Whole Genome Sequencing Analysis of Porcine Faecal Commensal Escherichia coli Carrying Class 1 Integrons from Sows and Their Offspring

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Field Animal Trial
2.2. Faecal Sample Collection, E. coli Isolation and Identification
2.3. Isolate Identifiers
2.4. DNA Extraction and Whole Genome Sequencing
2.5. Assembly Statistics
2.6. Gene Identification, Serotyping, Phylogrouping, Phylogenetic Analysis and Multilocus Sequence Typing (MLST)
2.7. Ethical Statement
2.8. Data Availability
- One hundred and seventeen short read pairs and corresponding draft genome assemblies of E. coli as described in this project were deposited in GenBank under BioProject PRJNA509690. Individual sample accession numbers can be found in Table S1. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA509690
- Maximum likelihood phylogenetic tree of 185 faecal E. coli whole genome sequences from Australian pigs. https://doi.org/10.6084/m9.figshare.12233744
- Recombination filtered alignment of 185 faecal E. coli whole genome sequences from Australian pigs. Aligned to K12-MG1655 complete genome with Snippy and recombination filtered with Gubbins. https://doi.org/10.6084/m9.figshare.12233747
3. Results
3.1. Class 1 Integrase Gene Presence
3.2. Phylogroups, Sequence Types and Serotypes
3.3. Phylogenetic Analysis
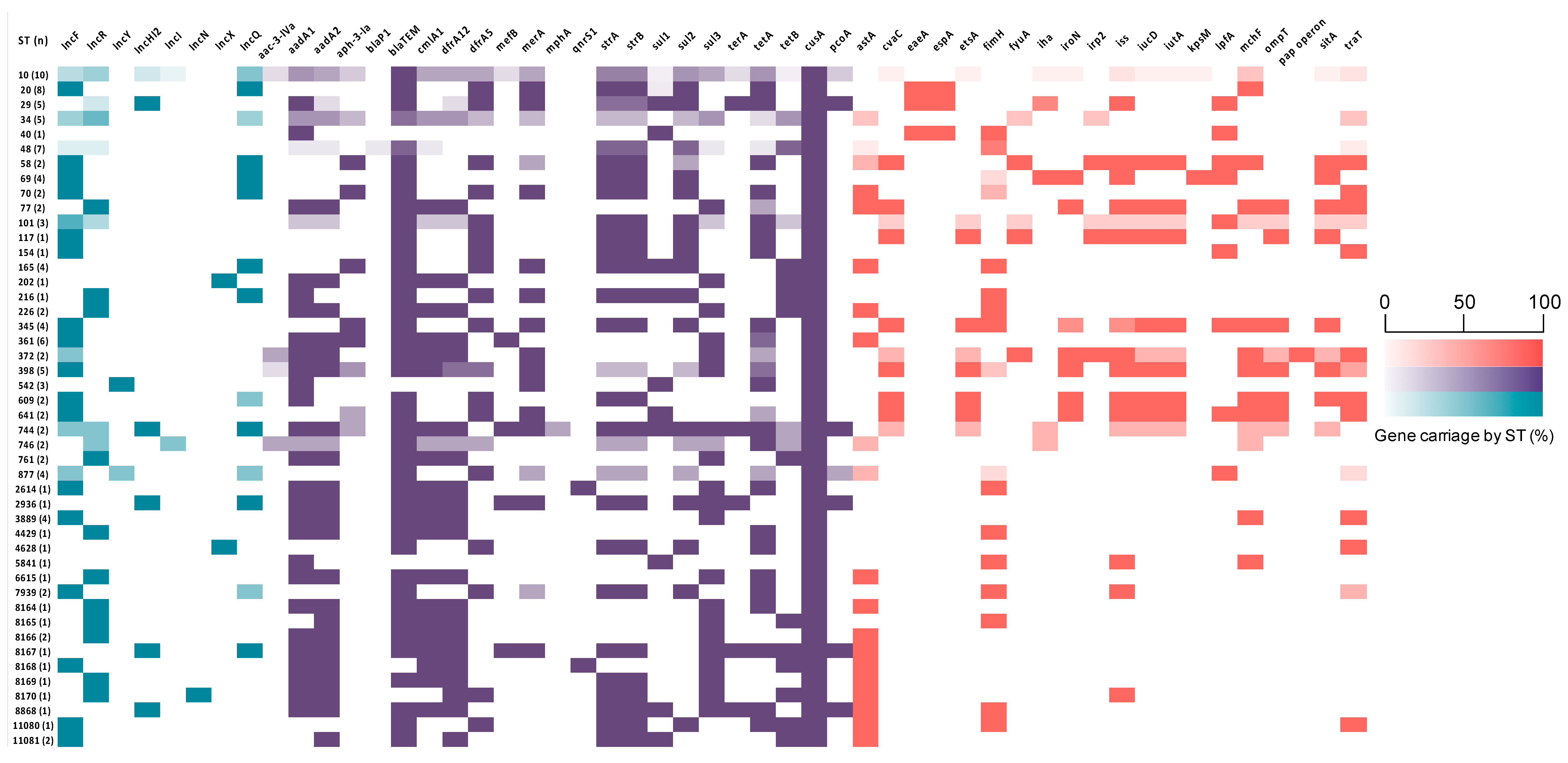
3.4. Antimicrobial Resistance Genes (ARGs)
3.5. Class 1 Integron Structures
3.6. Virulence Associated Genes (VAGs)
3.7. Plasmid Incompatibility Groups
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stokes, H.W.; Gillings, M.R. Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiol. Rev. 2011, 35, 790–819. [Google Scholar]
- Armstrong, T.A.; Cook, D.R.; Ward, M.M.; Williams, C.M.; Spears, J.W. Effect of dietary copper source (cupric citrate and cupric sulfate) and concentration on growth performance and fecal copper excretion in weanling pigs. J. Anim. Sci. 2004, 82, 1234–1240. [Google Scholar]
- Jacela, J.Y.; DeRouchey, J.M.; Tokach, M.D.; Goodband, R.D.; Nelssen, J.L.; Renter, D.G.; Dritz, S.S. Feed additives for swine: Fact sheets – high dietary levels of copper and zinc for young pigs, and phytase. J. Swine Health Prod. 2010. [Google Scholar]
- Thacker, P.A. Alternatives to antibiotics as growth promoters for use in swine production: A review. J. Anim. Sci. Biotechnol. 2013, 4, 35. [Google Scholar]
- Case, C.L.; Carlson, M.S. Effect of feeding organic and inorganic sources of additional zinc on growth performance and zinc balance in nursery pigs. J. Anim. Sci. 2002, 80, 1917–1924. [Google Scholar]
- Van Boeckel, T.P.; Brower, C.; Gilbert, M.; Grenfell, B.T.; Levin, S.A.; Robinson, T.P.; Teillant, A.; Laxminarayan, R. Global trends in antimicrobial use in food animals. Proc. Natl. Acad. Sci. USA 2015, 112, 5649–5654. [Google Scholar]
