
Abstract
The methanol extract form the leaves of Phytolacca icosandra L., afforded the unprecedented artificial triterpenoid fatty acid ester 1 derived from the new natural triterpenoid phytolaccagenic acid 3-O-myristate (1a), along with the three known triterpenoids serjanic, acinosolic and phytolaccagenic acid (2 – 4). Their structures were stablished by HR-EI-MS, 1D and 2D NMR techniques. The possible mechanistic formation of 1 is proposed, and the in vitro toxicity of all compounds was assessed using the brine shrimp lethality assay (BSLA).
Graphic Abstract

Similar content being viewed by others

1 Introduction
The chemistry of Phytolacca ssp. is fairly wide and comprises a variety of secondary metabolites, composed mainly by triterpenoids, flavonoids and lignans [1]. Plants belonging to genus Phytolacca have been used in folk medicine for the treatment of several affections such as edema, rheumatism and dermatitis [1,2,3]; also as a molluscicidal plant in schistosomiasis prevention and control [4, 5]. Several studies on P. icosandra have reported its antisecretory, anthelmintic, ovicidal and larvicidal activity [6,7,8].
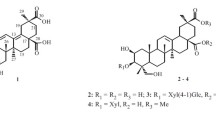
Phytochemical analysis of P. icosandra has lead to the isolation of several serjanic and spergulagenic acids [9, 10] and a previous investigation of the fruits yielded a novel peltogynoid, together with triterpenoids, neo-lignanes and 6′palmitoyl-α-d-glucoside sterols [11]. As part of our continuing search for new bioactive constituents from plants of the Phytolacca genus, the methanolic extract of the leaves of P. icosandra was investigated. As a result, a new artificial triterpenoid fatty acid ester (1) was isolated along with three other known pentacyclic triterpenoids 2–4 (Fig. 1). We also comment on the possible formation of 1, and the in vitro toxicity of all compounds against brine shrimps (Artemia salina).

Chemical structure of compounds 1—4
2 Results and Discussion
2.1 Structure Elucidation of Isolated Compounds
Compound 1 was isolated as a white wax. A molecular formula of C48H80O8 was assigned from its HR-MS spectra, which showed a molecuar ion peak [M]+ at m/z 784.5859 (calcd. 784.5853) accounting for nine degrees of unsaturation. The IR spectrum exhibited absorption bands due to the presence of hydroxyl and carboxylic acid groups (2900–3400 cm−1), carbonyl groups (1701–1705 cm−1), olefinic bond (1472 cm−1) and long chain alkanes band (728 cm−1). The 1H NMR spectrum in conjunction with the HMQC spectrum, revealed the presence of five tertiary methyl groups at δH/C 0.69/16.8 (H-26/C-26), 0.93/16.5 (H-25/C-25), 1.01/12.4 (H-24/C-24), 1.12/25.9 (H-27/C-27), 1.13/28.3 (H-29/C-29); a 2,2-dioxy-propane group [δH/C 1.39/29.7 (H-3′'/C-3′') and 1.42/19.3 (H-2′'/C-2′') and δC 99.0 (O > C < O, C-1′'), one methoxy group at δH 3.67 / δC 51.8, one oxymethine proton at δH 3.48 (1H, dd, J = 3.7, 11.7 Hz, H-3)/δC 77.6 (C-3), two oxymethylene protons at δH 3.41, 3.50 (2H, d, J = 10.7 Hz, H-23)/δC 72.6 (C-23) and one vinylic proton at δH 5.32 (1H, t, J = 3.5 Hz, H-12)/δC 123.2. Additional data showed that compound 1 was esterified with a long chain fatty acid, due the presence of several peaks between δH 1.20 – 1.32 (14H, m, H-5′ – H-11′)/δC 29.2 – 29.7, (C-5′ – C-11′), two multiplets at δH 1.22 (H-4′)/δC 29.0 (C-4′) and δH 1.59 (H-3′)/δC 24.7 (C-3′), one methylene triplet at δH 2.31 (2H, t, J = 7.4 Hz, H-2′)/δC 34.0 (C-2′) and one primary methyl at δH 0.85 (3H, t, J = 6.9 Hz, H-14′)/δC 14.4 (C-14′).
