Thermodynamics of Uranium Tri-Iodide from Density-Functional Theory



Lawrence Livermore National Laboratory, Livermore, CA 94550, USA
*
Author to whom correspondence should be addressed.
Appl. Sci. 2020, 10(11), 3914; https://doi.org/10.3390/app10113914
Submission received: 29 April 2020
/
Revised: 20 May 2020
/
Accepted: 3 June 2020
/
Published: 5 June 2020
(This article belongs to the Special Issue Experimental and Theoretical Studies on the Physical Properties of Lanthanides, Actinides, and Their Compounds)
Abstract
:Density-functional theory (DFT) is employed to investigate the thermodynamic and ground-state properties of bulk uranium tri-iodide, UI3. The theory is fully relativistic and electron correlations, beyond the DFT and generalized gradient approximation, are addressed with orbital polarization. The electronic structure indicates anti-ferromagnetism, in agreement with neutron diffraction, with band gaps and a non-metallic system. Furthermore, the formation energy, atomic volume, crystal structure, and heat capacity are calculated in reasonable agreement with experiments, whereas for the elastic constants experimental data are unavailable for comparison. The thermodynamical properties are modeled within a quasi-harmonic approximation and the heat capacity and Gibbs free energy as functions of temperature agree with available calculation of phase diagram (CALPHAD) thermodynamic assessment of the experimental data.
1. Introduction
Uranium tri-iodide, UI3, is a rare anti-ferromagnetic inorganic compound that forms in the PuBr3-type, Cmcm, orthorhombic structure [1,2]. This structure has the same space group number (#63) as the ground-state phase of uranium metal (α-uranium). From an applications standpoint, it has been used as a starting material for organometallic uranium chemistry [3,4,5] and for separation of uranium metal [6]. Aside from its crystal structure and magnetic configuration, UI3’s physical properties have not received a lot of attention compared to those of other uranium compounds. As with most uranium compounds, UI3 has substantial magnetic moments that have been reported in the range 1.97–3.65 µB on the uranium atoms [2,3]. These moments are reported [2] to order in a complex non-intuitive anti-ferromagnetic fashion. In addition to its structure and magnetic properties, the energy of formation (or formation enthalpy) has been measured (−98.8 [7] and −111.6 kcal/mol [8]).
From a fundamental science perspective, first-principles approaches have not yet been applied for the study of UI3 as a bulk material that we are aware of, but in the form of a two-dimensional monolayer [9]. In that investigation, the magnetic properties were the primary interest and the fact that this 2-D material may be a possible candidate for spintronic applications. The research [9] involved conventional density-functional theory with a pseudopotential approach that neglected spin–orbit coupling. In contrast to the bulk, the monolayer was predicted to favor ferromagnetism over anti-ferromagnetism and the spin moment was reported to be 3.0 μB. It is expected that the bulk spin moment is quenched somewhat relative to that of the monolayer and should therefore be less than 3.0 μB. The spin–orbit interaction was also ignored [9] and there was no consideration of an orbital moment. In reality, however, uranium compounds typically have rather large orbital moments with a magnitude comparable to that of the spin moment but aligned anti-parallel (uranium atom has less than half filled 5f shell). The total magnetic moment (sum of spin and orbital contributions) is thus significantly reduced when full relativity and orbital moments are addressed.
The primary focus of the present work on UI3 is not magnetism but the fundamental thermodynamic properties at elevated temperatures. In general, the high-temperature thermodynamic information often provides useful guidance for controlling thermally induced phase evolution and/or reaction kinetics of the chemical compounds (e.g., thermal decomposition at high temperatures) [10]. In addition, the temperature-dependent thermodynamics is the important basis for modeling phase transformations and associated microstructure evolution upon temperature variation since it determines the driving force for the kinetic processes [11,12,13]. To this end, accurate computational approaches for investigating the thermodynamics at elevated temperatures can play a critical role in establishing essential thermodynamic databases of chemical compounds. As mentioned above, the fundamental bulk properties of uranium tri-iodide have not been investigated with any ab initio method so far. Therefore, we apply our well-established density-functional theory (DFT) method to the bulk UI3 compound to compute fundamental thermodynamic properties in this study.
Contrary to the previous study [9] of the UI3 monolayer, we do not limit ourselves to the pseudopotential approximation. Furthermore, we go beyond that approach [9] and address all relativistic effects including spin-orbit coupling with an extension to allow for orbital polarization (orbital–orbital coupling). Because very little is known regarding the strength of the electron correlations in UI3 we have refrained, as in the treatment in [9], from applying additional onsite Coulomb repulsion (effective Hubbard U; DFT + U method) that is often advised for uranium oxides.
Our thermodynamical investigation is limited to the solid state ranging from zero up to 1000 K (UI3 melts at 1039 K [14]), and because of this relatively low maximum temperature, we expect that a quasi-harmonic treatment of the thermal-lattice excitations is appropriate. In our recent study of uranium mononitride [15] we discovered that anharmonic effects (phonon–phonon coupling) were very important for the thermodynamical properties, but only for temperatures above about 1000 K. For comparison, and to confirm our first-principles model for UI3, we carry out phase diagram (CALPHAD) calculations of the Gibbs free energies and heat capacities using the SSUB6 (see below) thermodynamic database and the Thermo-Calc software.
