The Epidemiological Characteristics of the Korean Bat Paramyxovirus between 2016 and 2019

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Reverse Transcription Semi-Nested PCR (RT-Semi-Nested PCR) and Reverse Transcription PCR (RT-PCR) Screening
2.3. Phylogenetic Analysis
2.4. Bat Species Identification
3. Results
3.1. PCR-Based Detection of Bat Paramyxoviruses in Bat Feces
3.2. PCR-Based Identification of Bat Species in Bat Feces
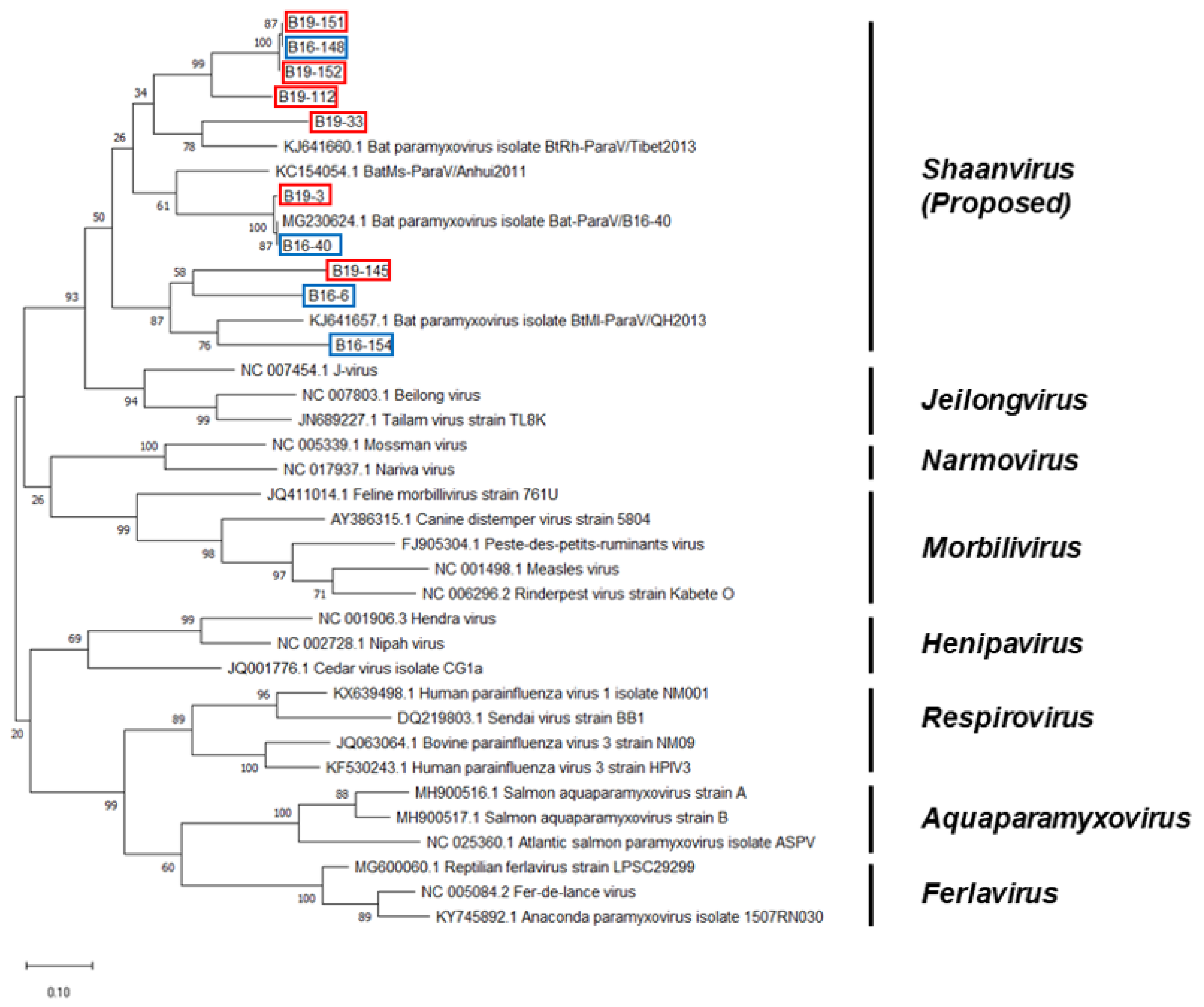
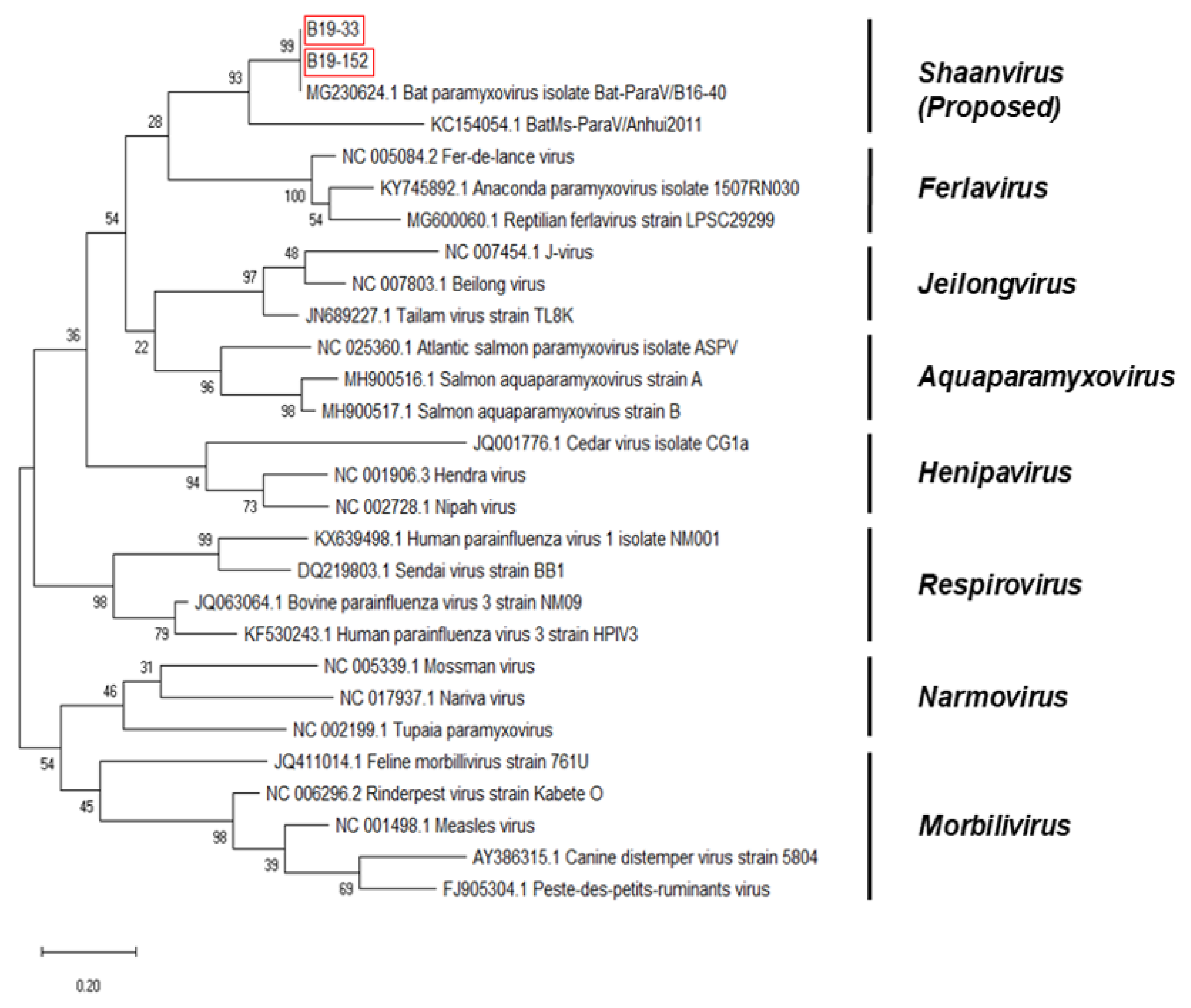
3.3. Phylogenetic Analysis Based on the Genomic Nucleotide Sequence of the Paramyxovirus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Tan, Y.Y.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The Origin, Transmission and Clinical Therapies on Coronavirus Disease 2019 (COVID-19) Outbreak - An Update on the Status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Ge, X.; Wang, L.F.; Shi, Z. Bat origin of human coronaviruses. Virol. J. 2015, 12, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.; Lau, S.; Woo, P.; Yuen, K.Y. Bats as a continuing source of emerging infections in humans. Rev. Med. Virol. 2007, 17, 67–91. [Google Scholar] [CrossRef]
- Rahman, M.; Chakraborty, A. Nipah virus outbreaks in Bangladesh: A deadly infectious disease. WHO South East Asia J. Public Health 2012, 1, 208–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.S.; Sazzad, H.M.; Satter, S.M.; Sultana, S.; Hossain, M.J.; Hasan, M.; Rahman, M.; Campbell, S.; Cannon, D.L.; Stroher, U.; et al. Nipah Virus Transmission from Bats to Humans Associated with Drinking Traditional Liquor Made from Date Palm Sap, Bangladesh, 2011–2014. Emerg. Infect. Dis. 2016, 22, 664–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luby, S.P.; Rahman, M.; Hossain, M.J.; Blum, L.S.; Husain, M.M.; Gurley, E.; Khan, R.; Ahmed, B.N.; Rahman, S.; Nahar, N.; et al. Foodborne transmission of Nipah virus, Bangladesh. Emerg. Infect. Dis. 2006, 12, 1888–18894. [Google Scholar] [CrossRef] [PubMed]