- National Research Council. National Research Council Nutrient requirements of Swine: Eleventh Revised Edition; National Academies Press: Washington, DC, USA, 2012. [Google Scholar]
- Herrero-Fresno, A.; Larsen, I.; Olsen, J.E. Genetic relatedness of commensal Escherichia coli from nursery pigs in intensive pig production in Denmark and molecular characterization of genetically different strains. J. Appl. Microbiol. 2015, 119, 342–353. [Google Scholar]
- Binh, C.T.T.; Heuer, H.; Kaupenjohann, M.; Smalla, K. Piggery manure used for soil fertilization is a reservoir for transferable antibiotic resistance plasmids. FEMS Microbiol. Ecol. 2008, 66, 25–37. [Google Scholar]
- Binh, C.T.T.; Heuer, H.; Gomes, N.C.M.; Kotzerke, A.; Fulle, M.; Wilke, B.-M.; Schloter, M.; Smalla, K. Short-term effects of amoxicillin on bacterial communities in manured soil. FEMS Microbiol. Ecol. 2007, 62, 290–302. [Google Scholar]
- Peng, S.; Wang, Y.; Zhou, B.; Lin, X. Long-term application of fresh and composted manure increase tetracycline resistance in the arable soil of eastern China. Sci. Total Environ. 2015, 506–507, 279–286. [Google Scholar]
- Topp, E.; Larsson, D.G.J.; Miller, D.N.; Van den Eede, C.; Virta, M.P.J. Antimicrobial resistance and the environment: Assessment of advances, gaps and recommendations for agriculture, aquaculture and pharmaceutical manufacturing. FEMS Microbiol. Ecol. 2018, 94, fix185. [Google Scholar]
- Gillings, M.R. Class 1 integrons as invasive species. Curr. Opin. Microbiol. 2017, 38, 10–15. [Google Scholar]
- Jechalke, S.; Kopmann, C.; Rosendahl, I.; Groeneweg, J.; Weichelt, V.; Krögerrecklenfort, E.; Brandes, N.; Nordwig, M.; Ding, G.-C.; Siemens, J.; et al. Increased abundance and transferability of resistance genes after field application of manure from sulfadiazine-treated pigs. Appl. Environ. Microbiol. 2013, 79, 1704–1711. [Google Scholar]
- Gillings, M.R. Integrons: Past, present, and future. Microbiol. Mol. Biol. Rev. 2014, 78, 257–277. [Google Scholar]
- Partridge, S.R. Analysis of antibiotic resistance regions in Gram-negative bacteria. FEMS Microbiol. Rev. 2011, 35, 820–855. [Google Scholar]
- Gillings, M.R. DNA as a pollutant: The clinical class 1 integron. Curr. Pollut. Rep. 2018, 4, 49–55. [Google Scholar]
- Zhu, Y.-G.; Zhao, Y.; Li, B.; Huang, C.-L.; Zhang, S.-Y.; Yu, S.; Chen, Y.-S.; Zhang, T.; Gillings, M.R.; Su, J.-Q. Continental-scale pollution of estuaries with antibiotic resistance genes. Nat. Microbiol. 2017, 2, 16270. [Google Scholar]
- Zhu, Y.-G.; Johnson, T.A.; Su, J.-Q.; Qiao, M.; Guo, G.-X.; Stedtfeld, R.D.; Hashsham, S.A.; Tiedje, J.M. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc. Natl. Acad. Sci. USA 2013, 110, 3435–3440. [Google Scholar]