The 13C NMR spectra showed that compound 1 has an 3,23-dihydroxy-olean-12(13)-en-28,30-dioic acid-30-methyl ester triterpene skeleton, because in addition to the presence of the peaks assigned to the five tertiary methyl mentioned above, it is possible to locate peaks corresponding to six sp3 quaternary carbons [δC 36.8 (C-4), 37.2 (C-10), 39.4 (C-8), 41.4 (C-14), 43.7 (C-20), 45.8 (C-17)], three sp3 methines [δC 42.2 (C-18), 47.7 (C-9), 51.5 (C-5)], one oxymethylene carbon [δC 72.6 (C-23)], one oximethyne [δC 77.6 (C-3)], two carbons from a tri-substituted double bond [δC 123.2 (C-12), 142.8 (C-13)], and two carbonyl carbons [δC 176 (C-30), 183.4 (C-28) ppm]. An acyclic acetonide moiety, previously described as a 2,2-dioxy-propane group, esterified by the aforementionated long chain fatty acid were also elucidated in the molecule, across the 13C NMR spectra.
The HMBC correlations of the oxymethylene protons at δH 3.41/3.50 with carbons at δC 12.4 (C-24), 36.8 (C-4), 51.5 (C-5) and 77.6 (C-3), permitted to assign this protons to H-23. The acetonide moiety was located on C-23 due to the 3 J HMBC interaction of H-23 protons with the carbon at δC 99.0 (C-1") and the interaction of this carbon with two tertiary methyl protons at δH 1.39 (H-3") and 1.42 (H-2"). Proton H-3 (δH 3.48) was assigned by its HMBC correlations with carbon peaks at δC 12.4 (C-24), 23.2 (C-2) and 72.6 (C-23). Surprisingly, there was no HMBC correlation between proton H-3 and the carbonyl group of the fatty acid at δC 179.7 (C-1′). The fact that the chemical shift of this proton at δH 3.48 was unusually shielded in comparison to acylated oleanane triterpenes at H-3 position, which are observed between δH 4.46–4.57 [12,13,14,15,16], indicated that the ester moiety of the fatty acid was not located at C-3 position. In view of these observations, the only available position for the fatty acid chain previously stated, would be at the isopropylienedioxy carbon (C-1") attached to C-23. Thus, the acetonide triterpenoid fatty acid ester 1 was elucidated as 3β,23α-dihydroxy-olean-12(13)-en-28,30-dioic acid-30-methyl ester-23,1"-isopropyl enedioxy-1"-tetradecanoate. Complete stereochemistry of the triterpene was confirmed by analysis of its NOESY spectrum along with some biogenetic and chemotaxonomic considerations. NOE interactions where detected between H-3/H-5/H-23 and H-24/H-25/H-26, interaction between H-18/H-30 was not observed thus confirming configuration at C-3 3βOH, junctions of the B/C rings "trans" (8βMe, 9αH) and the D/E rings "cis" (18βH; 28βCOOH). This is also congruent with the configuration of all triterpenes previously isolated from Phytolacca genus.
Although few, there has been some reports on natural occurring triterpenoidal acetonides from plants [17,18,19]. Despite the fact that 1 is considered unusual being an acyclic acetonide ketal, it is assumed to be an artifact derived from phytolaccagenic acid 3β-O-myristate (1a) during the chromatographic process, in which acetone was used as solvent [20, 21]. The proposed mechanism in the formation of 1 from 1a involves firstly a nucleophilic attack of the C-23 hydroxyl to a protonated acetone molecule, followed by an intramolecular nucleophilic substitution at the fatty acid carbonyl at C-3 (Fig. 2).

Possible formation mechanism for compound 1
Finally, the toxicity of all compounds was assayed in the brine shrimp lethality assay [22] and compound 1 exhibited mild toxicity against Artemia; results are shown in Table 1.