2. Computational Methods
2.1. Electronic Structure Method
The electronic structure results we present here are obtained from density-functional theory. This theory is in principle exact, but it relies on practical simplifications and the foundational one is that for the electron interactions. For the actinides [16,17] the most robust formulation is the so-called generalized gradient approximation (GGA) [18].
Regarding the DFT implementation, we utilize an all-electron full-potential linear muffin-tin orbital (FPLMTO) method that is well known [19] and detailed [20]. FPLMTO makes no simplifications for the core electrons that occupy deeper energy levels than the valence states, as opposed to the pseudopotential method. The core approximation made in pseudopotential methods is computationally efficient but less accurate.
Basis functions, electron densities, and potentials are not subject to geometrical approximations and they are expanded in spherical harmonics inside non-overlapping (muffin-tin (MT)) spheres centered at each atomic site and in Fourier series in the region between these MTs. There is a need to determine the MT sphere radius (rMT) and here it is selected so that rMT ~ 0.7rWS, where rWS is the Wigner-Seitz (atomic-sphere) radius. The radial part of the basis functions inside the MT spheres is calculated from a wave equation for the l = 0 component of the potential that includes all relativistic corrections, including spin–orbit coupling for d and f states but not for the p states, as explained in [17]. The orbital–orbital interaction (orbital polarization (OP)) is only operating on the uranium f states [21]. This interaction is not explicit in conventional density-functional theory but has been shown to be important for some f-electron systems, particularly plutonium [17,22].
The crystal structure of the orthorhombic UI3 compound is optimized with respect to the total energy. In other words, all degrees of freedom, given the symmetry of the crystal, have been varied in order to produce the lowest (ground-state) total energy. This procedure is particularly important for an accurate calculation of the energy of formation of the compound. For this we also similarly relax the orthorhombic structure of α-uranium. Typically, structural relaxation from electronic structure calculations are guided by the forces on the atoms. However, in the presence of strong spin–orbit interaction and orbital polarization these forces are difficult to compute accurately [23]. The relaxation is therefore here accomplished without an explicit need to compute forces on the atoms but by separately moving them small amounts from their equilibrium positions, followed, iteratively, by optimizing the unit cell (b/a and c/a axial ratios). Although a somewhat tedious and demanding scheme, it is very robust for relaxing crystal structures and it was developed for our study of ε-uranium phonons [23]. The obtained lattice constants for UI3 are a = 4.48 Å, b = 15.94 Å, and c = 10.26 Å. Both a and c are good while the b lattice constant is overestimated relative to the experimental data (4.33, 14.01, and 10.0 Å) [2]. We should mention that atomic forces can be evaluated resulting from this structural-relaxation procedure (they approach zero at the equilibrium). Such forces were used for the calculation of temperature-dependent phonons for uranium mononitride recently [15], but for the low-symmetry orthorhombic phase relevant here, the applied phonon method requires further development.
For determining the formation enthalpy, we calculate the energies of UI3, α-uranium, and the iodine atom. For these energies to be consistent, the calculations need to be set up as similarly as possible, using the same computational tool, and for the iodine atom we thus construct a simple-cubic cell with the atoms far apart. The atoms are gradually separated until the total energy of the cell converges and we define the result as the atomic energy. One also needs the formation energy of the iodine molecule and because our computational code is not efficient for molecules, we take that energy from experiments.
UI3 is an anti-ferromagnet [2] with a somewhat complex magnetic configuration. Here, we compute the energies assuming a simplified anti-ferromagnetic arrangement of the spin moments on the uranium atoms along the (001) direction in the orthorhombic cell. This configuration turns out to be slightly lower in energy (~0.5 mRy/atom) than the ferromagnetic state and DFT thus confirms anti-ferromagnetism in UI3.
For the 16-atom per unit cell UI3 structure we apply 144 k-points in the irreducible part of the Brillouin zone and each energy eigenvalue is broadened with a Gaussian distribution having a 20 mRy energy width. For the elastic constants the total energies need to be very carefully converged and for this purpose we utilize up to 288 k points.
The thermodynamic properties (heat capacity and Gibbs free energy) are obtained from quasi-harmonic Debye–Grüneisen theory up to 1000 K. This methodology has been thoroughly detailed in the literature [24]. In the present implementation [25] of the model only the energy–volume relationship is needed, which we obtain from the DFT electronic structure calculations. We recently applied this straightforward framework for uranium mononitride. For UN, we found that for lower temperatures (<1000 K) it produces heat capacities that compare well with the anharmonic treatment while above that temperature one definitely needs to consider anharmonic lattice dynamics [15] that go beyond the quasi-harmonic assumption.
2.2. CALPHAD Method
The CALPHAD method is utilized here to calculate the heat of formation, heat capacity, and Gibbs energy of UI3 as functions of temperature for comparison with our first-principle-theory data to validate our model. Generally, the major function of the CALPHAD scheme is to model the Gibbs energy of individual phases pertaining to binary and ternary systems to best reproduce critically reviewed phase diagrams and thermodynamic properties, and then leverage the assessments of these “building blocks” to predict phase stability and thermodynamic properties of multicomponent systems [26,27,28]. CALPHAD works iteratively to generate a set of functions and parameters describing the Gibbs energy of appropriate phases and when such functions and parameters have been determined, they are compiled in a database that can be applied to calculated thermodynamical properties of multi-component systems.
