
Abstract
Direct massively parallel sequencing of SARS-CoV-2 genome was undertaken from nasopharyngeal and oropharyngeal swab samples of infected individuals in Eastern India. Seven of the isolates belonged to the A2a clade, while one belonged to the B4 clade. Specific mutations, characteristic of the A2a clade, were also detected, which included the P323L in RNA-dependent RNA polymerase and D614G in the Spike glycoprotein. Further, our data revealed emergence of novel subclones harbouring nonsynonymous mutations, viz. G1124V in Spike (S) protein, R203K, and G204R in the nucleocapsid (N) protein. The N protein mutations reside in the SR-rich region involved in viral capsid formation and the S protein mutation is in the S2 domain, which is involved in triggering viral fusion with the host cell membrane. Interesting correlation was observed between these mutations and travel or contact history of COVID-19 positive cases. Consequent alterations of miRNA binding and structure were also predicted for these mutations. More importantly, the possible implications of mutation D614G (in SD domain) and G1124V (in S2 subunit) on the structural stability of S protein have also been discussed. Results report for the first time a bird’s eye view on the accumulation of mutations in SARS-CoV-2 genome in Eastern India.
Similar content being viewed by others


1 Introduction
SARS-CoV-2 is the causative agent of current pandemic of novel coronavirus disease (COVID-19) which has infected millions of people and is responsible for more than 200,000 deaths worldwide in a span of just 4 months. The virus has a positive sense, single-stranded RNA genome, which is around 30 kb in length. The genome codes for four structural and multiple non-structural proteins (Astuti and Ysrafil 2020). While the structural proteins form capsid and envelope of the virus, non-structural proteins are involved in various steps of viral life cycle such as replication, translation, packaging and release (Lai and Cavanagh 1997). Although at a slower rate, mutations are emerging in the SARS-CoV-2 genome which might modulate viral transmission, replication efficiency and virulence in different regions of the world (Jia et al. 2020; Pachetti et al. 2020) .
The genome sequence data has revealed that SARS-CoV-2 is a member of the genus Betacoronavirus and belongs to the subgenus Sarbecovirus that includes SARS-CoV while MERS-CoV belongs to a separate subgenus, Merbecovirus (Lu et al. 2020; Wu et al. 2020; Zhu et al. 2020). SARS-CoV-2 is approximately 79% similar to SARS CoV at the nucleotide sequence level. Epidemiological data suggests that SARS-CoV-2 had spread widely from the city of Wuhan in China (Chinazzi et al. 2020) after its zoonotic transmission originating from bats via the Malayan pangolins (Zhang et al. 2020). Global sequence and epidemiological data reveals that since its emergence, SARS-CoV-2 has spread rapidly to all parts of the globe, facilitated by its ability to use the human ACE2 receptor for cellular entry (Hoffmann et al. 2020). The accumulating mutations in the SARS-CoV-2 genome have resulted in the evolution of 11 clades out of which the ancestral clade O originated in Wuhan.
Since the first report of sequence of SARS-CoV-2 from India, there have been multiple sequence submissions in Global Initiative on Sharing All Influenza Data (GISAID, https://www.gisaid.org/). Extensive sequencing of the viral genome from different regions in India is required urgently. This will provide information on the prevalence of various viral clades and any regional differences therein, which might lead to improved understanding of the transmission patterns, tracking of the outbreak and formulation of effective containment measures. The mutation data might provide important clues for development of efficient vaccines, antiviral drugs and diagnostic assays. We have initiated a study on sequencing of SARS-CoV-2 genome from swab samples obtained from infected individuals from different regions of West Bengal in Eastern India and report here the first nine sequences and the results of analysis of the sequence data with respect to other sequences reported from the country until date. We have detected unique mutations in the RNA-dependent RNA polymerase (RdRp), Spike (S) and Nucleocapsid (N) coding viral genes. It appears that the mutation in nucleocapsid gene might lead to alterations in local structure of the N protein. Also the putative sites of miRNA binding could be affected, which might have major consequences. The possible implications of the mutations have been discussed, which will provide important insights for functional validation to understand the molecular basis of differential disease severity.
2 Methods
2.1 Clinical sample collection
The Regional Virus Research & Diagnostic Laboratory (VRDL) in Indian Council of Medical research- National Institute of Cholera and Enteric Diseases (ICMR-NICED) is a Government-designated laboratory for providing laboratory diagnosis for SARS-CoV-2 (COVID19) in Eastern India. Nasopharyngeal and oropharyngeal swabs in Viral transport media (VTM) (Himedia labs, India) collected from suspect cases with acute respiratory symptoms/travel history to affected countries or contacts of the COVID-19 confirmed cases were referred to the laboratory for diagnosis. The test reports were provided to the health authorities for initiating treatment and quarantine measures. Residual deidentified positive samples for SARS-Cov-2 were used for RNA isolation and sequencing in accordance with ethics guidelines of Govt. of India.
