
Abstract
Specific activating missense HRAS variants cause Costello syndrome (CS), a RASopathy with recognizable facial features. The majority of these dominant disease causing variants affect the glycine residues in position 12 or 13. A clinically suspected CS diagnosis can be confirmed through identification of a dominant pathogenic HRAS variant. A novel HRAS variant predicting p.(Glu62_Arg68dup) was identified in an individual with hypertrophic cardiomyopathy, Chiari 1 malformation and ectodermal findings consistent with a RASopathy. Functional studies showed that the p.Glu62_Arg68dup alteration affects HRAS interaction with effector protein PIK3CA (catalytic subunit of phosphoinositide 3-kinase) and the regulator neurofibromin 1 (NF1) GTPase-activating protein (GAP). HRASGlu62_Arg68dup binding with effectors rapidly accelerated fibrosarcoma (RAF1), RAL guanine nucleotide dissociation stimulator (RALGDS) and phospholipase C1 (PLCE1) was enhanced. Accordingly, p.Glu62_Arg68dup increased steady-state phosphorylation of MEK1/2 and ERK1/2 downstream of RAF1, whereas AKT phosphorylation downstream of PI3K was not significantly affected. Growth factor stimulation revealed that expression of HRASGlu62_Arg68dup abolished the HRAS’ capacity to modulate downstream signaling. Our data underscore that different qualities of dysregulated HRAS-dependent signaling dynamics determine the clinical severity in CS.
Similar content being viewed by others

Introduction
Costello syndrome (CS) can be clinically suspected based on its characteristic multiorgan presentation with distinctive facial features, failure-to-thrive, developmental delay, cardiac manifestations, and a history of polyhydramnios [1]. The diagnosis is molecularly confirmed through the identification of a dominant pathogenic HRAS variant [2]. Approximately, 90% of CS-causative HRAS variants involve amino acid glycine at position 12 and result in a recognizable, typical or, severe phenotype [3]. A more variable, milder or “attenuated” phenotype occurs with rarer dominant pathogenic HRAS variants [3]. HRAS c.187_207dup p.(Glu63_Asp69dup) causes an attenuated phenotype with milder intellectual disability, fewer feeding issues, and a lower tumor risk [4, 5].
HRAS acts as molecular switch by alternating between an active guanosine triphosphate (GTP)-bound and inactive guanosine diphosphate (GDP)-bound state. Guanine nucleotide exchange factors (GEFs) catalyze the exchange from HRAS-bound GDP to GTP and turn on signaling, whereas GTPase-activating proteins (GAPs) terminate signaling by stimulating GTP hydrolysis. Active HRAS binds to various effectors, such as serine/threonine RAF kinases, the catalytic subunits (PIK3CA) of phosphoinositide 3-kinases (PI3K), phospholipase C1 (PLCE1) and RAL guanine nucleotide dissociation stimulator (RALGDS), and promotes signal transduction.
Disease-associated changes of HRAS amino acids 12, 13, and 61 affect GTPase activity and trap HRAS in its active state independently from incoming signals. Consequently, increased activation of HRAS-dependent downstream signaling describes CS pathology. However, several studies demonstrated a more diverse spectrum of functional consequences [5,6,7]. Notably, the CS-associated HRAS p.Glu63_Asp69dup variant did not increase PI3K-Akt signaling.
Material and methods
Consent to publish the proband’s data and photographs was provided by her parents. A description of the laboratory methods is available in Supporting information.
Results
Clinical summary
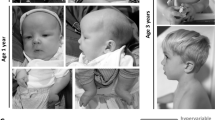
The proband (Fig. 1a) was born at 38 weeks gestation after a pregnancy complicated by large fetal size. The girl weighed 4.4 kg (>97th centile), length was 50 cm (50th–75th centile) and OFC was 35 cm (75th–90th centile). She had slow weight gain and low-normal length with relative macrocephaly. Gross motor development was delayed with sitting independently at age 11 months and walking at 14 months. Her language development was age appropriate with pronunciation difficulties. Moderate concentric left ventricular hypertrophy was identified at age 15 months and treated with propranolol. Progression of the septal hypertrophy and mild mitral valve regurgitation was noted at age 3 years 2 months, when status epilepticus occurred in the setting of cellulitis. Imaging showed a Chiari 1 malformation with 8 mm cerebellar tonsil protrusion and syringomyelia. At age 3.5 years, her height was 93 cm (10th–25th centile), weight was 14.2 kg (25th–50th centile) and OFC 50.7 cm (75th–90th centile). She was very interactive and age appropriate, with at times difficult to understand speech. She had short sparse hair that had never been cut, full and flaring eye brows and unusually long eye lashes. Her facial features were notable for hypertelorism, a low nasal bridge and low-set ears. Her neck was wide. She had deep palmar creases with mild redundancy of the soft tissues and hyperkeratosis on palms and soles. She had multiple lentigines scattered over her head and extremities, mild generalized hyperpigmentation with some more notably darkened skin regions. Clinical features are summarized in Fig. 1b.