In addition to comparing CALPHAD data and first-principles modeling, we realize that the coupling of CALPHAD and the theoretical results enhances the capability of thermodynamic modeling to consider material systems with many thermodynamic unknowns. First, ab initio data (e.g., the heats of formation and transformation) can directly provide important parameters to the CALPHAD modeling framework when lacking experimental data. Second, it is worth mentioning that the procedure of parameter optimization and error minimization within in the CALPHAD approach is an inverse problem with infinite degrees of freedom [29]. Therefore, a large number of combinations of values of parameters determined by the user can result in phase diagrams that coincide. Therefore, the use of DFT-derived properties associated with the CALPHAD assessment constrains the optimization and validates the final thermodynamical database, not only from a phase-diagram standpoint, but also from an energetics standpoint. In the literature, one readily finds first-principles-informed CALPHAD assessments for actinides [30,31,32].
There is no available experimental phase diagram for the U-I system, thus no CALPHAD assessment can be performed across the whole composition range. However, thermochemical data of the UI3 compound have been compiled by the Scientific Group Thermodata Europe (SGTE) and the corresponding CALPHAD functions are used in the present study through the SSUB6 Substances Database of the Thermo-Calc software (2019b version) [33,34]. Following the present work, ab initio predictions of specific heat and free energy versus temperature for actinide systems are extremely useful to the CALPHAD community.
3. Results
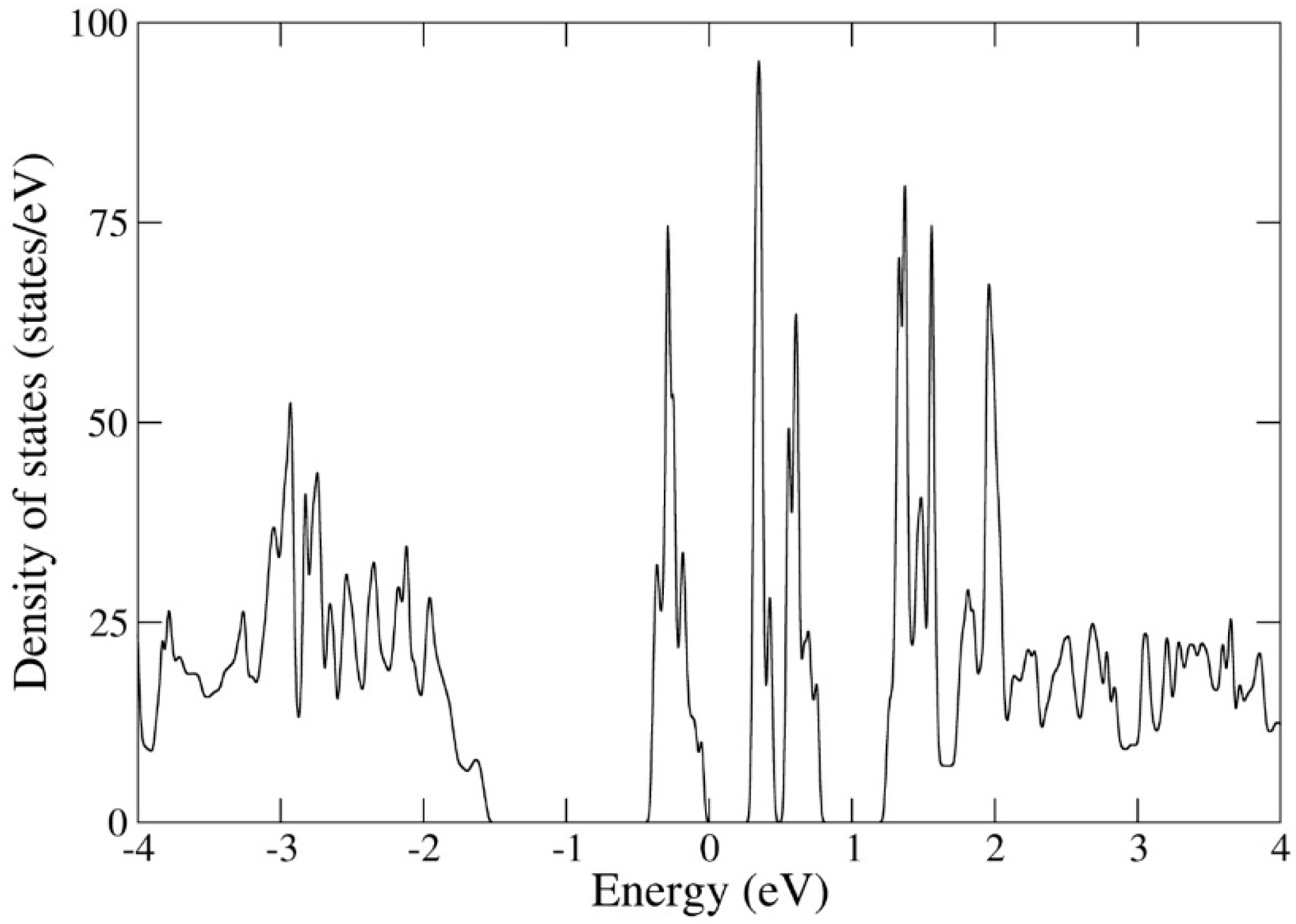
The calculated (DFT) electronic density of states for UI3, shown in in Figure 1, displays several band gaps, including at the highest occupied energy. It is not uncommon that uranium compounds form band gaps and perhaps the uranium oxides are best known for this behavior. DFT typically underestimates or even fails to predict these band gaps, whereas theory that incorporates Mott-Hubbard electron correlations (DFT + U method) can correct the size and position of the band gap [35] given an appropriately chosen Hubbard U parameter. However, photoemission or other experiments illuminating the existence of band gaps have not been undertaken for UI3 and therefore accurately determining the U parameter is impossible. Without knowledge of this central parameter, the DFT + U approach becomes less practical. Fortunately, we are not focused on band gaps or spectroscopical properties in this report, but on high-temperature thermodynamical properties that can be derived from energetics that are less sensitive to unoccupied energy levels associated with band gaps.