2.2 Viral RNA extraction and diagnostic test of SARS-CoV-2 (COVID-19) virus
Extraction of viral RNA from the clinical sample (200 μl) was performed using the QIAamp viral RNA mini kit as per manufacturer’s protocol (Qiagen, Germany). The extracted RNA was tested for SARS-CoV-2 (COVID-19) by Real Time Reverse Transcription PCR (qRT-PCR) (ABI 7500, Applied Biosystems, USA) using the protocol provided by NIV-Pune, India (https://www.icmr.gov.in/pdf/covid/labs/1_SOP_for_First_Line_Screening_Assay_for_2019_nCoV.pdf; https://www.icmr.gov.in/pdf/covid/labs/2_SOP_for_Confirmatory_Assay_for_2019_nCoV.pdf).
Briefly, first line screening was done for Envelope E gene and RNase P (Internal control). Clinical samples positive for E gene (Ct ≤ 35.0) were subjected to confirmatory test with primers specific for RdRp and HKU ORF (HKU-orf1-nsp14). Positive Control and No Template Control were run for all genes. A specimen was considered confirmed positive for SARS-CoV-2 if reaction growth curves crossed the threshold line within 35 cycles (Ct cut off ≤ 35.0) for E gene, and both RdRp, ORF or either RdRp or ORF.
2.3 Viral genome sequencing
RNA isolated from nasopharyngeal and oropharyngeal swabs were depleted of ribosomal RNA using Ribo-Zero rRNA removal Kit (Illumina, USA). The residual RNA was then converted to double stranded cDNA and sequencing libraries prepared using TruSeq Stranded Total RNA Library Preparation Kit (Illumina Inc, USA) according to the manufacturer’s instructions. The sequencing libraries were checked using high sensitivity D1000 ScreenTape in 2200 TapeStation system (Agilent Technologies, USA) and quantified by Real Time PCR using Library Quantitation Kit (Kapa Biosystems, USA). The libraries were sequenced using MiSeq Reagent Kit v3 in MiSeq system (Illumina Inc, USA) to generate 2x100 bp paired end sequencing reads. For viral genome amplification in samples which did not generate sufficient viral reads, the RNA samples were converted to double stranded cDNA and amplified using QIAseq SARS-CoV-2 Primer Panel (Qiagen GmbH, Germany) according to the manufacturer’s instructions. The multiplexed amplicon pools were then converted to sequencing libraries by enzymatic fragmentation, end repair and ligation to adapters. The sequencing libraries were checked and quantified as above and sequenced using Miseq reagent Kit v2 Nano in Miseq system (Illumina Inc, USA) to generate 2x150 bp paired end sequencing reads.
2.4 Analysis of sequence data
The sequencing reads obtained in shotgun RNA-Seq experiment were mapped to reference viral sequence, variants detected and consensus sequence for each sample built using Dragen RNA pathogen detection software (version 9) in BaseSpace (Illumina Inc, USA). For amplified whole genome sequencing, the viral sequences were assembled using CLC Genomics Workbench v20.0.3 (Qiagen GmbH, Germany). In both cases, the Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1 as reference genome (Accession NC_045512.2) was used as the reference sequence. Each variant call generated in either pipeline was manually verified in Integrated Genome Viewer IGV v7.8.2 (JT Robinson et al. 2017). Clustal Omega was used to display the mutations in the context of the sequence alignments. BioEdit software (v7.2) was used to extract the CDS from consensus sequence and to check codon usage. Nucleotide to amino acid conversion was done in Emboss Transeq online tool (F Madeira et al. 2019).
2.5 Phylogenetic analysis
To generate the clustering patterns of the viral sequences from West Bengal, a subset of representative virus sequence data (n = 310) were downloaded from GISAID global database (supplementary table 1). Only high coverage data (where the entries have less than 1% N’s and less than 0.05% amino acid mutations), complete genome (entries with base pair greater than 29,000) and excluding low coverage entries (entries having more than 5% N’s) were used in the analysis. All of the sequences were aligned using MAFFT (Multiple alignment using fast Fourier transform). We used the Nextstrain pipeline to process the sequence data. Nextstrain with the augur pipeline was used to build phylogenetic tree based on the IQTREE method, which is a fast and effective stochastic algorithm to infer phylogenetic trees by maximum likelihood. The tree building process involves the use of these subtypes ‘Wuhan-Hu-1/2019’, ‘Wuhan/WH01/2019’ to generate the root of the tree. The tree is refined using RAxML (Randomized Axelerated Maximum Likelihood). A web-based visualization program, auspice was then used to present and interact with phylogenetic and phylogeographic data.
2.6 Structural and function impact of mutations
We investigated the potential miRNA binding site in the region coding for N protein, found to be mutated in our samples. STarMir (http://sfold.wadsworth.org/cgi-bin/starmirtest2.pl) software was used for this purpose. The whole human mature miRNA library was obtained from miRbase database. The sequence in query was taken 50 nt upstream and 50 nt downstream from the site of mutation. The miRNAs which bind to the mutation site through seed sequence were shortlisted. The change in bases can prevent certain miRNA binding and support the binding of others. Therefore, miRNA binding was checked for both, original and mutated site. We checked the levels of miRNAs in the cancer conditions around the upper respiratory tract in the dBDEMC2 database (https://www.picb.ac.cn/dbDEMC/). The TissueAtlas database (https://ccb-web.cs.uni-saarland.de/tissueatlas/) was used to analyse the presence and correlation of miRNAs in body fluids.