a Photographs of the patient at age 3 years. Note short hair, bitemporal narrowing and dolichocephaly, hypertelorism, and low-set ears. Multiple scattered lentigines are present. b Comparison of phenotypic findings in the patient reported here to those of the individuals reported in Lorenz et al. [5] and Xu et al. [4]; and findings typically seen in individuals with Costello syndrome as reported in Grant et al. [2].
Analysis of the facial photograph (Fig. 1a, frontal image) by Face2Gene (FDNA, Boston, MA) (version 19.1.7) without additional molecular or clinical information provided, ranked CS as the top match, followed by Noonan syndrome with loose anagen hair (data not shown) [8].
Testing for gene changes associated with hypertrophic cardiomyopathy was performed during her hospitalization for status epilepticus and identified a novel HRAS variant, c.186_206dup (NM_005343.4); p.(Glu62_Arg68dup) (NP_005334.1), leading to a suspected diagnosis of CS. Genotyping of parental DNAs did not identify this alteration and confirmed paternity. Delineation of parental origin was not possible due to lack of informative markers.
Functional characterization
The HRAS c.186_206dup variant was predicted to cause the duplication of HRAS amino acids 62–68, or p.(Glu62_Arg68dup). Six of these residues are an integral part of the switch II domain (amino acids 59–67, Fig. S1) mediating HRAS binding with regulator and effector proteins [9,10,11]. The p.(Glu62_Arg68dup) variant is the second HRAS germline duplication affecting multiple amino acid. Another duplication, HRAS c.187_207dup p.(Glu63_Asp69dup) was identified in two patients with a milder phenotype [4, 5]. We used GST-fusion proteins of interacting motifs from RAS effectors RAF1, RALGDS, PIK3CA and PLCE1 and precipitated activated hemagglutinin (HA)-tagged HRAS protein variants from HEK293T cell extracts (Fig. 2a). CS-typical HA-HRASGly12Ser and oncogenic HA-HRASGly12Val strongly co-precipitated with any effector under all cell culture condition tested (Fig. 2a). As a negative control, we used the dominant negative variant HA-HRASSer17Asn; this protein variant was pulled down only minimally (Fig. 2a). Similar to HA-HRASGly12Ser and HA-HRASGly12Val the amounts of HA-HRASGlu62_Arg68dup and HA-HRASGlu63_Asp69dup were elevated compared to HA-HRASWT in the RAF1, PLCE1, and RALGDS precipitates under all culture conditions. However, as already shown for HA-HRASGlu63_Asp69dup [5], HA-HRASGlu62_Arg68dup was only slightly enriched in precipitates using PIK3CA (the catalytic subunit of PI3K) as bait when compared to HA-HRASWT (Fig. 2a). These data suggest (1) that HA-HRASGlu62_Arg68dup is trapped in an active conformation resulting in stable binding to RAF1, PLCE1, and RALGDS and (2) that the p.Glu62_Arg68dup alteration affects binding to PIK3CA.

a HRASGlu62_Arg68dup co-precipitates with RAF1, RALGDS, and PLCE1, but not with PIK3CA. HRAS variants were transiently expressed in HEK293T cells under culture conditions as indicated, precipitated from cell extracts by using GST-fused effector peptides, and subjected to immunoblotting; n = 3. b p.Glu62_Arg68dup affects binding efficiency with NF1. HRAS variants were transiently expressed under culture conditions as indicated, precipitated from cell extracts by using GST-fused NF1333 peptide, and subjected to immunoblotting; n = 3. Pcpt precipitates; tcl total cell lysates; RBD RAS binding domain; RA Ras association; RALGDS RAL guanine nucleotide dissociation stimulator; PLCE1 phospholipase C1; PIK3CA phosphatidylinositol 3-kinase catalytic subunit alpha; NF1 neurofibromin 1; HA hemagglutinin.