In terms of the crystal structure, our DFT-relaxed (predicted) structure compares reasonably well with experiments, particularly in regard to the a and c lattice parameters of the orthorhombic structure. Our calculated b/a and c/a axial ratios are 3.56 and 2.29, respectively. These values agree fairly with experiments (3.24 and 2.31) [2] but indicate a non-negligible overestimation of b in the model. The reason for this overestimation of b is unclear but could be due to thermal softening that our zero-temperature DFT is not accounting for.
The formation energy (or formation enthalpy) can be estimated in our model. From the DFT total energy of UI3, α-uranium, and the isolated iodine atom we calculate the formation enthalpy of the UI3 compound, knowing the enthalpy of formation of the iodine molecule. With our computational technique the molecule is not practical to compute so we adopt the experimental value [36]. From these energies we obtain a zero temperature UI3 formation enthalpy of −320 mRy/at (−100 kcal/mol) in good accord with the experimental numbers −98.8 and −111.6 kcal/mol [7,8] and the room-temperature CALPHAD data (−110 kcal/mol). The experimental formation enthalpies were obtained at 370 °C [7] and room temperature [8], respectively. This level of agreement suggests that the chemical bonding in the DFT model is rather appropriate and we proceed to investigate the single-crystal elastic constants for UI3.
The orthorhombic symmetry of the crystal implies nine independent elastic constants (Cij) that are obtained from the total-energy response to different strains [37]. Our calculated elastic moduli for UI3 are summarized in Table 1. It is illuminating to investigate the mechanical stability criteria to establish stability of UI3 in the model. For an orthorhombic crystal, necessary and sufficient Born criteria for stability have been published [38] and our calculated elastic constants fulfill those (not shown). However, to categorically demonstrate full phase stability one needs to investigate all phonons, and this is beyond the scope of this study. Unfortunately, there are no experimental single-crystal elastic moduli with which we can verify our results. The calculated moduli thus serve as predictions and can be used to constrain semi-empirical models or interatomic potentials.
We mentioned that UI3 is an anti-ferromagnet with substantial magnetic moments on the uranium atoms. Experimentally, there is a rather large spread between the quoted magnetic moments, ranging from 1.97 to 3.65 μB [2,3]. Our calculations adopt a simplified anti-ferromagnetic configuration relative to the observed one and we find spin and orbital moments on the uranium atoms that are 2.7, and −3.3 μB, respectively. The calculated magnitude of the moments is presumably not very sensitive to the actual magnetic configuration (a ferromagnetic treatment results in the same moment magnitudes) and we believe our simplification with anti-ferromagnetic moments along (001) is sensible. There is an experimental uncertainty in the exact magnitudes of the moments, but it does appear that the theory predicts smaller total moments than what is observed.