3 Results
3.1 Clinical status and contact/travel history
All patients were diagnosed positive for SARS-CoV-2 RNA by Real Time PCR as described above. Five of the patients suffered from fever, while seven patients exhibited some symptoms of infection like sore throat, cough with sputum, running nose or breathlessness. One patient suffered from Acute Respiratory Distress Syndrome (ARDS). Two patients did not exhibit any symptom (table 1). Five individuals had contact with COVID-19 patients in particular; both S2 and S3 had contact with the same patient (table 1). One individual had history of international travel while another had history of domestic travel.
3.2 Sequencing
The shotgun RNA-Seq data resulted in high coverage (greater than 100X median depth of coverage) of complete genome sequences of the SARS-CoV-2 in five samples (S2, S3, S5, S6 and S11) in which greater than 96% of the viral genome was covered at greater than 5X and greater than 99% of the viral genome was covered at greater than 1X. A negative correlation was found between viral load (represented by the Threshold Cycle or Ct value of the RNA samples in the Real Time PCR based diagnostic assay) and the number of reads mapped to the viral genome in the RNA-Seq library. Even with 9 samples, the Pearson Correlation Coefficient was found to be −0.63 (P value = 0.036) (table 1). In particular, it was observed that samples with Ct values greater than 25 mostly resulted in generation of low counts of viral sequence reads leading to less than 15X median depth of coverage of the viral genome. In the remaining four samples (S1, S8, S10 and S12), the median depth of coverage was less than 15X and hence the viral genome sequencing was achieved after amplification of the viral genome by a multiplex PCR approach. All the nine sequences have been submitted in the Global Initiative on Sharing All Influenza Data (GISAID) database.
3.3 Phylogenetic analysis
Phylogenetic tree analysis of the sequences, along with other complete viral genome sequences submitted from India in GISAID, revealed that seven of these sequences belonged to the A2a clade while only one sequence belonged to clade B4 (figure 1 and table 1). We were unable to classify one of the nine sequences, S1, into any clade due to low sequence coverage. To understand transmission histories of these nine SARS-CoV-2 isolates from West Bengal, we aligned these sequences with more than 6000 global sequences, including thirty sequences submitted in GISAID from India (at the time of our analysis) to identify specific mutations that occur at the highest level of the tip in a branch leading to the specific subtype. The predicted origin of the transmitted subtype in each case was identified with 98-100% confidence from the branch in which our samples were located in the phylogenetic tree (table 1).

Radial phylogenetic timetree of the sequences from West Bengal, Rest of India and the World. The sequences from West Bengal included in this report are marked.
3.4 Mutation analysis
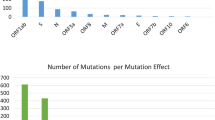
The list of mutations detected in the sequences from nine samples are provided (table 2). Seven sequences harboured the important signature mutations of A2a clade. These consisted of the 14408 C/T mutation resulting in a change of P323L in the RdRp and the 23403 A/G mutation resulting in a change of D614G in the Spike glycoprotein of the virus. In addition to these, 24933 G/T mutation in the gene coding for Spike glycoprotein (G1124V) and triple base mutations of 2881-2883 GGG/AAC in the gene coding for nucleocapsid resulting in two consecutive amino acid changes R203K and G204R were detected in S2, S3 and S2, S3, S5 respectively. While the 24933 G/T S gene mutation was unique to these samples and could not be found in any other sequence from India or the rest of the World, the nucleocapsid mutations could be detected in only three other sequences from India (figure 2). Out of these, two sequences were obtained from individuals with contact history of a COVID-19 patient who had travelled from Italy. Interestingly, two out of three sequences harbouring these mutations obtained by us belonged to Kolkata and with contact history with one COVID-19 patient who had travelled from London (UK). The third sequence was obtained from a COVID-19 patient from Darjeeling, India who had history of travel from Chennai, India. These mutations have been found in 16% of SARS-CoV-2 sequences reported World-wide from countries like UK, Netherlands, Iceland, Belgium, Portugal, USA, Australia, Brazil, etc.

(A) 24933 G/T (G1124V) mutation in the Spike protein coding gene in Clustal Omega. Only the two samples from West Bengal (S2 and S3) harbour this mutation. (B) 28881-3 GGG/AAC (R203K and G204R)) mutations in the nucleocapside protein coding gene in Clustal Omega. Six samples from India, including three samples from West Bengal (S2, S3 and S5) harbour this mutation. (C) 13730 C/T (A88V) mutation in the RdRp gene in Clustal Omega. Only two samples from West Bengal (S11 and S12) harbour this mutation. (D) 14323 C/T, 14326 C/A and 14331 T/C mutations in the RdRp gene in Clustal Omega. Only one sample from West Bengal (S10) harbour these mutations.
RdRp (NSP12) gene of the SARS-CoV-2 codes for the RNA-dependent RNA polymerase and is vital for the replication machinery of the virus. We detected a total of six mutations in this gene in the nine samples, out of which four were nonsynonymous, including the A2a clade specific 14408 C/T (RdRp: P323L) mutation. Two individuals, S11 and S12, harboured viral genome sequences that shared a unique 13730 C/T (A88V) mutation which was not found in any other sequence reported from India or rest of the World. One individual S10, whose viral sequence belonged to B4 clade, harboured 3 mutations in RdRp, which appear to be clade specific, out of which 2 were nonsynonymous.