- Chen, C.-M.; Yu, W.-L.; Huang, M.; Liu, J.-J.; Chen, I.-C.; Chen, H.-F.; Wu, L.-T. Characterization of IS26-composite transposons and multidrug resistance in conjugative plasmids from Enterobacter cloacae. Microbiol. Immunol. 2015, 59, 516–525. [Google Scholar]
- Dolejska, M.; Papagiannitsis, C.C.; Medvecky, M.; Davidova-Gerzova, L.; Valcek, A. Characterization of the Complete Nucleotide Sequences of IMP-4-Encoding Plasmids, Belonging to Diverse Inc Families, Recovered from Enterobacteriaceae Isolates of Wildlife Origin. Antimicrob. Agents Chemother. 2018, 62, e02434-17. [Google Scholar]
- Mangat, C.S.; Bekal, S.; Irwin, R.J.; Mulvey, M.R. A Novel Hybrid Plasmid Carrying Multiple Antimicrobial Resistance and Virulence Genes in Salmonella enterica Serovar Dublin. Antimicrob. Agents Chemother. 2017, 61, e02601-16. [Google Scholar]
- Tseng, S.-P.; Wang, S.-F.; Ma, L.; Wang, T.-Y.; Yang, T.-Y.; Siu, L.K.; Chuang, Y.-C.; Lee, P.-S.; Wang, J.-T.; Wu, T.-L.; et al. The plasmid-mediated fosfomycin resistance determinants and synergy of fosfomycin and meropenem in carbapenem-resistant Klebsiella pneumoniae isolates in Taiwan. J. Microbiol. Immunol. Infect. 2017, 50, 653–661. [Google Scholar]
- Venturini, C.; Hassan, K.A.; Roy Chowdhury, P.; Paulsen, I.T.; Walker, M.J.; Djordjevic, S.P. Sequences of two related multiple antibiotic resistance virulence plasmids sharing a unique IS26-related molecular signature isolated from different Escherichia coli pathotypes from different hosts. PLoS ONE 2013, 8, e78862. [Google Scholar]
- Reid, C.J.; Wyrsch, E.R.; Roy Chowdhury, P.; Zingali, T.; Liu, M.; Darling, A.E.; Chapman, T.A.; Djordjevic, S.P. Porcine commensal Escherichia coli: A reservoir for class 1 integrons associated with IS26. Microb. Genom. 2017, 3. [Google Scholar]
- Reid, C.J.; Roy Chowdhury, P.; Djordjevic, S.P. Tn6026 and Tn6029 are found in complex resistance regions mobilised by diverse plasmids and chromosomal islands in multiple antibiotic resistant Enterobacteriaceae. Plasmid 2015, 80, 127–137. [Google Scholar]
- Partridge, S.R.; Zong, Z.; Iredell, J.R. Recombination in IS26 and Tn2 in the evolution of multiresistance regions carrying blaCTX-M-15 on conjugative IncF plasmids from Escherichia coli. Antimicrob. Agents Chemother. 2011, 55, 4971–4978. [Google Scholar]
- Liu, B.-T.; Song, F.-J.; Zou, M.; Hao, Z.-H.; Shan, H. Emergence of Colistin Resistance Gene mcr-1 in Cronobacter sakazakii Producing NDM-9 and in Escherichia coli from the Same Animal. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar]
- Wang, Z.; Fu, Y.; Du, X.-D.; Jiang, H.; Wang, Y. Potential transferability of mcr-3 via IS26-mediated homologous recombination in Escherichia coli. Emerg. Microbes Infect. 2018, 7, 55. [Google Scholar]