Based on our aforementioned results, which exhibit reasonable accuracy, let us now focus on the thermodynamical properties at elevated temperatures. Because UI3 has a relatively low melting temperature of about 1039 K [14], we assume that anharmonic effects on the phonons are limited and that a quasi-harmonic scheme for the lattice dynamics may suffice. The most sensitive test for this assumption is to compare our quasi-harmonic Gibbs energy with the CALPHAD database, which results from the assessment of existing experimental data.
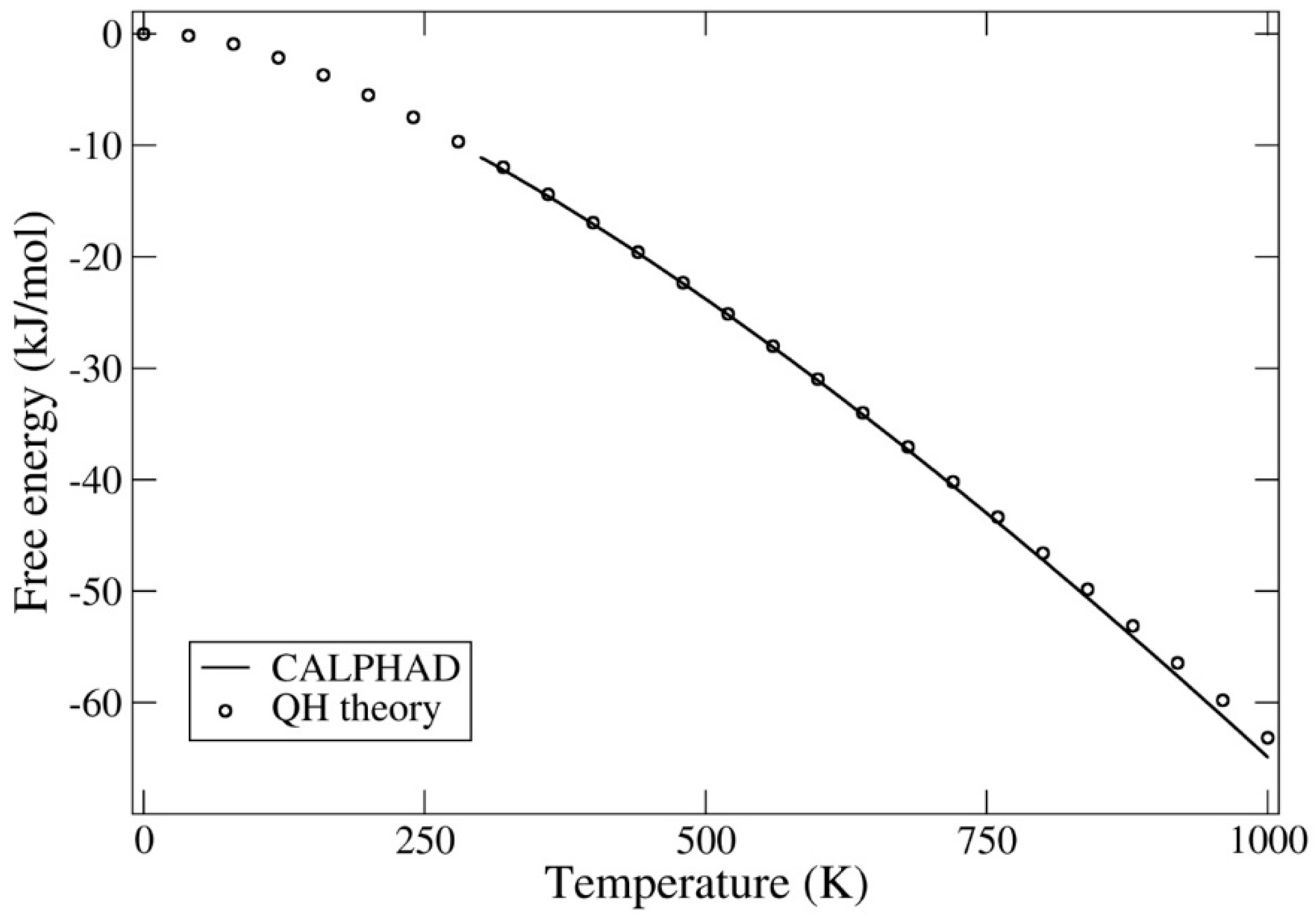
In Figure 2 we contrast our DFT-quasi-harmonic (QH) free energy with that of the CALPHAD (from room temperature and up). Generally, the free energy contains contributions from the electronic structure (here obtained from the DFT calculations), lattice vibrations (Debye–Grüneisen model), electronic excitations, electron–phonon coupling, and finally magnetic entropy. Because we are limiting the model to not too high temperatures, we make the assumption that the latter three contributions can be ignored, considering the overall level of accuracy dictated by the Debye–Grüneisen model. Certainly, that assumption was true for uranium mononitride that we studied recently [15].
Clearly, Figure 2 suggests that the QH result is consistent with CALPHAD for most temperatures. Noticeable deviation between the two approaches is observed only at relatively high temperatures, approaching 1000 K. Apparently, the quasi-harmonic assumption for the lattice vibrations begins to break down at those temperatures.
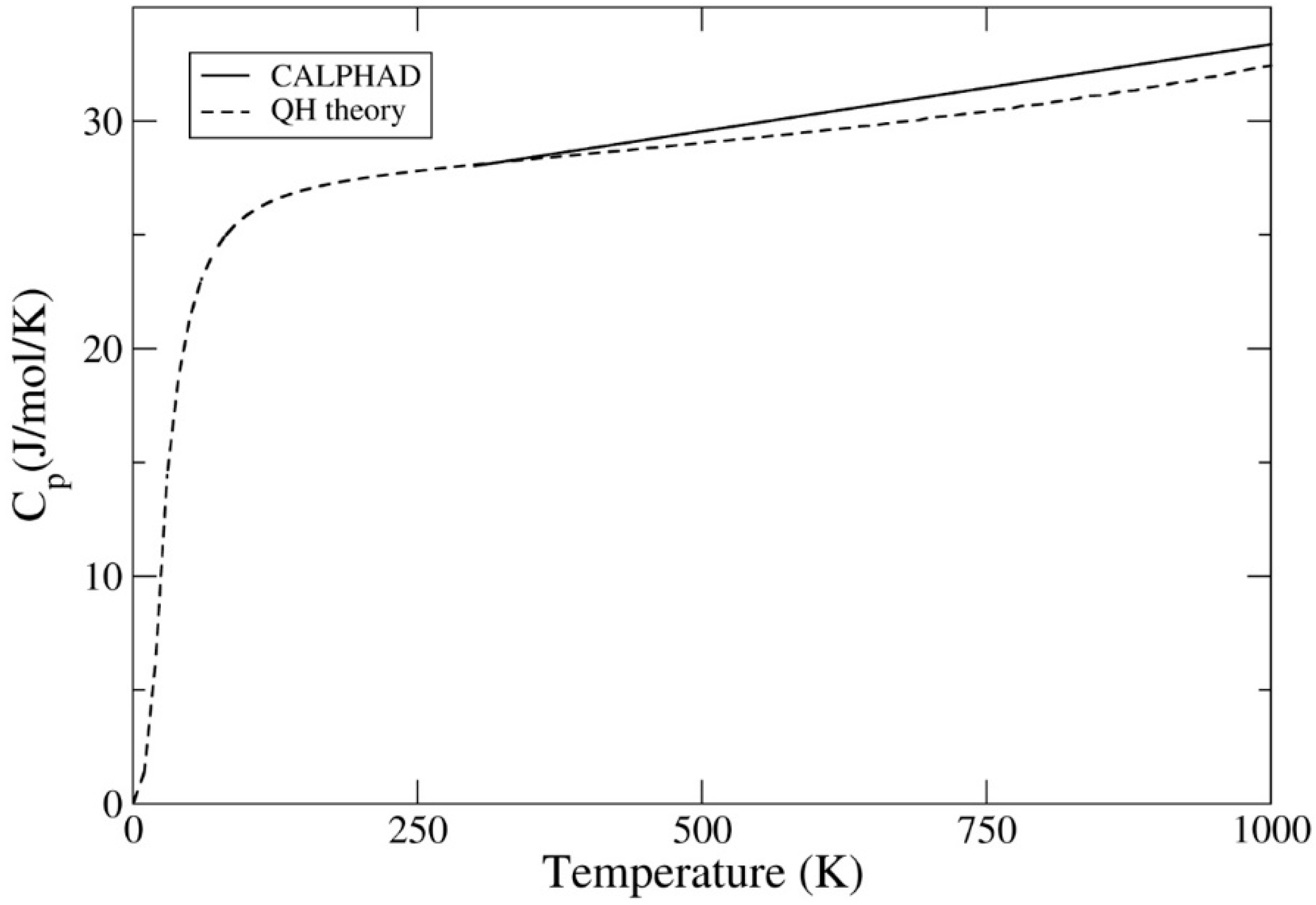
Next, encouraged by the consistency of the QH theory and CALPHAD for the UI3 free energy, we show their respective heat capacity at constant pressure, CP, in Figure 3. As in the case of the free energy, no electron-thermal contribution to the heat capacity is considered in the QH theory. We find in Figure 3 that at lower temperatures the agreement is good, but we notice, consistent with that of the free energies, that CP begins to deviate between the two methods at temperatures approaching 1000 K. We interpret this discrepancy as a need to address anharmonic phonons at temperatures nearing 1000 K or to perform more dedicated experiments on the U-I system to further validate and/or refine the CALPHAD data. In the CALPHAD data, there may also be implicit effects related to formation of defects such as vacancies at high temperatures that are not addressed in the first-principles theory.