3.5 Impact of mutations in nucleocapsid gene on miRNA binding
To study the functional relevance of the mutations, we investigated the alteration in miRNA binding in the nucleocapsid coding region, predicted to be caused by the 28881-3 GGG/AAC mutations. We found seven miRNAs which bind to the original sequence and three which bind the mutated sequence exclusively (table 3 and figure 3). The number of bases in the sequence (GGG/AAC) which bind the seed sequence of miRNA were also identified. The strength of miRNA prediction is reflected by the ∆G value mentioned in the figure 3. Lesser the value, stronger is the binding. The values are comparable to some of the experimentally validated miRNA bindings like miR122 binding to HCV RNA has ∆G value of −18.3 kcal/mol for S1 binding site and −22.6 kcal/mol for S2 binding site (data not shown). The values of ∆G obtained for the miRNAs binding to N protein coding region are comparable to these values, suggesting their relevance in the in vivo conditions.

Predicted binding of miRNAs to the orignial (A) and mutated (B) nucleocapsid gene sequences. The sequence in red denotes the seed sequence of miRNAs and the sequence underline in magenta denotes original/mutated site of N- gene involved in miRNA binding.
We checked the levels of these miRNAs in cancer conditions around the upper respiratory tract in the dBDEMC2 database. We found that miR-24-1-5p and miR-299-5p were downregulated in most of the cancers. miR-24-2-5p was found to be upregulated in Esophageal Cancer (ESCA), Head and neck cancer (HNSC), Lung cancer (LUCA) and downregulated in Nasopharyngeal cancer (NSCA) (supplementary figure 1). Assuming that the binding of miRNAs would inhibit the viral replication/stability, higher abundance of that miRNA would be protective against infection and lower abundance would increase the susceptibility towards infection. To comprehend the results, we have found that if a patient suffering from ESCA, HNSC, LUCA is infected with the original virus containing GGG sequence, the upregulated miR-24-2-5p would be protective against the infection. But, if the same patient is infected with the mutated virus containing AAC sequence, miR-24-2-5p will not be functional anymore and miR-299-5p which targets the mutated site is also downregulated. This could make the patients suffering from described cancers, highly susceptible to infection with the mutant virus.
We also checked if these miRNAs are associated with other disease conditions and found that miR-299-5p is down regulated in Type 2 Diabetes Mellitus (T2DM) and hence could serve as one of factors for increased susceptibility of T2DM patients for the mutated viral subtype and increase the risk of co-morbidity (Huang et al. 2018). Another miR-3162-3p, targeting original subtype, is reported to be higher in Asthma patients (Fang et al. 2016). This could be one of the factors limiting the original viral propagation, but the loss of its targeting site in mutated viral subtype could increase the host susceptibility towards viral infection.
We further checked if there are some other conditions that could alter the availability of these miRNAs at the site of infection. Therefore, we used the TissueAtlas database to analyse the presence and correlation of these miRNAs in body fluids. We found that there is differential expression of certain miRNAs in the saliva of patients suffering from pancreatic cancer. miR-642b-5p, miR-3162-3p and miR-299-5p were found to be upregulated in the saliva of pancreatic cancer patients which could provide similar protective/susceptible effect as mentioned of miRNAs before (supplementary figure 2). miRNAs have been known to affect viral replication and stability by binding to protein coding regions of the genome of H1N1, EV71, CVB3 and many more viruses (Bruscella et al. 2017; Trobaugh and Klimstra 2017). In most of the cases, binding of miRNAs leads to translational repression of the targeted protein and hence directly affects viral RNA replication. Targeting by miRNAs could decrease the levels of N protein, which is involved in various steps of viral life cycle including replication, translation and coating of viral RNA to form the nucleocapsid. Hence, altered levels of the shortlisted miRNA could regulate various viral processes and severity of SARS-CoV-2 infection. The effect of miRNAs would be opposite if they assist in viral replication/stability, but that needs to be experimentally confirmed and still holds the importance of miRNAs targeting the original and mutated sites.
3.6 Structural impact of mutations in nucleocapsid
We analysed the 28881-3 GGG/AAC mutations in the nucleocapsid gene which results in contiguous amino acid changes of R203K and G204R for their potential role in alteration of structure of the encoded protein. The sites of these mutations at position are located in the SR-rich region which is known to be intrinsically disordered (Chang et al. 2014). In addition, this region is known to encompass a few phosphorylation sites (Surjit et al. 2005), notably the GSK3 phosphorylation site at Ser202 and a CDK phosphorylation site at Ser206 which are in close proximity to these mutations. The sequence motifs ‘SRGTS’ (202-206) and ‘SPAR’ (206-209) are entirely consistent with GSK3 and CDK phosphorylation motifs, respectively. When Ser202 is phosphorylated which incorporates a large negative group tethered to the sidechain of Ser, as seen in many other substrates of kinases, it is likely that charge neutralization takes place involving positively charged sidechains in the sequential and spatial vicinity. Arg203 is a part of GSK3 phosphorylation motif and its sidechain could potentially contribute to charge neutralization at P-Ser202. Given the sequential, and therefore spatial proximity of Arg203 to P-Ser206 the sidechain of Arg203 could potentially be involved in interaction also with phosphate group at position 206. This interaction would contribute to reduction of conformational entropy. Similarly, Arg209, a part of CDK phosphorylation motif, would contribute to charge neutralization at P-Ser206. Arg203 and Gly204 are mutated to Lys and Arg respectively (figure 4).

