(Pt 10):2411–2418
Johnson PR, Collins PL (1988) The fusion glycoproteins of human respiratory syncytial virus of subgroups A and B: sequence conservation provides a structural basis for antigenic relatedness. J Gen Virol 69(Pt 10):2623–2628
Eshaghi A, Duvvuri VR, Lai R, Nadarajah JT, Li A, Patel SN, Low DE, Gubbay JB (2012) Genetic variability of human respiratory syncytial virus A strains circulating in Ontario: a novel genotype with a 72 nucleotide G gene duplication. PLoS ONE 7:e32807
Yoshihara K et al (2016) Association of RSV-A ON1 genotype with increased pediatric acute lower respiratory tract infection in Vietnam. Sci Rep 6:27856
Bromley SK et al (2001) The immunological synapse. Annu Rev Immunol 19:375–396
Werdelin O, Meldal M, Jensen T (2002) Processing of glycans on glycoprotein and glycopeptide antigens in antigen-presenting cells. Proc Natl Acad Sci U S A 99:9611–9613
Bukreyev A, Yang L, Fricke J, Cheng L, Ward JM, Murphy BR, Collins PL (2008) The secreted form of respiratory syncytial virus G glycoprotein helps the virus evade antibody-mediated restriction of replication by acting as an antigen decoy and through effects on fc receptor-bearing leukocytes. J Virol 82:12191
Roberts SR, Lichtenstein D, Ball LA, Wertz GW (1994) The membrane-associated and secreted forms of the respiratory syncytial virus attachment glycoprotein G are synthesized from alternative initiation codons. J Virol 68:4538–4546
Tripp RA, Moore D, Jones L, Sullender W, Winter J, Anderson LJ (1999) Respiratory syncytial virus G and/or SH protein alters Th1 cytokines, natural killer cells, and neutrophils responding to pulmonary infection in BALB/c mice. J Virol 73:7099–7107
Arnold R, Konig B, Werchau H, Konig W (2004) Respiratory syncytial virus deficient in soluble G protein induced an increased proinflammatory response in human lung epithelial cells. Virology 330:384–397
Bukreyev A, Yang L, Collins PL (2012) The secreted G protein of human respiratory syncytial virus antagonizes antibody-mediated restriction of replication involving macrophages and complement. J Virol 86:10880–10884
Chirkova T, Boyoglu-Barnum S, Gaston KA, Malik FM, Trau SP, Oomens AG, Anderson LJ (2013) Respiratory syncytial virus G protein CX3C motif impairs human airway epithelial and immune cell responses. J Virol 87:13466–13479
McLellan JS et al (2013) Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340:1113–1117
Anderson LJ, Dormitzer PR, Nokes DJ, Rappuoli R, Roca A, Graham BS (2013) Strategic priorities for respiratory syncytial virus (RSV) vaccine development. Vaccine 31(Suppl 2):B209–B215
Hashimoto K, Hosoya M (2017) Neutralizing epitopes of RSV and palivizumab resistance in Japan. Fukushima J Med Sci 63:127–134
Kolokoltsov AA, Deniger D, Fleming EH, Roberts NJ Jr, Karpilow JM, Davey RA (2007) Small interfering RNA profiling reveals key role of clathrin-mediated endocytosis and early endosome formation for infection by respiratory syncytial virus. J Virol 81:7786–7800
Tayyari F, Marchant D, Moraes TJ, Duan W, Mastrangelo P, Hegele RG (2011) Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat Med 17:1132–1135
Zhao X, Singh M, Malashkevich VN, Kim PS (2000) Structural characterization of the human respiratory syncytial virus fusion protein core. Proc Natl Acad Sci U S A 97:14172–14177
Krzyzaniak MA, Zumstein MT, Gerez JA, Picotti P, Helenius A (2013) Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the F protein. PLoS Pathog 9:e1003309
Weisshaar M, Cox R, Plemper RK (2015) Blocking respiratory syncytial virus entry: a story with twists. DNA Cell Biol 34:505–510
McLellan JS, Yang Y, Graham BS, Kwong PD (2011) Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J Virol 85:7788–7796
Zhivaki D et al (2017) Respiratory syncytial virus infects regulatory B Cells in human neonates via chemokine receptor CX3CR1 and promotes lung disease severity. Immunity 46:301–314
Perez M, Garcia-Barreno B, Melero JA, Carrasco L, Guinea R (1997) Membrane permeability changes induced in Escherichia coli by the SH protein of human respiratory syncytial virus. Virology 235:342–351