- Cain, A.K.; Liu, X.; Djordjevic, S.P.; Hall, R.M. Transposons related to Tn1696 in IncHI2 plasmids in multiply antibiotic resistant Salmonella enterica serovar Typhimurium from Australian animals. Microb. Drug Resist. 2010, 16, 197–202. [Google Scholar]
- Baquero, F.; Martínez, J.-L.; Cantón, R. Antibiotics and antibiotic resistance in water environments. Curr. Opin. Biotechnol. 2008, 19, 260–265. [Google Scholar]
- Martinez, J.L. Environmental pollution by antibiotics and by antibiotic resistance determinants. Environ. Pollut. 2009, 157, 2893–2902. [Google Scholar]
- Birkegård, A.C.; Halasa, T.; Folkesson, A.; Clasen, J.; Græsbøll, K.; Toft, N. Persistence of antimicrobial resistance genes from sows to finisher pigs. Prev. Vet. Med. 2018, 149, 10–14. [Google Scholar]
- Callens, B.; Faes, C.; Maes, D.; Catry, B.; Boyen, F.; Francoys, D.; de Jong, E.; Haesebrouck, F.; Dewulf, J. Presence of antimicrobial resistance and antimicrobial use in sows are risk factors for antimicrobial resistance in their offspring. Microb. Drug Resist. 2015, 21, 50–58. [Google Scholar]
- Mathew, A.G.; Garner, K.N.; Ebner, P.D.; Saxton, A.M.; Clift, R.E.; Liamthong, S. Effects of antibiotic use in sows on resistance of E. coli and Salmonella enterica Typhimurium in their offspring. Foodborne Pathog. Dis. 2005, 2, 212–220. [Google Scholar]
- Hammerum, A.M.; Larsen, J.; Andersen, V.D.; Lester, C.H.; Skovgaard Skytte, T.S.; Hansen, F.; Olsen, S.S.; Mordhorst, H.; Skov, R.L.; Aarestrup, F.M.; et al. Characterization of extended-spectrum β-lactamase (ESBL)-producing Escherichia coli obtained from Danish pigs, pig farmers and their families from farms with high or no consumption of third- or fourth-generation cephalosporins. J. Antimicrob. Chemother. 2014, 69, 2650–2657. [Google Scholar]
- Chen, J.; Griffiths, M.W. PCR differentiation of Escherichia coli from other Gram-negative bacteria using primers derived from the nucleotide sequences flanking the gene encoding the universal stress protein. Lett. Appl. Microbiol. 1998, 27, 369–371. [Google Scholar]
- Sallen, B.; Rajoharison, A.; Desvarenne, S.; Mabilat, C. Molecular epidemiology of integron-associated antibiotic resistance genes in clinical isolates of Enterobacteriaceae. Microb. Drug Resist. 1995, 1, 195–202. [Google Scholar]
- Maguire, A.J.; Brown, D.F.; Gray, J.J.; Desselberger, U. Rapid screening technique for class 1 integrons in Enterobacteriaceae and nonfermenting gram-negative bacteria and its use in molecular epidemiology. Antimicrob. Agents Chemother. 2001, 45, 1022–1029. [Google Scholar]