- Homaira, N.; Rahman, M.; Hossain, M.J.; Epstein, J.H.; Sultana, R.; Khan, M.S.; Podder, G.; Nahar, K.; Ahmed, B.; Gurley, E.S.; et al. Nipah virus outbreak with person-to-person transmission in a district of Bangladesh, 2007. Epidemiol. Infect. 2010, 138, 1630–1636. [Google Scholar] [CrossRef] [Green Version]
- Yaiw, K.C.; Crameri, G.; Wang, L.; Chong, H.T.; Chua, K.B.; Tan, C.T.; Goh, K.J.; Shamala, D.; Wong, K.T. Serological Evidence of Possible Human Infection with Tioman Virus, a Newly Described Paramyxovirus of Bat Origin. J. Infect. Dis. 2007, 196, 884–886. [Google Scholar] [CrossRef] [Green Version]
- Mahy, B.W.J.; van Regenmortel, M.H.V. Desk Encyclopeida of Animal and Bacterial Virology; Academic Press: Cambridge, MA, USA, 2009; p. 175. [Google Scholar]
- Lamb, R.A.; Kolakofsky, D. Paramyxoviridae: The Viruses and Their Replication; Lippincott-Raven Press: Philadelphia, PA, USA, 2006; pp. 1449–1496. [Google Scholar]
- Amarasinghe, G.K.; Ayllon, M.A.; Bao, Y.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales: Update 2019. Arch. Virol. 2019, 164, 1967–1980. [Google Scholar] [CrossRef] [Green Version]
- Drexler, J.F.; Corman, V.M.; Muller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [Green Version]
- Halpin, K.; Hyatt, A.D.; Fogarty, R.; Middleton, D.; Bingham, J.; Epstein, J.H.; Rahman, S.A.; Hughes, T.; Smith, C.; Field, H.E.; et al. Pteropid Bats are Confirmed as the Reservoir Hosts of Henipaviruses: A Comprehensive Experimental Study of Virus Transmission. Am. J. Trop. Med. Hyg. 2011, 85, 946–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, B.T.; Broder, C.C.; Middleton, D.; Wang, L.F. Hendra and Nipah viruses: Different and dangerous. Nat. Rev. Microbiol. 