Schematic representation to depict the possible implications of mutations in the nucleocapsid (N protein) of SARS-CoV-2. (Top) This panel shows the domain organization of the wild type SARS-CoV-2 N-protein with Arg and Gly at position 203 and 204, respectively. (Bottom) This panel shows the domain organization of the variant SARS-CoV-2 N-protein with mutations at position 203 and 204 (R203K, G204R). The RNA binding domain (RBD; residues 44-180) in the N-terminal region (N) of the protein is shown as blue rectangle, the Dimerization domain (DD; residues 247-364) in the C-terminal region (C) is shown as red rectangle. The disordered regions (residues 1-43, 181-246, 365-419) are shown as grey wavy lines. The different pattern of wavy lines in the top and bottom panel corresponding to region 181-246 indicates the proposed local structural changes in the two genotypes. Domain boundaries have been adopted from Chang et al. 2006. The phosphate groups on Ser residues at position 202 and 206 are denoted as red circles on black sticks. The potential electrostatic interactions between the positively charged Arg/Lys and the negatively charged P-Ser are shown as black dotted lines. Only the sequence motifs (top: 202-209, SRGTSPAR/ bottom: 202-209, SKRTSPAR) of interest in the disordered region are explicitly shown. These residues are colour-coded based on their side-chain chemistry (hydroxyl group: orange, S/T; positively charged: blue, K/R; aliphatic: pink, A; imino acid: cyan, P). Gly (green circle) lacks a side-chain and offers maximum flexibility to the backbone.
3.7 Possible implications of D614G mutation (in SD domain) on protein structural stability
Spike protein (S) of coronaviruses is a class I viral fusion protein which is synthesized as a single chain precursor that trimerizes upon folding. It is composed of two subunits: S1 (in the amino terminal) containing the receptor binding domain (RBD) and S2 (in the carboxy terminal) that drives membrane fusion (figure 5). While the S1 N-terminal region comprises of domain A, the S1 C-terminal half folds as three spatially distinct domains: B, C and D (Walls et al. 2016; Wrapp et al. 2020). The S protein pre-dominantly exists in two structurally distinct conformations: pre-fusion and post-fusion (Li 2016). The pre-fusion state is metastable. Interactions between the protomers facilitated through interlocking of the S1 subunit around the S2 trimer in a crown-like fashion stabilizes the pre-fusion conformation (Walls et al. 2016). Transition from pre-fusion to post-fusion state involves large conformational change to fuse the viral membrane with host cell membrane. This process is triggered when S binds to hACE2 protein via the SB domain to enter the host cell. The ectodomain trimer of S protein in coronaviruses are known to adopt multiple SB conformations (Walls et al. 2020). At the time of preparing this manuscript, three pre-fusion state structures of SARS-CoV-2 S protein solved by cryo-EM are available which encompass the two sites of mutation (D614 and G1124) of our interest. While one of these three structures (PDB code: 6VXX) (Walls et al. 2020) is in a perfectly closed state (i.e., the SB domain in all the three protomers of the trimeric S protein are in a closed conformation), the other two structures (PDB codes: 6VYB and 6VSB) (Walls et al. 2020; Wrapp et al. 2020) are in partially open state. In 6VYB, the SB domain in chain B is in open state whereas it has a closed conformation in chain A and C. In 6VSB, the SB domain in chain A is in open state whereas it has a closed conformation in the other two chains (B and C). In all the three structure, D614 lies in a loop at the interface between any two out of the three protomers. The co-ordinates for the D614 side-chain in chain A and C of 6VYB are available only up to Cβ-atom and the orientation of these atoms are similar to that observed in the respective atoms of D614 in 6VXX. The co-ordinates of all the side-chain atoms of D614 in chain B of 6VYB are available and they are similar to that observed in chain B of 6VXX. The side-chain of D614 in all the protomers of 6VXX and chain B of 6VYB point outward from the core of the protein toward the solvent. The side-chain orientation of D614 in all the three chains of 6VSB is different from the former two structures. This differential orientation of D614 side-chain in 6VSB facilitates formation of hydrogen bond between D614 (present in S1 subunit) and T859 (present in S2 subunit) from the neighbouring chain in two out of the three interfaces found in 6VSB (figure 6).

Structure of S protein. (a) The trimeric structure of SARS-CoV-2 s protein is shown in surface representation with the S1 and S2 subunits. The figure has been generated using PDB code 6VXX. The three chains have been shown in three different colours. (b) One of the protomers from 6VXX is shown as green cartoon. The residues at the respective sites of mutation (D614 and G1124) have been shown as spheres (orange carbon). The zoomed-in views of these sites are shown in the insets. (c) One of the protomer of mouse coronavirus S protein is shown in violet cartoon representation. The image has been generated using the PDB entry: 3JCL. The general topology of mouse coronavirus (MHV-A59) S protein is similar to that of SARS-CoV-2 S protein. The residue equivalent to D614 of SARS-CoV-2 is N655 in MHV-A59. It is shown in sphere representation (light brown carbon). In (b) and (c), The nitrogen and oxygen atoms are shown in blue and red, respectively. Protein rendering has been done using PyMOL (Schrödinger, LLC).