3 Experimental Section
3.1 General Procedures
Optical rotation was measured in Karl-ZEISS, Model 93,772 equipment. IR spectra were obtained from KBr pellets with Shimadzu IR-408 equipment. Solution 1H, 13C{1H}, 1H,1H-COSY, HSQC, HMBC and NOESY NMR spectra were recorded on Bruker Avance 600 MHz spectrometer at Laboratorio Nacional de Resonancia Magnética Nuclear, Instituto Venezolano de Investigaciones Cientificas (IVIC), using CDCl3 and MeOD as solvent. Peak positions are relative to tetramethylsilane for 1H and 13C{1H}. The chemical shifts (δ) were measured according to IUPAC [23], expressed in parts per million (ppm) and were calibrated against the residual solvent resonance (1H) or the deuterated solvent triplet (13C). Coupling constants J are given in Hertz (Hz) as absolute values. The multiplicity of the signals are indicated as s, d, t, q, or m for singlets, doublets, triplets, quartets or multiplets respectively. All NMR spectra were recorded at room temperature (25 °C) in CDCl3 dried over molecular sieves. ESI–MS was run on a TSQ QUANTUM, Ultra AM, ThermoScientific Spectro-photometer and the HR-EI-MS analysis was conducted in a JEOL JMS-AX505WA spectrometer with direct inlet and dual approach mass analyzer, using electron impact (EI) method. Solvents were obtained from Sigma-Aldrich (Milwaukee, Wisconsin, USA) and Merck (Kenilworth, NJ, USA), and were used without any purification. Analytical thin layer chromatography (TLC) was performed on silica gel (15–40 µm PF254) 0.25 mm and 0.5 mm thick plates respectively (supplied by Merck), and the spots were visualized by spraying with AcOH/H2O/H2SO4 (37:8:5) mixture, followed by heating to 100 °C. Column chromatography was performed using silica gel 230–400 Mesh.
3.2 Plant Material
Phytolacca icosandra leaves were collected in Mucuhies-Gavidia, Municipio Rangel, Estado Mérida-Venezuela, in August 2008 and identified by Ing. For. Juan Carmona Arzola, Universidad de Los Andes (Mérida-Venezuela). A voucher specimen (Amaro et al. N° 2322) was deposited in the MERF herbarium, Faculty of Pharmacy, ULA.
3.3 Extraction and Isolation
Air-dried and powdered leaves of P. icosandra (≅ 2 kg) were exhaustively extracted at room temperature with MeOH in a Soxhlet for 48 h. After vacuum evaporation of the solvent, the crude extract (≅ 300 g) was pre-absorbed on normal phase silica gel and submitted to a chromatographic process (CC), using Hex/CHCl3 (0% up to 100%), Hex/EtOAc (30% up to 100%) and CHCl3/MeOH (20% up to 100%) mixture solvents, to afford 13 sub-fractions (A-M). Sub-fraction “E” (12.7 g, Hex/CHCl3 80%) was submitted to further CC, using solvent mixture CHCl3/Acetone (19:1 v/v) as eluent, to afford six sub-fractions E1–E6. Compound 1 was isolated from sub-fraction E2 (1.16 g) trough chromatographic column process on silica gel and eluted with CHCl3/Acetone (9:1 v/v) solvent mixture, to yield a white wax (35.2 mg). A portion of fraction “I” (1.23 g, Hex/EtOAc 75%), was further fractioned and purified by several chromatographic processes (CC) on silica gel to afford 2 (36.7 mg) from sub-fraction I2 (CHCl3/Acetone, 17:3 v/v), 3 (28.7 mg) from sub-fraction I3(CHCl3/Acetone, 4:1 v/v), and 4 (60.2 mg) from sub-fraction I5 (CHCl3/Acetone 3:1 v/v).
3.4 Identification of Known Compounds
Known compounds were identified by comparison of their physical constants and NMR spectroscopic data with those reported in the literature [24,25,26].