2006, 4, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Noh, J.Y.; Jeong, D.G.; Yoon, S.W.; Kim, J.H.; Choi, Y.G.; Kang, S.Y.; Kim, H.K. Isolation and characterization of novel bat paramyxovirus B16–40 potentially belonging to the proposed genus Shaanvirus. Sci. Rep. 2018, 8, 12533. [Google Scholar] [CrossRef]
- Tong, S.; Chern, S.W.; Li, Y.; Pallansch, M.A.; Anderson, L.J. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J. Clin. Microbiol. 2008, 46, 2652–22658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT Nucl. Nucleic Acids Symp. 1999, 41, 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Han, S.; Jung, C.W.; Choi, Y.G.; Kim, S.S. Sounds of the Bats in Korea. In National Institute of Biological; Resources Press: Incheon, Korea, 2012. [Google Scholar]
- Schlegel, M.; Ali, H.S.; Stieger, N.; Groschup, M.H.; Wolf, R.; Ulrich, R.G. Molecular Identification of Small Mammal Species Using Novel Cytochrome B Gene-Derived Degenerated Primers. Biochem. Genet. 2012, 50, 440–447. [Google Scholar] [CrossRef]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Morse, S.S.; Mazet, J.A.; Woolhouse, M.; Parrish, C.R.; Carroll, D.; Karesh, W.B.; Zambrana-Torrelio, C.; Lipkin, W.I.; Daszak, P. Prediction and prevention of the next pandemic zoonosis. Lancet 2012, 380, 1956–1965. [Google Scholar] [CrossRef]
- Kim, H.K.; Yoon, S.W.; Kim, D.J.; Koo, B.S.; Noh, J.Y.; Kim, J.H.; Choi, Y.G.; Na, W.; Chang, K.T.; Song, D.; et al. Detection of Severe Acute Respiratory Syndrome-Like, Middle East Respiratory Syndrome-Like Bat Coronaviruses and Group H Rotavirus in Faeces of Korean Bats. Transbound. Emerg. Dis. 2016, 63, 365–372. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primer | Sequence (5′-3′) | PCR Method | Amplicon Size (bp) | Reference |