Conformation of D614 in three structures (6VXX, 6VYB, 6VSB). (a), (b), (c) Overlay of D614 (6VXX: yellow carbon; 6VYB: white carbon; 6VSB: dark pink carbon) from chain A, B and C of the three structures, respectively. To maintain visual clarity, only the backbone of respective chain of 6VXX is shown in cartoon representation. (d), (e), (f) Orientation of D614 (green carbon) from chain A (purple cartoon) and T859 (dark blue carbon) from chain B (teal cartoon) in 6VXX, 6VYB and 6VSB, respectively. Hydrogen bond is depicted as black dashed line. (g), (h), (i) Orientation of D614 (green carbon) from chain C (orange cartoon) and T859 (dark blue carbon) from chain A (purple cartoon) in 6VXX, 6VYB and 6VSB, respectively. Hydrogen bond is depicted as black dashed line. The side-chain co-ordinates for D614 in chain A and C of 6VYB are unavailable. Protein rendering has been done using PyMOL (Schrödinger, LLC).
Taken together, these facts suggest that D614 is highly flexible and support the wobbly nature of the inter-protomeric hydrogen bond observed between D614 and T859. Contribution of this transient hydrogen bond toward stability of the pre-fusion state cannot be negated. Interestingly, S protein of mouse coronavirus (MHV-A59) which has a similar structural topology as that of the SARS-CoV-2 S protein but shares a low overall sequence identity (~32%), has a conservative substitution at the position equivalent to D614 of the latter. The Asn (N655) of mouse coronavirus (MHV-A59) is replaced with Asp (D614) in SARS-CoV-2 (figure 5 and figure 7). In earlier literature, N655 has been suggested to offer inter-protomeric interactions that contribute toward maintenance of the S2 fusion machinery in its metastable state (AC Walls et al. 2016). Given the conservation of Asp at this position in closely related coronaviruses (Bat coronaviruses: BtCoV-RaTG13 and BtCoV-HKU3; SARS-CoV) and its conservative substitution in mouse coronavirus (MHV-A59), it is likely that D614 is important for structural stability of S protein.

Multiple sequence alignment (MSA) of S protein. The MSA has been done using MUSCLE (Edgar 2004) and the view has been generated using ESPript server (http://espript.ibcp.fr) (Robert and Gouet 2014). Only the two blocks containing the mutation site of our interest are shown. The numbers above the blocks denote the residue position of corresponding to SARS-CoV-2 S protein sequence in the particular columns. The numbers at the extreme right indicate the overall percentage identity of the full-length sequence of S protein from respective organism with respect to SARS-CoV-2.
As Gly lacks a side-chain, the transient hydrogen bond as observed in the wild-type S protein would be lost in the variant with D614G mutation. This can potentially compromise on the structural stability of pre-fusion state of S protein possibly interfering with conformational transitions. Moreover, replacement of Asp with Gly at this position would come with higher conformational freedom at the backbone (C Ramakrishnan and GN Ramachandran 1965) of the polypeptide resulting in enhancement of local conformational entropy.
3.8 Possible implications of G1124V mutation (in S2 subunit) on protein structural stability
The Gly at this position is solvent exposed and is present at the tip of the C-terminal end of a β-strand. This position is proximal to the region where the S protein attaches itself to the viral membrane (figure 5). It is to be noted that the Gly at this position is conserved among the closely related coronaviruses (Bat coronavirus RaTG13 and HKU3, SARS-CoV) hinting toward its possible role in maintenance of structure and function of the S protein (figure 7). In general, as explained above, Gly backbone has higher conformational freedom than any other amino acid residues (Ramakrishnan and Ramachandran 1965). Therefore, substitution of Gly with Val would impart rigidity to the local region. The possible implication of such rigidity on the association of S protein with viral membrane could be understood from a structure of S protein in association with the viral membrane. However, such a structure is currently unavailable.
4 Discussion
Substantial uncertainties surround the trajectory of the recent epidemic of COVID-19 in India. It is extremely important to track the outbreak by analysing the phylogenetic relationships between different SARS-CoV-2 genomes prevalent in India and compare them with genomes reported from rest of the world. The error-prone replication process of all RNA viruses in general, results in introduction of mutations in their genomes which behave as a molecular clock that can provide insights into the emergence and evolution of the virus. The data till date suggests that SARS-CoV-2 emerged not long before the first cases of pneumonia in Wuhan occurred (Wu et al. 2020).
In this study, direct massively parallel sequencing of the viral genome was undertaken on nasopharyngeal and oropharyngeal swab samples collected from infected individuals from different districts of West Bengal. We have analysed the first nine sequences in this report. Recent analysis of SARS-CoV-2 sequences from all over the globe has revealed that the outbreaks have been initially triggered in most countries by the original strain from Wuhan, clade O, which thereafter have diversified into multiple clades (Yadav et al. 2020; Biswas and Majumder 2020). Temporal sweeps leading to replacement of the ancestral O and other clades by A2a, have been detected. Until our report, initial sequences from samples obtained from individuals with travel history to China, reported genetic similarity to the clade O, which was obtained at the beginning of the outbreak in Wuhan, China. Rest of the sequences reported from India mostly belonged to either clade A3 (18%) or A2a (44%) (supplementary table 3), with evidence of the temporal sweep where the A2a is emerging as the predominant clade (Biswas and Majumder 2020). The A2a clade is characterized by the signature nonsynonymous mutations leading to amino acid changes of P323L in the RdRp which is involved in replication of the viral genome and the change of D614G in the Spike glycoprotein which is essential for the entry of the virus in the host cell by binding to the ACE2 receptor. Notably, the D614G mutation is close to the Furin recognition site for cleavage of the Spike protein, which plays an important role in virus entry. Whether both these mutations have resulted in the evolution of a more transmissible viral subtype i.e. the A2a clade, is yet to be verified by in vitro and in silico analyses. Interestingly, we also found that one of viral sequences in our study belonged to the B4 clade, which originated in China (Gonzalez-Reiche et al. 2020). B4 clade sequences have not been reported from India earlier and are only less than 1% of sequences reported worldwide. Probably, the individual S10 was transmitted this subtype by contact with others who had travel history to China although this information was not available in the patient clinical history.