3.5 Phytolaccagenic Acid 23α-O-Isopropyl Tetradecanoate (1)
White wax; [α]23D + 23.8 (c 0.13, CHCl3); Rf: 0.24 (CHCl3/Acetone, 9:1);. IR (KBr): 3500–2600, 2917, 1703–1705, 1472, 1206, 728 cm−1; 1H NMR (600 MHz, CDCl3) δ (ppm) 0.69 (3H, s, H-26), 0.75 (1H, m, H-5), 0.85 (3H, t, J = 6.9 Hz, H-14′), 0.93 (3H, s, H-25), 1.01 (3H, s, H-24), 1.12 (3H, s, H-27), 1.13 (3H, s, H-29), 1.20 – 1.39 (20H, m, H-4′/H-13′), 1.39 (3H, s, -CH3), 1.42 (3H, s, -CH3), 1.56 (1H, m, H-9), 1.59 (2H, m, H-3′), 2.31 (2H, t, J = 7.4, Hz, H-2′), 2.65 (1H, dd, J = 13.9, 13.7 Hz, H-18), 3.41 (1H, d, J = 10.7 Hz, H-23a), 3.48 (1H, t, J = 3.7, 11.7 Hz, H-3), 3.50 (1H, d, J = 10.7 Hz, H-23b), 3.65 (3H, s, -OCH3), 5.32 (1H, t, J = 3.5 Hz, H-12). 13C NMR (150 MHz, CDCl3) δ (ppm) 12.4 (CH3, C-24), 14.4 (CH3, C-14′), 16.5 (CH3, C-25), 16.8 (CH3, C-26), 17.6 (CH2, C-6), 19.3 (CH3, C-2′'), 22.7 (CH2, C-11), 23.0 (CH2, C-16), 23.2 (CH2, C-2), 23.4 (CH2, C-13′), 24.7 (CH2, C-3′), 25.9 (CH3, C-27), 27.5 (CH2, C-15), 28.3 (CH3, C-29), 29.0 (CH2, C-4′), 29.2 – 29.7 (CH2, C-5′/C-11′), 29.7 (CH3, C-3′'), 30.3 (CH2, C-21), 31.9 (CH2, C-12′), 32.7 (CH2, C-7), 33.4 (CH2, C-22), 34.0 (CH2, C-2′), 36.4 (C, C-4), 37.2 (C, C-10), 38.8 (CH2, C-1), 39.4 (C, C-8), 41.4 (C, C-14), 42.0 (CH2, C-19), 42.2 (C, C-18), 43.7 (C, C-20), 45.8 (C, C-17), 47.7 (CH, C-9), 51.5 (CH, C-5), 51.8 (-OCH3), 72.6 (CH2, C-23), 77.6 (CH, C-3), 99.0 (O > C < O, C-1′'), 123.2 (CH, C-12), 142.8 (C, C-13), 176.9 (C, C-30), 179.7 (C, C-1′), 183.4 (C, C-28). HR-MS m/z 784.5859 [M+] (calcd for C48H80O8, 784.5853).
3.6 Brine Shrimp Lethality Assay
The assay was performed as described previously by Meyer et al. [22] with some minor modifications. Brine shrimp eggs (Gulf Breeze®) were hatched in artificial sea water prepared with commercial salt mixture (Instant Ocean®), illuminated and oxygenated with an aquarium pump. After 48 h incubation at 27 °C, 10 shrimps were transferred with a Pasteur pipette to three sample vials for each of three doses (100, 50, 10 μg/mL) for a total of nine vials. The sample was prepared by dissolving the compound 1 (3 mg) in CHCl3 (5 mL) and transferring the solution to each vial (833, 417 or 83 μL solution for 100, 50 or 10 ppm doses) followed by high vacuum for 1 h. After the solvent was evaporated, the compound was redissolved in 20 μL of Tween 80® and 5 mL of artificial sea water were added to achieve the correct concentration. Survivors were counted and the percent deaths at each dose and control were determined. Tween 80® at this concentration did not affect this bioassay. The LC50 and 95% confidence intervals were calculated from 24 h counts, using the Probit analysis method [27].