4. Summary and Conclusions
We have explored the rare uranium tri-iodide compound from first-principles theory and CALPHAD. In the bulk form, UI3 has not before been investigated from first principles. Tangible quantities that are directly measurable by experiments, such as formation enthalpy, atomic volume, crystal structure, and magnetism, are reasonably well reproduced by our parameter-free DFT approach. The theory furthermore predicts the existence of band gaps in the electronic structure that may suggest a need for methodologies accounting for stronger electron correlation. However, no experiments that we are aware of have measured the band gaps and the state of the electron correlations (e.g., the degree of uranium 5f-electron localization) remains an open question. The band gaps are important for understanding the details of the electronic structure and spectroscopical properties but have limited impact on the energetics and associated quantities that we are primarily focused on here.
Furthermore, we have calculated the nine single-crystal elastic constants for UI3. These are predictions that may prove difficult to validate experimentally because of issues sourcing ample-sized samples of single-crystal UI3. However, the calculated moduli appear realistic and support mechanical stability of the orthorhombic phase. These moduli may also help to constrain future semi-empirical modeling or interatomic potentials for UI3.
High-temperature thermodynamics of UI3 is the principal target of the present investigation and we have leveraged the thermodynamical CALPHAD approach to provide guidance and consistency for these properties. DFT quantum molecular-dynamics simulations would predict the most accurate thermodynamical quantities, but it is computationally too exhaustive for a complex material such as UI3. Instead, we have coupled our DFT to a quasi-harmonic Debye–Grüneisen scheme that is used to more efficiently estimate the thermal properties. Because the melting temperature is relatively low for UI3, the QH treatment turns out to be fairly accurate in our estimation. We have compared the formation enthalpy, Gibbs energy, and specific heat with CALPHAD and the agreement is satisfactory, suggesting good accuracy. Our conclusion, for these properties at least, is that anharmonic phonons, beyond the quasi-harmonic limit, are not significant in UI3.
Author Contributions
Conceptualization, P.S.; methodology, P.S., A.P., E.E.M., A.L.; writing-review and editing, P.S., A.P., E.E.M., A.L., T.W.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partly funded by the Laboratory Directed Research and Development Program at LLNL under Project Tracking Code No. 18-SI-001.