- Gillings, M.R.; Gaze, W.H.; Pruden, A.; Smalla, K.; Tiedje, J.M.; Zhu, Y.-G. Using the class 1 integron-integrase gene as a proxy for anthropogenic pollution. ISME J. 2015, 9, 1269–1279. [Google Scholar]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar]
- Clermont, O.; Bonacorsi, S.; Bingen, E. Rapid and simple determination of theEscherichia coli phylogenetic group. Appl. Environ. Microbiol. 2000, 66, 4555–4558. [Google Scholar]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar]
- Mobley, H.L.; Chippendale, G.R.; Tenney, J.H.; Hull, R.A.; Warren, J.W. Expression of type 1 fimbriae may be required for persistence of Escherichia coli in the catheterized urinary tract. J. Clin. Microbiol. 1987, 25, 2253–2257. [Google Scholar]
- Langermann, S.; Palaszynski, S.; Barnhart, M.; Auguste, G.; Pinkner, J.S.; Burlein, J.; Barren, P.; Koenig, S.; Leath, S.; Jones, C.H.; et al. Prevention of mucosal Escherichia coli infection by FimH-adhesin-based systemic vaccination. Science 1997, 276, 607–611. [Google Scholar]
- Nolan, L.K.; Horne, S.M.; Giddings, C.W.; Foley, S.L.; Johnson, T.J.; Lynne, A.M.; Skyberg, J. Resistance to serum complement, iss, and virulence of avian Escherichia coli. Vet. Res. Commun. 2003, 27, 101–110. [Google Scholar]
- Jerse, A.E.; Yu, J.; Tall, B.D.; Kaper, J.B. A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7839–7843. [Google Scholar]
- Neves, B.C.; Knutton, S.; Trabulsi, L.R.; Sperandio, V.; Kaper, J.B.; Dougan, G.; Frankel, G. Molecular and ultrastructural characterisation of EspA from different enteropathogenicEscherichia coli serotypes. FEMS Microbiol. Lett. 1998, 169, 73–80. [Google Scholar]
- Johnson, J.R.; Russo, T.A. Molecular epidemiology of extraintestinal pathogenic (uropathogenic) Escherichia coli. Int. J. Med. Microbiol. 2005, 295, 383–404. [Google Scholar]
- Micenková, L.; Štaudová, B.; Bosák, J.; Mikalová, L.; Littnerová, S.; Vrba, M.; Ševčíková, A.; Woznicová, V.; Šmajs, D. Bacteriocin-encoding genes and ExPEC virulence determinants are associated in human fecal Escherichia coli strains. BMC Microbiol. 2014, 14, 109. [Google Scholar]
- Kidsley, A.K.; Abraham, S.; Bell, J.M.; O’Dea, M.; Laird, T.J.; Jordan, D.; Mitchell, P.; McDevitt, C.A.; Trott, D.J. Antimicrobial Susceptibility of Escherichia coli and Salmonella spp. Isolates From Healthy Pigs in Australia: Results of a Pilot National Survey. Front. Microbiol. 2018, 9, 1207. [Google Scholar]
- Gerhold, G.; Schulze, M.H.; Gross, U.; Bohne, W. Multilocus sequence typing and CTX-M characterization of ESBL-producing E. coli: A prospective single-centre study in Lower Saxony, Germany. Epidemiol. Infect. 2016, 144, 3300–3304. [Google Scholar]
- Ding, Y.; Tang, X.; Lu, P.; Wu, B.; Xu, Z.; Liu, W.; Zhang, R.; Bei, W.; Chen, H.; Tan, C. Clonal analysis and virulent traits of pathogenic extraintestinal Escherichia coli isolates from swine in China. BMC Vet. Res. 2012, 8, 140. [Google Scholar]