GAP proteins stimulate HRAS GTPase activity and the switch II region in HRAS is essential for binding active HRAS with GAPs [9]. To analyze binding between HRAS and the RAS-specific NF1 GAP we pulled down ectopically expressed HRAS protein variants with the GAP-related domain of NF1 (fused with GST; GST-NF1333). HA-HRASWT co-precipitated only weakly with GST-NF1333 reflecting a relatively small fraction of active HA-HRASWT molecules (Fig. 2b). Consistently, constitutively active HA-HRASGly12Ser and HA-HRASGly12Val but not dominant negative HA-HRASSer17Asn strongly co-precipitated with GST-NF1333 (Fig. 2b). Although HA-HRASGlu62_Arg68dup and HA-HRASGlu63_Asp69dup were pulled down stronger than HA-HRASWT, co-precipitation efficiency was low compared to HA-HRASGly12Val and HA-HRASGly12Ser (Fig. 2b). Because RAF1, PLCE1 and RALGDS (i.e. effector) pulldowns showed similar activation of HA-HRASGlu62_Arg68dup, HA-HRASGlu63_Asp69dup, HA-HRASGly12Val, and HA-HRASGly12Ser (Fig. 2a), these data suggest that duplication of HRAS amino acids 62–68 or 63-69 interferes with NF1 GAP binding.
To assess signaling consequences of p.Glu62_Arg68dup, we measured phosphorylated MEK1/2, ERK1/2, and AKT in cells expressing HRAS variants. Expression of HA-HRASGly12Val, HA-HRASGly12Ser, or HA-HRASGlu63_Asp69dup but not HA-HRASSer17Asn promoted MEK1/2 and ERK1/2 phosphorylation in HEK293T cells under all culture conditions (Fig. 3a). Likewise, HA-HRASGlu62_Arg68dup enhanced phosphorylation of MEK1/2 and ERK1/2 compared to HA-HRASWT. We detected no significant differences in AKT phosphorylation between cells expressing HA-HRASWT and HRASGly12Val, HA-HRASGly12Ser, HA-HRASGlu63_Asp69dup, or HRASGlu62_Arg68dup; indicating that AKT signaling is very robust in HEK293T cells and their dependence on HRAS as well as responsiveness to serum factors is limited (Fig. S2). Consequently, we applied another cell line, MCF7 cells, and found that expression of HA-HRASGly12Val or HA-HRASGly12Ser induced a detectable AKT phosphorylation response compared with HA-HRASWT (Fig. 3b) On the other hand, in cells expressing HA-HRASGlu63_Asp69dup or HRASGlu62_Arg68dup the increase of AKT phosphorylation was very weak (Fig. 3b). Our data suggest that the p.Glu62_Arg68dup alteration preferentially intensifies MAPK signal flux.

a Expression of HRASGlu62_Arg68dup enhances downstream signaling via MEK1/2 and ERK1/2 in HEK293T cells. HRAS variants were transiently expressed in HEK293T cells. For control, cells were transfected with empty vector. Cells were cultured under serum-starved condition (0.1% serum), normal growth condition (10% serum), or serum-starved condition followed by 20 min stimulation with EGF (10 ng/ml EGF stimulation). Total cell lysates (tcl) were subjected to immunoblotting as indicated; n = 3. b Expression of HRASGlu62_Arg68dup marginally affects AKT phosphorylation in MCF7 cells. HRAS variants were transiently expressed in MCF7 cells and samples were treated as described above. Total cell lysates (tcl) were subjected to immunoblotting as indicated; n = 3. c p.Glu62_Arg68dup impairs epidermal growth factor sensitivity of HRAS. HEK293T cells transiently expressing various HA-HRAS variants were stimulated with EGF for various times (5, 15, and 30 min) or left untreated (0 min). Active HA-HRAS was precipitated from cell lysates using GST-fused effector peptides RAF1[RBD]. Precipitates (pcpt) and total cell lysates (tcl) were subjected to immunoblotting using specific antibodies as indicated; n = 3. d HRAS activation and MEK1/2, ERK1/2 and AKT phosphorylation dynamics. Activation and phosphorylation levels upon 0, 5, 15, and 30 min EGF stimulation relative to maximum levels in cells expressing HRASWT, HRASGly12Ser HRASGlu62_Arg68dup. The number of independent experiments (n) is indicated, and the data represent the mean of n independent experiments ± standard deviation (for untreated cells, 0 min EGF). Significance levels are specified between data points [∗P < 0.05; ns, statistically not significant (two tailed t test)].