References
L.A.D. Williams, H. Rösner, J. Conrad, W. Möller, U. Beifuss, K. Chiba, J.P. Nkurunziza, W. Kraus, Rec. Res. Devel. Phytochem. 6, 13–68 (2002)
G. Jolliffe, British Homeopath J. 71, 31–34 (1982)
G. Ravikiran, A.B. Raju, Y. Venugopal, Int. J. Res. Pharm. Biomed. Sci. 2, 942–946 (2011)
A. Lemma, Bull. World Health Org. 42, 597–612 (1970)
J.D.H. Lambert, L. Wolde-Johannas, L. Makhubu, Bioscience 35, 364–366 (1985)
J.A. Santos-López, J.R. Villagómez-Ibarra, A. López-Ramirez, G. Montiel-Jarillo, M. Bautista-Ávila, J.A. Gayosso-de Lucio, C. Velázquez-González, Rev. CENIC Cienc. Biol. 41, 1–5 (2010)
M.M. Hernández-Villegas, R. Borges-Argáez, R.I. Rodriguez-Vivas, J.F.J. Torres-Acosta, M. Méndez-Gonzalez, M. Cáceres-Farfan, Vet. Parasitol. 179, 100–106 (2011)
M.M. Hernández-Villegas, R. Borges-Argáez, R.I. Rodríguez-Vivas, J.F.J. Torres-Acosta, M. Méndez-González, M. Cáceres-Farfán, Vet. Parasitol. 189, 284–290 (2012)
V. Treyvaud, A. Marston, W. Dyatmiko, K. Hostettmann, Phytochemistry 55, 603–609 (2000)
E. Galarraga, J.M. Amaro-Luis, L.B. Rojas, A.C. Mitaine-Offer, M.A. Lacaille-Dubois, Ciencia (Maracaibo) 22, 53–66 (2014)
E. Galarraga Montes, J.M. Amaro-Luis, Nat. Prod. Res. 30, 89–94 (2016)
M. Ukiya, T. Akihisa, K. Yasukawa, Y. Kasahara, Y. Kimura, K. Koike, T. Nikaido, M. Takido, J. Agric. Food Chem. 49, 3187–3197 (2001)
J. Wandji, F. Tillequin, D.A. Mulholland, J.D. Wansi, T.Z. Fomum, V. Fuendjiep, F. Libot, N. Tsabang, Planta Med. 68, 822–826 (2002)
Y.C. Shen, C.V.S. Prakash, L.T. Wang, C.T. Chien, M.C. Hung, J. Chin. Chem. Soc. 50, 297–302 (2003)
K.W. Wang, Nat. Prod. Res. 21, 669–674 (2007)
H. Maza, P. Mkounga, S.L. Fenkama, S.K. Sado, H. Ishikawac, H. Nishino, E.A. Nkengfack, Phytochem. Lett. 19, 23–29 (2017)
F.J. Arriaga-Giner, J.M. Rullkötter, T.M. Peakman, E. Wollenweber, Z. Naturforsch. 46c, 507–512 (1991)
D.A. Iskenderov, I.M. Isaev, M.I. Isaev, Chem. Nat. Comp. 45, 656–659 (2009)
S.X. Yang, Z.C. Yu, Q.Q. Lu, W.Q. Shi, H. Laatsch, J.M. Gao, Phytochem. Lett. 5, 576–580 (2012)
F. Maltese, F. van der Kooy, R. Verpoorte, Nat. Prod. Commun. 4, 447–454 (2009)
J.R. Hanson, J. Chem. Res. 41, 497–503 (2017)
B.N. Meyer, N.R. Ferrigni, J.E. Putnam, L.B. Jacobsen, D.E. Nichols, J.L. McLaughlin, Planta Med. 45, 31–34 (1982)
R.K. Harris, E.D. Becker, S.M. Cabral de Menezes, R. Goodfellow, P. Granger, Pure & Appl. Chem. 73, 1795–1818 (2001)
K.W. Glombitza, W. Gielsdorf, G. Eckhardt, M.L. Koch, Planta Med. 27, 367–371 (1975)
W.S. Woo, Natural Products Research Institute, Seoul Natural University. (1978)
S. Harkar, T.K. Razdan, E.S. Waigth, Phytochemistry 23, 2893–2898 (1984)
D. Finney, Probit Analysis, A Statistical Treatment of the Sigmoids Response Curve, 3rd edn. (Cambridge University Press, New York, 1971), pp. 21–49
Acknowledgements
The authors would like to thank USB-DID (project S1-IC-CB-004–15) for financial support. Also thanks are expressed to Ing. For. Juan A. Carmona Arzola (ULA) for plant identification.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Galarraga, E., Mavares, A., Urdaneta, N. et al. Artificial Triterpenoid Fatty Acid Ester Isolated From the Leaves of Phytolacca icosandra L. Nat. Prod. Bioprospect. 10, 221–225 (2020). https://doi.org/10.1007/s13659-020-00249-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-020-00249-x