Acknowledgments
This work was performed under the auspices of the U.S. DOE by LLNL under Contract DE-AC52-07NA27344.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Levy, J.H.; Taylor, J.C.; Wilson, P.W. The structure of uranium (III) triiodide by neutron diffraction. Acta Crystallogr. 1975, B31, 880–882. [Google Scholar] [CrossRef] [Green Version]
- Murasik, A.; Fisher, P.; Szczepaniak, W. Neutron diffraction study of long-range antiferromagnetic order and crystal structure of uranium (III) tri-iodide. J. Phys. C Solid State Phys. 1981, 14, 1847–1854. [Google Scholar] [CrossRef]
- Edelstein, N.M.; Lander, G.H. Magnetic properties. In The Chemistry of the Actinide and Transactinide Elements; Morss, L.R., Edelstein, N.M., Fuger, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Rudel, S.S.; Deubner, H.L.; Scheibe, B.; Conrad, M.; Kraus, F. Facile syntheses of pure uranium(III) halides: UF3, UCl3, UBr3, UI3. Z. Anorg. Allg. Chem. 2018, 644, 323–329. [Google Scholar] [CrossRef]
- Evans, W.J.; Kozimor, S.A.; Ziller, J.W.; Fagin, A.A.; Bochkarev, M.N. Facile syntheses of unsolvated UI3 and tetramethylcyclopentadienyl uranium halides. Inorg. Chem. 2005, 44, 3993–4000. [Google Scholar] [CrossRef] [PubMed]
- Idell, Y.S.; Holliday, K.S.; Stillwell, R.L.; Jeffries, J.R. Reduction of uranium triiodide to metal by thermal decomposition. J. Radional. Nucl. Chem. 2019, 320, 793–800. [Google Scholar] [CrossRef]
- Tveekrem, J.O.; Chandraseharaiah, M.S. The standard free energy of formation of UI3 from EMF measurements on a solid electrolyte galvanic cell. J. Electrochem. Soc. 1968, 115, 1021–1023. [Google Scholar] [CrossRef]
- Guillaumont, R.; Fanghanel, T.; Neck, V.; Fuger, J.; Palmer, D.A.; Grenthe, I.; Rand, M.H. Update on the Chemical Thermodynamics of Uranium, Neptunium, Plutonium, Americium, and Technetium; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Li, S.; Wang, Z.; Zhou, M.; Zheng, F.; Shao, X.; Zhang, P. UI3: A high Curie temperature f-electron two-dimensional magnet. J. Phys. D Appl. Phys. 2020, 53, 185301. [Google Scholar] [CrossRef]
- Childs, B.C.; Martin, A.A.; Perron, A.; Moore, E.E.; Idell, Y.; Heo, T.W.; Rosas, D.L.; Schaeffer-Cuellar, C.; Stillwell, R.L.; Söderlind, P.; et al. Formation of high purity uranium via laser induced thermal decomposition of uranium nitride. Mater. Des. 2020, 192, 108706. [Google Scholar] [CrossRef]
- Heo, T.W.; Colas, K.B.; Motta, A.T.; Chen, L.Q. A phase-field model for hydride formation in polycrystalline metals: Application to delta-hydride in zirconium alloys. Acta Mater. 2019, 181, 262–277. [Google Scholar] [CrossRef]
- Perron, A.; Roehling, J.D.; Turchi, P.E.A.; Fattebert, J.-L.; McKeown, J.T. Matching time and spatial scales of rapid solidification: Dynamic TEM experiments coupled to CALPHAD-informed phase-field simulations. Model. Simul. Mater. Sci. Eng. 2018, 26, 014002. [Google Scholar] [CrossRef]
- Roehling, J.D.; Perron, A.; Fattebert, J.-L.; Haxhimali, T.; Guss, G.; Li, T.T.; Bober, D.; Stokes, A.W.; Clarke, A.J.; Turchi, P.E.A.; et al. Rapid solidification in bulk Ti-Nb alloys by single-track laser melting. JOM 2018, 70, 1589–1597. [Google Scholar] [CrossRef]
- Haynes, W.M. CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Söderlind, P.; Landa, A.; Perron, A.; Sadigh, B.; Heo, T.W. Ground-state and thermodynamical properties of uranium mononitride from anharmonic first-principles theory. Appl. Sci. 2019, 9, 3914. [Google Scholar] [CrossRef] [Green Version]
- Söderlind, P.; Eriksson, O.; Johansson, B.; Wills, J.M. Electronic properties of f-electron metals using the generalized gradient approximation. Phys. Rev. B 1994, 50, 7291–7294. [Google Scholar] [CrossRef]
- Sadigh, B.; Kutepov, A.; Landa, A.; Söderlind, P. Assessing relativistic effects and electron correlation in the actinide metals Th to Pu. Appl. Sci. 2019, 9, 5020. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Lejaeghere, K.; Bihlmayer, G.; Björkman, T.; Blaha, P.; Blügel, S.; Blum, V.; Caliste, D.; Castelli, I.E.; Clark, S.J.; Dal Corso, A.; et al. Reproducibility in density functional theory calculations for solids. Science 2016, 351, aad3000. [Google Scholar] [CrossRef] [Green Version]
- Wills, J.M.; Alouani, M.; Andersson, P.; Delin, A.; Eriksson, O.; Grechnyev, O. Full-Potential Electronic Structure Method; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Eriksson, O.; Brooks, M.S.S.; Johansson, B. Orbital polarization in narrow-band systems: Application to volume collapses in light lanthanides. Phys. Rev. B 1990, 41, 7311–7314. [Google Scholar] [CrossRef]
- Söderlind, P.; Landa, A.; Sadigh, B. Density-functional theory for plutonium. Adv. Phys. 2019, 68, 1–47. [Google Scholar] [CrossRef]
- Söderlind, P.; Grabowski, B.; Yang, L.; Landa, A.; Björkman, T.; Souvatzis, P.; Eriksson, O. High-temperature phonon stabilization of γ-uranium from relativistic first-principles theory. Phys. Rev. B 2012, 85, 060301. [Google Scholar] [CrossRef] [Green Version]