|---|---|---|---|---|---|
| RdRp | PAR-F1 | GAAGGITATTGTCAIAARNTNTGGAC | RT-semi-nested PCR | 580 | [17] |
| PAR-F2 | GTTGCTTCAATGGTTCARGGNGAYAA | ||||
| PAR-R | GCTGAAGTTACIGGITCICCDATRTTNC | ||||
| F | F-F2 | ACATCAGCCCAGATTACTGC | RT-PCR | 320 | This study |
| F-R2 | AGCTTGAATTGACAAGGTCT | ||||
| HN | HN-F2 | CCTAATAAACTCAGCATCAAG | RT-PCR | 350 | This study |
| HN-R2 | GCTATCTGGTTGTGAGTGTA |
| Month | Jan | Feb | Mar | Apr | May | Jun | Jul | Aug | Sep | Oct | Nov | Dec | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Year | ||||||||||||||
| 2016 | - | - | 1/13 | 1/5 | 0/12 | 0/31 | 2/30 | 0/8 | 0/6 | 0/8 | 0/7 | 0/1 | 4/121 | |
| 2017 | - | - | 0/1 | 0/21 | 0/26 | 0/18 | 0/3 | 0/13 | 0/5 | 0/10 | 0/4 | 0/6 | 0/107 | |
| 2018 | - | 0/2 | 0/2 | 0/13 | 0/36 | 0/20 | 0/2 | 0/9 | 0/10 | 0/4 | - | 0/9 | 0/107 | |
| 2019 | - | 1/8 | 0/19 | 1/15 | 0/20 | 0/19 | 1/32 | 3/25 | - | - | - | - | 6/138 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, S.S.; Noh, J.Y.; Lo, V.T.; Choi, Y.G.; Yoon, S.-W.; Jeong, D.G.; Kim, H.K. The Epidemiological Characteristics of the Korean Bat Paramyxovirus between 2016 and 2019. Microorganisms 2020, 8, 844. https://doi.org/10.3390/microorganisms8060844
Jang SS, Noh JY, Lo VT, Choi YG, Yoon S-W, Jeong DG, Kim HK. The Epidemiological Characteristics of the Korean Bat Paramyxovirus between 2016 and 2019. Microorganisms. 2020; 8(6):844. https://doi.org/10.3390/microorganisms8060844
Chicago/Turabian StyleJang, Seong Sik, Ji Yeong Noh, Van Thi Lo, Yong Gun Choi, Sun-Woo Yoon, Dae Gwin Jeong, and Hye Kwon Kim. 2020. "The Epidemiological Characteristics of the Korean Bat Paramyxovirus between 2016 and 2019" Microorganisms 8, no. 6: 844. https://doi.org/10.3390/microorganisms8060844