- Reid, C.J.; DeMaere, M.Z.; Djordjevic, S.P. Australian porcine clonal complex 10 (CC10) Escherichia coli belong to multiple sublineages of a highly diverse global CC10 phylogeny. Microb. Genom. 2018, 5. [Google Scholar]
- Spencer, B.T.; Howell, P.G. Some husbandry factors influencing weaning stresses in piglets. J. S. Afr. Vet. Assoc. 1989, 60, 62–64. [Google Scholar]
- Chen, L.; Xu, Y.; Chen, X.; Fang, C.; Zhao, L.; Chen, F. The Maturing Development of Gut Microbiota in Commercial Piglets during the Weaning Transition. Front. Microbiol. 2017, 8, 1688. [Google Scholar]
- Herrero-Fresno, A.; Ahmed, S.; Hansen, M.H.; Denwood, M.; Zachariasen, C.; Olsen, J.E. Genotype variation and genetic relationship among Escherichia coli from nursery pigs located in different pens in the same farm. BMC Microbiol. 2017, 17, 5. [Google Scholar]
- Ahmed, S.; Olsen, J.E.; Herrero-Fresno, A. The genetic diversity of commensal Escherichia coli strains isolated from non-antimicrobial treated pigs varies according to age group. PLoS ONE 2017, 12, e0178623. [Google Scholar]
- Jensen, A.N.; Hansen, L.L.; Baggesen, D.L.; Mølbak, L. Effects of feeding finisher pigs with chicory or lupine feed for one week or two weeks before slaughter with respect to levels of Bifidobacteria and Campylobacter. Animal 2013, 7, 66–74. [Google Scholar]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar]
- Dawes, F.E.; Kuzevski, A.; Bettelheim, K.A.; Hornitzky, M.A.; Djordjevic, S.P.; Walker, M.J. Distribution of class 1 integrons with IS26-mediated deletions in their 3′-conserved segments in Escherichia coli of human and animal origin. PLoS ONE 2010, 5, e12754. [Google Scholar]
- McKinnon, J.; Roy Chowdhury, P.; Djordjevic, S.P. Genomic analysis of multidrug-resistant Escherichia coli ST58 causing urosepsis. Int. J. Antimicrob. Agents 2018, 52, 430–435. [Google Scholar]
- Perreten, V.; Boerlin, P. A new sulfonamide resistance gene (sul3) in Escherichia coli is widespread in the pig population of Switzerland. Antimicrob. Agents Chemother. 2003, 47, 1169–1172. [Google Scholar]
- Billman-Jacobe, H.; Liu, Y.; Haites, R.; Weaver, T.; Robinson, L.; Marenda, M.; Dyall-Smith, M. pSTM6-275, a Conjugative IncHI2 Plasmid of Salmonella enterica That Confers Antibiotic and Heavy-Metal Resistance under Changing Physiological Conditions. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar]
- Moran, R.A.; Holt, K.E.; Hall, R.M. pCERC3 from a commensal ST95 Escherichia coli: A ColV virulence-multiresistance plasmid carrying a sul3-associated class 1 integron. Plasmid 2016, 84–85, 11–19. [Google Scholar]