Emergence of viral subclones in an outbreak can affect the transmission patterns and disease severity, which are immensely important for public health (Harvala et al. 2017; Jones et al. 2019). Given the large size of the infected population in India, with the possibility of regional differences in the population and host-related factors, this can have the potential to affect the course of the outbreak. Population surveillance is essential for early detection of emergence of such subclones. We analysed the mutations detected in each sequence that we generated and found preliminary evidence of this. We found that three individuals of this study, viz. S2, S3 and S5, shared rare set of three contiguous mutations in their genome which resulted in the consecutive alterations of R203K and G204R. These mutations were also found to be shared with 3 other sequences reported from Western India. Interestingly, while two out the three sequences harbouring these mutations were from individuals who shared contact history with a COVID-19 patient with history of travel from Italy, two out of the three samples from West Bengal shared contact history with the same COVID-19 patient with history of travel from UK. The third individual whose sample harboured these mutations, viz. S5, was found to have history of travel from Chennai, India, but the possibility of the patient having contact in transit with an individual with international travel history cannot be excluded. Additionally, origin of the viral subtypes infecting S5 and S6 has also been predicted by phylogenetic analysis to be Europe (UK). S6 had been infected in Delhi, India where he had contact with an infected individual who travelled from Europe. One of the individuals S10, harboured a viral subtype which is predicted to have been transmitted in China. S2 and S3, who shared an identical sequence of the virus, also harboured one unique mutation resulting in the amino acid alteration of G1124V in the Spike protein. This correlates with the fact that these two individuals had also been known to have contact with the same COVID-19 patient. Viral RNA sequences obtained from two samples S11 and S12 shared all mutations except a V32L mutation at ORF8 harboured by S11 and not by S12. Interestingly, both these individuals belonged to the same district of East Medinipur, had history of contact with COVID-19 patients and did not exhibit any clinical symptom. Thus our findings indicate that the viral subtypes transmitted in the eastern region of India, in particular West Bengal, have mostly originated from Europe and also China. Sequencing of large number of samples are being presently undertaken to confirm and elaborate these initial findings.
RdRp is essential for replication of viral RNA genome and hence this gene is expected to be conserved. Interestingly, we detected multiple mutations in this gene, the majority of which were non synonymous and hence result in alteration of protein sequence. In particular, the P323L was present in all A2a sequences in our samples. This mutation is located adjacent to a hydrophobic cleft in RdRp which is a promising target for potential drugs (Pachetti et al. 2020). Sequences from two samples, S11 and S12, shared a unique RdRp mutation at A88V which has not been detected until date in rest of the sequences submitted from India or Worldwide. As observed earlier, these two samples harbour viral subtypes whose genomes are strikingly similar. Sequence obtained from one of the samples S10, which belonged to the clade B4, did not possess the P323L mutation. Instead, it harboured three different mutations resulting in two non-synonymous changes of H286Y, P287T and a synonymous mutation which were not found in any other sequences reported from India until date and are specific for the B4 clade. It remains to be seen whether these amino acid alterations result in substantial changes in structure or function of RdRp, resulting in emergence of drug resistant subtypes or enhancement in mutation rate in the viral genome.
We investigated the potential of the mutations detected in the nucleocapsid region to effect alterations in the viral and host processes. We found that this mutation results in considerable alterations in the predicted binding of miRNAs, which might play a role in the establishment and progress of infection in the patient. We also found that some of the miRNAs which are predicted to bind to the mutated subtype might be downregulated in multiple cancer types. This raises the possibility that cancer patients might have higher susceptibility to the mutated sub-clone due to the reduced ability to contain the virus in vivo, compared to infection by the original virus of the same clade. The leads obtained from this study need to be pursued to develop miRNA based novel therapeutic approaches.