Araujo GC, Silva RH, Scott LP, Araujo AS, Souza FP, de Oliveira RJ (2016) Structure and functional dynamics characterization of the ion channel of the human respiratory syncytial virus (hRSV) small hydrophobic protein (SH) transmembrane domain by combining molecular dynamics with excited normal modes. J Mol Model 22:286
Karron RA, Wright PF, Crowe JE Jr, Clements-Mann ML, Thompson J, Makhene M, Casey R, Murphy BR (1997) Evaluation of two live, cold-passaged, temperature-sensitive respiratory syncytial virus vaccines in chimpanzees and in human adults, infants, and children. J Infect Dis 176:1428–1436
Russell RF, McDonald JU, Ivanova M, Zhong Z, Bukreyev A, Tregoning JS (2015) Partial attenuation of respiratory syncytial virus with a deletion of a small hydrophobic gene is associated with elevated interleukin-1beta responses. J Virol 89:8974–8981
Rostad CA et al (2016) A recombinant respiratory syncytial virus vaccine candidate attenuated by a low-fusion F protein is immunogenic and protective against challenge in cotton rats. J Virol 90:7508–7518
Bakker SE, Duquerroy S, Galloux M, Loney C, Conner E, Eleouet JF, Rey FA, Bhella D (2013) The respiratory syncytial virus nucleoprotein-RNA complex forms a left-handed helical nucleocapsid. J Gen Virol 94:1734–1738
Cespedes PF et al (2014) Surface expression of the hRSV nucleoprotein impairs immunological synapse formation with T cells. Proc Natl Acad Sci U S A 111:E3214–E3223
Lifland AW, Jung J, Alonas E, Zurla C, Crowe JE Jr, Santangelo PJ (2012) Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J Virol 86:8245–8258
Shi T et al (2017) Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet 390:946–958
Yamaji Y, Yasui Y, Nakayama T (2016) Development of acquired immunity following repeated respiratory syncytial virus infections in cotton rats. PLoS ONE 11:e0155777
Ochola R, Sande C, Fegan G, Scott PD, Medley GF, Cane PA, Nokes DJ (2009) The level and duration of RSV-specific maternal IgG in infants in Kilifi Kenya. PLoS ONE 4:e8088
Chu HY, Englund JA (2017) Maternal immunization. Birth Defects Res 109:379–386
Kissling RE, Reese DR (1963) Anti-rabies vaccine of tissue culture origin. J Immunol 91:362–368
Wilton T, Dunn G, Eastwood D, Minor PD, Martin J (2014) Effect of formaldehyde inactivation on poliovirus. J Virol 88:11955–11964
Werzberger A et al (1992) A controlled trial of a formalin-inactivated hepatitis A vaccine in healthy children. N Engl J Med 327:453–457
Kapikian AZ, Mitchell RH, Chanock RM, Shvedoff RA, Stewart CE (1969) An epidemiologic study of altered clinical reactivity to respiratory syncytial (RS) virus infection in children previously vaccinated with an inactivated RS virus vaccine. Am J Epidemiol 89:405–421
Hogan AB, Campbell PT, Blyth CC, Lim FJ, Fathima P, Davis S, Moore HC, Glass K (2017) Potential impact of a maternal vaccine for RSV: a mathematical modelling study. Vaccine 35:6172–6179
Novavax (2018) A study to determine the safety and efficacy of the RSV F vaccine to protect infants via maternal immunization. In: Clinical Trials (ed) https://clinicaltrials.gov/ct2/show/NCT02624947 Accessed 28 May 2018
Beeler JA, van Wyke Coelingh K (1989) Neutralization epitopes of the F glycoprotein of respiratory syncytial virus: effect of mutation upon fusion function. J Virol 63:2941–2950
Tripp RA, Jones LP, Haynes LM, Zheng H, Murphy PM, Anderson LJ (2001) CX3C chemokine mimicry by respiratory syncytial virus G glycoprotein. Nat Immunol 2:732–738
Boyoglu-Barnum S et al (2014) Prophylaxis with a respiratory syncytial virus (RSV) anti-G protein monoclonal antibody shifts the adaptive immune response to RSV rA2-line19F infection from Th2 to Th1 in BALB/c mice. J Virol 88:10569–10583
Rixon HW, Brown G, Aitken J, McDonald T, Graham S, Sugrue RJ (2004) The small hydrophobic (SH) protein accumulates within lipid-raft structures of the Golgi complex during respiratory syncytial virus infection. J Gen Virol 85:1153–1165
Funding
Funding was provided by The National Children’s Hospital Foundation (Grant no: 1719).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Efstathiou, C., Abidi, S.H., Harker, J. et al. Revisiting respiratory syncytial virus’s interaction with host immunity, towards novel therapeutics. Cell. Mol. Life Sci. 77, 5045–5058 (2020). https://doi.org/10.1007/s00018-020-03557-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03557-0