- Gündoğdu, A.; Long, Y.B.; Vollmerhausen, T.L.; Katouli, M. Antimicrobial resistance and distribution of sul genes and integron-associated intI genes among uropathogenic Escherichia coli in Queensland, Australia. J. Med. Microbiol. 2011, 60, 1633–1642. [Google Scholar]
- Cummins, M.L.; Reid, C.J.; Roy Chowdhury, P.; Bushell, R.N.; Esbert, N.; Tivendale, K.A.; Noormohammadi, A.H.; Islam, S.; Marenda, M.S.; Browning, G.F.; et al. Whole genome sequence analysis of Australian avian pathogenic Escherichia coli that carry the class 1 integrase gene. Microb. Genom. 2019, 5. [Google Scholar]
- Djordjevic, S.P.; Stokes, H.W.; Roy Chowdhury, P. Mobile elements, zoonotic pathogens and commensal bacteria: Conduits for the delivery of resistance genes into humans, production animals and soil microbiota. Front. Microbiol. 2013, 4, 86. [Google Scholar]
- Fang, L.; Li, X.; Li, L.; Li, S.; Liao, X.; Sun, J.; Liu, Y. Co-spread of metal and antibiotic resistance within ST3-IncHI2 plasmids from E. coli isolates of food-producing animals. Sci. Rep. 2016, 6, 25312. [Google Scholar]
- Reid, C.J.; McKinnon, J.; Djordjevic, S.P. Clonal ST131-H22 Escherichia coli strains from a healthy pig and a human urinary tract infection carry highly similar resistance and virulence plasmids. Microb. Genom. 2019, 5, e000295. [Google Scholar]
- Lillehoj, H.; Liu, Y.; Calsamiglia, S.; Fernandez-Miyakawa, M.E.; Chi, F.; Cravens, R.L.; Oh, S.; Gay, C.G. Phytochemicals as antibiotic alternatives to promote growth and enhance host health. Vet. Res. 2018, 49, 76. [Google Scholar]
- Johnson, J.R.; Stell, A.L. Extended virulence genotypes of Escherichia coli strains from patients with urosepsis in relation to phylogeny and host compromise. J. Infect. Dis. 2000, 181, 261–272. [Google Scholar]
- Baranzoni, G.M.; Fratamico, P.M.; Gangiredla, J.; Patel, I.; Bagi, L.K.; Delannoy, S.; Fach, P.; Boccia, F.; Anastasio, A.; Pepe, T. Characterization of Shiga Toxin Subtypes and Virulence Genes in Porcine Shiga Toxin-Producing Escherichia coli. Front. Microbiol. 2016, 7, 574. [Google Scholar]
- Jarocki, V.M.; Reid, C.J.; Chapman, T.A.; Djordjevic, S.P. Escherichia coli ST302: Genomic Analysis of Virulence Potential and Antimicrobial Resistance Mediated by Mobile Genetic Elements. Front. Microbiol. 2019, 10, 3098. [Google Scholar]
- Wyrsch, E.R.; Reid, C.J.; DeMaere, M.Z.; Liu, M.Y.; Chapman, T.A.; Roy Chowdhury, P.; Djordjevic, S.P. Complete Sequences of Multiple-Drug Resistant IncHI2 ST3 Plasmids in Escherichia coli of Porcine Origin in Australia. Front. Sustain. Food Syst. 2019, 3, 18. [Google Scholar]
- Wyrsch, E.; Roy Chowdhury, P.; Abraham, S.; Santos, J.; Darling, A.E.; Charles, I.G.; Chapman, T.A.; Djordjevic, S.P. Comparative genomic analysis of a multiple antimicrobial resistant enterotoxigenic E. coli O157 lineage from Australian pigs. BMC Genomics 2015, 16, 1382. [Google Scholar]