For CS and related RASopathies, impaired signaling dynamics are more relevant than a simple static hyperactivation of RAS-dependent signaling [5, 7, 12]. Therefore, we compared the intensity of EGF-induced HRAS activation and downstream signaling over time. RAS pull-down assays showed activation of HA-HRASWT after 5 min, followed by signal decrease at 15–30 min after EGF addition (Fig. 3c, d). In contrast, EGF had no stimulating effect on constitutively active HA-HRASGly12Ser, HA-HRASGly12Val, or HA-HRASGlu62_Arg68dup (Fig. 3c, d), suggesting that HRASGlu62_Arg68dup is constitutively active and uncoupled from growth factor stimulation. EGF stimulation induced a strong MEK1/2 and ERK1/2 phosphorylation response in HA-HRASWT cells, whereas in cells expressing HA-HRASGly12Ser, HA-HRASGly12Val, or HA-HRASGlu62_Arg68dup, basic phosphorylation (0 min EGF) was enhanced with little or no further increase (Fig. 3c, d). AKT phosphorylation was slightly induced upon EGF treatment in cells expressing HRASWT, but only marginally altered in cells expressing HA-HRASGly12Ser, HA-HRASGly12Val, or HA-HRASGlu62_Arg68dup (Fig. 3c, d). These data suggest that the p.Glu62_Arg68dup duplication impairs HRAS’ capacity to modulate signaling.
Discussion
Phenotype associated with HRAS c.186_206dup
The proband’s phenotype of large birth weight, relative macrocephaly, mild motor delay, Chiari 1 malformation with syringomyelia, hypertrophic cardiomyopathy, and ectodermal findings is consistent with CS (Fig. 1b). An objective digital assessment of her facial features using Face2Gene most closely matched CS. Limited genotype–phenotype correlations have been established for CS [1]. This novel intragenic duplication is most similar to a duplication reported in two unrelated individuals [4, 5]. The individuals with intragenic duplications share multiple lentigines, and short and slow growing hair, without the tight curls characteristic for CS (Fig. 1b). None shows the wide mouth with prominent vermillion typical for CS. While three individuals do not allow for definitive genotype–phenotype conclusions, these reports establish intragenic HRAS duplications as a potential disease causing mechanism associated with attenuated CS.
Functional consequences of HRAS p.Glu62_Arg68dup
HRASGlu62_Arg68dup is characterized by two molecular aberrations. First, binding of HA-HRASGlu62_Arg68dup to effectors RAF1, PLCE1, and RALGDS is increased, and consequently, MAPK signaling is enhanced. This hyperactivation likely results from impaired GTP hydrolysis. Our experiments did not discriminate if p.Glu62_Arg68dup affects intrinsic or GAP-mediated GTP hydrolysis; however, compared with constitutively active HRASGly12Val and HRASGly12Ser, binding with NF1 GAP was reduced. The duplication (p.Glu62_Arg68dup) contains essential components of the HRAS switch II region (Fig. S1) involved in binding regulator proteins (GAPs and GEFs), effector molecules and GDP/GTP nucleotides [9,10,11]. We conclude that p.Glu62_Arg68dup interferes with the structure of the switch II region critical for interaction with NF1 GAP [9, 13]. Similarly, affected GAP binding occurred with other CS-associated HRAS variants including HRAS p.Glu37dup and HRAS p.Glu63_Asp69dup [5, 6].
Second, p.Glu62_Arg68dup does not cause more effective PIK3CA binding and marginally increases AKT phosphorylation. Given that HRASGlu62_Arg68dup is trapped in an active conformation—otherwise, it would not constitutively activate MAPK signaling—we conclude that p.Glu62_Arg68dup selectively affects PIK3CA binding. The HRAS switch II region is critical for interaction with PIK3CA [14, 15]. Accordingly, substitution of HRAS tyrosine 64 by a glycine eliminated the interaction with PIK3CA (and with NF1), whereas it had no effect on RAF1 binding [16]. RAF1 and RALGDS interact with RAS switch I, but not switch II [11]. PLCE1 makes contact with switch I and switch II, as does PI3K [17]. Our results indicate that PLCE1 interaction with switch I of HRASGlu62_Arg68dup is sufficient for effective binding. Identical results were reported for HRASGlu63_Asp69dup [5]. In this context, data from this and other reports suggest that the activation status of PI3K-AKT signaling is a significant factor determining the severity of CS manifestation [5, 12, 18]. The absence of PI3K-AKT hyperactivation may be associated with a phenotype at the milder end of the CS spectrum.
HRASGlu62_Arg68dup stimulates MAPK signaling under static cell culture conditions and independently of growth factor stimulation in HEK293T cells. Similar to HRASGly12Ser and HRASGly12Val, HRASGlu62_Arg68dup is constitutively active and lacks the ability to respond to upstream signals. Notably, HRASGlu63_Asp69dup showed residual reagibility to stimuli in COS7 cells, a finding we attribute to the different cell lines used. Importantly, the molecular property of reduced reagibility to stimuli is common to all CS-associated HRAS variants.