- Moruzzi, V.L.; Janak, J.F.; Schwarz, K. Calculated thermal properties of solids. Phys. Rev. B 1988, 37, 790–799. [Google Scholar] [CrossRef]
- Söderlind, P.; Nordström, L.; Yongming, L.; Johansson, B. Relativistic effects on the thermal expansion of the actinide elements. Phys. Rev. B 1990, 42, 4544–4552. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, L.; Bernstein, H. Computer Calculation of Phase Diagrams with Special Reference to Refractory Metals; Academic Press: Cambridge, MA, USA, 1970. [Google Scholar]
- Saunders, N.; Miodownik, A. CALPHAD Calculation of Phase Diagrams: A Comprehensive Guide; Elsevier Science: Amsterdam, The Netherlands, 1998. [Google Scholar]
- Lukas, H.; Fries, S.; Sundman, B. Computational thermodynamics: The CALPHAD method; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Bajaj, S.; Landa, A.; Söderlind, P.; Turchi, P.E.A.; Arroyave, R. The U-Ti system: Strengths and weaknesses of the CALPHAD method. J. Nucl. Mater. 2011, 419, 177–185. [Google Scholar] [CrossRef]
- Perron, A.; Turchi, P.E.A.; Landa, A.; Söderlind, P.; Ravat, B.; Oudot, B.; Delaunay, F.; Kurata, M. Thermodynamic re-assessment of the Pu-U system and its application to the ternary Pu-U-Ga system. J. Nucl. Mater. 2014, 454, 81–95. [Google Scholar] [CrossRef] [Green Version]
- Perron, A.; Turchi, P.E.A.; Landa, A.; Söderlind, P.; Ravat, B.; Oudot, B.; Delaunay, F. The Pu-U-Am system: An ab initio informed CALPHAD thermodynamic study. J. Nucl. Mater. 2015, 458, 425–441. [Google Scholar] [CrossRef] [Green Version]
- Moore, E.E.; Turchi, P.E.A.; Landa, A.; Söderlind, P.; Oudot, B.; Belof, J.L.; Stout, S.A.; Perron, A. Development of a CALPHAD thermodynamic database for Pu-U-Fe-Ga alloys. Appl. Sci. 2019, 9, 5040. [Google Scholar] [CrossRef] [Green Version]
- Sundman, B.; Jansson, B.; Andersson, J.-O. The thermo-calk databank system. Calphad 1985, 9, 153–190. [Google Scholar] [CrossRef]
- Andersson, J.-O.; Helander, T.; Höglund, L.; Shi, P.; Sundman, B. Thermo-Calc and DICTRA, computational tools for material science. Calphad 2002, 26, 273–312. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Nguyen Mahn, D.; Sutton, A.P. Effect of Mott-Hubbard correlations on the electronic structure and structural stability of uranium dioxide. Philos. Mag. B 1997, 75, 613–628. [Google Scholar] [CrossRef]
- Bedjanian, Y.; Le Bras, G.; Poulet, G. Kinetic study of the Br+IO, I+BrO, and Br+I2 reactions. Chem. Phys. Lett. 1997, 266, 233–238. [Google Scholar] [CrossRef]
- Söderlind, P. First-principles elastic and structural properties of uranium metal. Phys. Rev. B 2002, 66, 085133. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Calculated total unit-cell electronic density of states (states/eV) for UI3. The energy scale on the x-axis is shifted so that the highest occupied level is located at zero energy.
Figure 1.
Calculated total unit-cell electronic density of states (states/eV) for UI3. The energy scale on the x-axis is shifted so that the highest occupied level is located at zero energy.

Figure 2.
Gibbs free energies (kJ/mol) calculated from density-functional theory (DFT) and Debye–Grüneisen quasi-harmonic (QH) theory together with results from the SSUB6 calculation of phase diagram (CALPHAD) database.
Figure 2.
Gibbs free energies (kJ/mol) calculated from density-functional theory (DFT) and Debye–Grüneisen quasi-harmonic (QH) theory together with results from the SSUB6 calculation of phase diagram (CALPHAD) database.

Figure 3.
UI3 specific heat at constant pressure, Cp (J/mol/K), calculated from our DFT-quasi-harmonic (QH) model and from the SSUB6 CALPHAD database.
Figure 3.
UI3 specific heat at constant pressure, Cp (J/mol/K), calculated from our DFT-quasi-harmonic (QH) model and from the SSUB6 CALPHAD database.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated elastic constants Cij for UI3 (GPa).
| C11 | C22 | C33 | C44 | C55 | C66 | C12 | C13 | C23 |
|---|---|---|---|---|---|---|---|---|
| 56.5 | 45.6 | 62.2 | 10.3 | 10.5 | 19.5 | 31.2 | 26.8 | 19.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Söderlind, P.; Perron, A.; Moore, E.E.; Landa, A.; Heo, T.W. Thermodynamics of Uranium Tri-Iodide from Density-Functional Theory. Appl. Sci. 2020, 10, 3914. https://doi.org/10.3390/app10113914
AMA Style
Söderlind P, Perron A, Moore EE, Landa A, Heo TW. Thermodynamics of Uranium Tri-Iodide from Density-Functional Theory. Applied Sciences. 2020; 10(11):3914. https://doi.org/10.3390/app10113914
Chicago/Turabian StyleSöderlind, Per, Aurélien Perron, Emily E. Moore, Alexander Landa, and Tae Wook Heo. 2020. "Thermodynamics of Uranium Tri-Iodide from Density-Functional Theory" Applied Sciences 10, no. 11: 3914. https://doi.org/10.3390/app10113914
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.