- Brilhante, M.; Perreten, V.; Donà, V. Multidrug resistance and multivirulence plasmids in enterotoxigenic and hybrid Shiga toxin-producing/enterotoxigenic Escherichia coli isolated from diarrheic pigs in Switzerland. Vet. J. 2019, 244, 60–68. [Google Scholar]
- García, V.; García, P.; Rodríguez, I.; Rodicio, R.; Rodicio, M.R. The role of IS26 in evolution of a derivative of the virulence plasmid of Salmonella enterica serovar Enteritidis which confers multiple drug resistance. Infect. Genet. Evol. 2016, 45, 246–249. [Google Scholar]
- Smith, M.G.; Jordan, D.; Chapman, T.A.; Chin, J.J.C.; Barton, M.D.; Do, T.N.; Fahy, V.A.; Fairbrother, J.M.; Trott, D.J. Antimicrobial resistance and virulence gene profiles in multi-drug resistant enterotoxigenic Escherichia coli isolated from pigs with post-weaning diarrhoea. Vet. Microbiol. 2010, 145, 299–307. [Google Scholar]
- Abraham, S.; Jordan, D.; Wong, H.S.; Johnson, J.R.; Toleman, M.A.; Wakeham, D.L.; Gordon, D.M.; Turnidge, J.D.; Mollinger, J.L.; Gibson, J.S.; et al. First detection of extended-spectrum cephalosporin- and fluoroquinolone-resistant Escherichia coli in Australian food-producing animals. J. Glob. Antimicrob. Resist. 2015, 3, 273–277. [Google Scholar]
- Wong, M.H.-Y.; Chan, E.W.-C.; Chen, S. IS26-mediated formation of a virulence and resistance plasmid in Salmonella Enteritidis. J. Antimicrob. Chemother. 2017, 72, 2750–2754. [Google Scholar]
- Australian Pork Limited Submission to Foreign Policy White Paper—Australian Pork Limited. Available online: https://australianpork.com.au/wp-content/uploads/2013/11/170228_-APL-Submission-to-Foreign-Policy-White-Paper.pdf (accessed on 3 August 2019).



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zingali, T.; Reid, C.J.; Chapman, T.A.; Gaio, D.; Liu, M.; Darling, A.E.; Djordjevic, S.P. Whole Genome Sequencing Analysis of Porcine Faecal Commensal Escherichia coli Carrying Class 1 Integrons from Sows and Their Offspring. Microorganisms 2020, 8, 843. https://doi.org/10.3390/microorganisms8060843
Zingali T, Reid CJ, Chapman TA, Gaio D, Liu M, Darling AE, Djordjevic SP. Whole Genome Sequencing Analysis of Porcine Faecal Commensal Escherichia coli Carrying Class 1 Integrons from Sows and Their Offspring. Microorganisms. 2020; 8(6):843. https://doi.org/10.3390/microorganisms8060843
Chicago/Turabian StyleZingali, Tiziana, Cameron J. Reid, Toni A. Chapman, Daniela Gaio, Michael Liu, Aaron E. Darling, and Steven P. Djordjevic. 2020. "Whole Genome Sequencing Analysis of Porcine Faecal Commensal Escherichia coli Carrying Class 1 Integrons from Sows and Their Offspring" Microorganisms 8, no. 6: 843. https://doi.org/10.3390/microorganisms8060843