References
Gripp KW, Morse LA, Axelrad M, Chatfield KC, Chidekel A, Dobyns W, et al. Costello syndrome: clinical phenotype, genotype and management guidelines. Am J Med Genet. 2019;179:1725–44.
Grant AR, Cushman BJ, Cavé H, Dillon MW, Gelb BD, Gripp KW, et al. Assessing the gene-disease association of 19 genes with the RASopathies using the ClinGen gene curation framework. Hum Mutat. 2018;39:1485–93.
Sol-Church K, Gripp KW The molecular basis of Costello syndrome. In: Zenker M (editor) Noonan syndrome and related disorders—a matter of deregulated Ras signaling. Monographs in human genetics, vol 17, Basel: Karger; 2009. p. 94–103
Xu F, Wang HJ, Lin ZM, Yu B. Recurrent duplication mutation in HRAS causing mild Costello syndrome in a Chinese patient. Clin Exp Dermatol. 2015;40:404–7. https://doi.org/10.1111/ced.12571
Lorenz S, Lissewski C, Simsek-Kiper PO, Alanay Y, Boduroglu K, Zenker M, et al. Functional analysis of a duplication (p.E63_D69dup) in the switch II region of HRAS: new aspects of the molecular pathogenesis underlying Costello syndrome. Hum Mol Genet. 2013;22:1643–53. https://doi.org/10.1093/hmg/ddt014
Gremer L, De Luca A, Merbitz-Zahradnik T, Dallapiccola B, Morlot S, Tartaglia M, et al. Duplication of Glu37 in the switch I region of HRAS impairs effector/GAP binding and underlies Costello syndrome by promoting enhanced growth factor-dependent MAPK and AKT activation. Hum Mol Genet. 2010;19:790–802. https://doi.org/10.1093/hmg/ddp548
Gripp KW, Bifeld E, Stabley DL, Hopkins E, Meien S, Vinette K, et al. A novel HRAS substitution (c.266C>G; p.S89C) resulting in decreased downstream signaling suggests a new dimension of RAS pathway dysregulation in human development. Am J Med Genet A. 2012;158A:2106–18. https://doi.org/10.1002/ajmg.a.35449
Gurovich Y, Hanani Y, Bar O, Nadav G, Fleischer N, Gelbman D, et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med. 2019;25:60–64.
Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–8.
Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. The structural basis of the activation of Ras by Sos. Nature. 1998;394:337–43.
Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–304.
Gripp KW, Kolbe V, Brandenstein LI, Rosenberger G. Attenuated phenotype of Costello syndrome and early death in a patient with an HRAS mutation (c.179G>T; p.Gly60Val) affecting signalling dynamics. Clin Genet. 2017;92:332–7. https://doi.org/10.1111/cge.12980
Ahmadian MR, Kiel C, Stege P, Scheffzek K. Structural fingerprints of the Ras-GTPase activating proteins neurofibromin and p120GAP. J Mol Biol. 2003;329:699–710.
Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 2000;103:931–43.
Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–20.
Moodie SA, Paris M, Villafranca E, Kirshmeier P, Willumsen BM, Wolfman A. Different structural requirements within the switch II region of the Ras protein for interactions with specific downstream targets. Oncogene. 1995;11:447–54.
Bunney TD, Harris R, Gandarillas NL, Josephs MB, Roe SM, Sorli SC, et al. Structural and mechanistic insights into ras association domains of phospholipase C epsilon. Mol Cell. 2006;21:495–507.
Gripp KW, Sol-Church K, Smpokou P, Graham GE, Stevenson DA, Hanson H, et al. An attenuated phenotype of Costello syndrome in three unrelated individuals with a HRAS c.179G>A (p.Gly60Asp) mutation correlates with uncommon functional consequences. Am J Med Genet A. 2015;167A:2085–97.
Acknowledgements
We thank the family for allowing us to share this information.
Funding
This work was supported by a grant from the Bundesministerium für Bildung und Forschung (BMBF, GeNeRARe, TP6 to GR, funding code 01GM1902E).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
KWG is the CMO for FDNA, the company providing the Face2Gene application.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Gripp, K.W., Baker, L., Robbins, K.M. et al. The novel duplication HRAS c.186_206dup p.(Glu62_Arg68dup): clinical and functional aspects. Eur J Hum Genet 28, 1548–1554 (2020). https://doi.org/10.1038/s41431-020-0662-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-0662-4
This article is cited by
-
A novel HRAS c.466C>T p.(Phe156Leu) variant in two patients with attenuated features of Costello syndrome
European Journal of Human Genetics (2022)