We also analysed the predicted structural alterations in the viral nucleocapsid protein, which might be caused by consecutive alterations of R203K and G204R. As a result of these mutations, we have two strong positively charged residues in close sequential positions as opposed to only one positively charged residue in the other genotype. Given the structural vicinity of P-Ser202 and P-Ser206 and the long sidechains of Lys and Arg with high positive charge and significant side-chain conformational freedom in this genotype, both these residues potentially could contribute to charge neutralization of the phosphorylated serine residues. This contributes to further reduction of conformational entropy compared to the other genotype. While Lys203 is likely to offer electrostatic interactions to P-Ser202, Arg204 (with a greater number of positively charged centres as compared to Lys) could potentially simultaneously interact with the phosphate groups at both P-Ser202 and P-Ser206. Together, these two positively charged residues (Lys203 and Arg204) have the potential to offer additional interactions to the phosphorylated serine residues at 202 and 206 positions as opposed to only one of them (Arg203) in the other genotype. Consequently, one can expect a significant difference in conformational entropy as well as in the inter-residue interaction structural network between the two genotypes especially when Ser202 and Ser206 are phosphorylated. Further, Gly at position 204 in one of the genotypes would confer significantly higher conformational freedom at the backbone (Ramakrishnan and Ramachandran 1965) of the polypeptide chain compared to Arg in the equivalent position in the other genotype. This mutation adds another dimension to the likely structural differences in this local region of the two genotypes. Subsequently, phosphorylation-mediated functional events might be different in the two genotypes (Surjit and Lal 2008; Surjit et al. 2006). These proposed differences in the inter-residue structural network between the two genotypes are depicted schematically in figure 4. Admittedly, the proposed network of interactions is fraught with uncertainty. However, given two positively charged residues in one genotype compared to only one in the other genotype, the charge neutralization structural interaction networks involving P-Ser202 and P-Ser206 has to be certainly different going by the highly established literature on kinase substrates (Kitchen et al. 2008; Krupa et al. 2004).
Interestingly, the mutations D614G (in SD domain) is supposed to confer flexibility in the SD domain and the mutation G1124V might impart partial rigidity in the conformation of S2 domain. Obvious question is whether such structural alterations in local region would have any consequence in receptor binding affinity of Spike protein. Since the mutation resides in RBD domain- S1 subunit of Spike protein, residue 614 is not directly involved in the interaction with ACE2. But the mutation might have some effect on the positioning of the residues involved in interaction.
Now to address the concerns whether these mutations are expected to affect the sensitivity of the existing diagnostic kit, we have again explored the implications of the structural changes. Most likely, the presence of mutation should not affect the Rapid detection kits because these kits detect the presence of specific IgG/IgM antibody against viral N protein or viral S protein. The whole protein is coated for the test and therefore polyclonal antibodies would provide the result here. Change in just one epitope might not affect the overall result. We have further checked if the mutation sites fall in immunodominant epitopes. This data is available for SARS proteins and the sites where we have found mutation have been shown to be conserved in SARS and SARS-CoV-2.
While the mutation site of N protein does not elicit much antibody response, region 603-634 of the S protein of SARS has been shown to be a major immunodominant epitope in S protein (He et al. 2004). Change in this epitope by mutation could alter the sensitivity of the IgG/IgM tests conducted.
Also, there are certain diagnostic kits being designed to check the presence of viral antigen in the clinical sample. The abundance of antibodies targeting the mutation sites needs to be checked in those kits, to be more effective across the viral strains harbouring different mutations.
We also detected interesting relationships between Ct value of diagnostic assay as a surrogate of viral copy number and viral sequence reads obtained. We recommend that for future sequencing studies, the shotgun RNA-Seq approach should be used for high viral copy number represented by low Ct values while for rest, a viral genome amplification method should be used. Although the sample size of our preliminary report is small, follow up studies are underway to confirm these observations for understanding the impact of the same in the ongoing outbreak of COVID-19 in India. We have not commented on the relationship of the viral sequence alterations with disease severity due to the limited sample size of this analysis. We hope to provide valuable information on this aspect based on the expanded number of samples being sequenced at present. Our findings provide leads which might benefit outbreak tracking and development of therapeutic and prophylactic strategies to contain the infection.
Finally, we conclude that the initial sequences generated by us from first nine samples in West Bengal in Eastern India indicate a selective sweep of the A2a clade of SARS-Cov-2. However, the viral population is not homogenous and other clades like B4 also exist. We have also detected emergence of mutations in the important regions of the viral genome including Spike, RdRP and nucleocapsid coding genes. Some of these mutations are predicted to have impact on viral and host factors, which might affect transmission and disease severity. This preliminary evidence of emergence of multiple subclones of SARS-CoV-2, which might have altered phenotypes, can have important consequences on the ongoing outbreak in India.
References
Astuti I and Ysrafil 2020 Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 14 407–412
Biswas NK and Majumder PP 2020 Analysis of RNA sequences of 3636 SARS-CoV-2 collected from 55 countries reveals selective sweep of one virus type. Indian J. Med. Res. https://doi.org/10.4103/ijmr.IJMR_1125_20
Bruscella P, Bottini S, Baudesson C, Pawlotsky J-M, Feray C, et al. 2017 Viruses and miRNAs: More friends than foes. Front. Microbiol. 8 824
Chang CK, Sue SC, Yu TH, et al. 2006 Modular organization of SARS coronavirus nucleocapsid protein. J. Biomed. Sci. 13 59–72
Chang C-k, Hou M-H, Chang C-F, Hsiao C-D and Huang T-H 2014 The SARS coronavirus nucleocapsid protein – Forms and functions. Antivir. Res. 103 39–50
Chinazzi M, Davis JT, Ajelli M, Gioannini C, Litvinova M et al. 2020 The effect of travel restrictions on the spread of the 2019 novel coronavirus (COVID-19) outbreak. Science 368 395–400
Fang C, Lu W, Li C, Peng X, Wang Y, et al. 2016 MiR-3162-3p Is a novel MicroRNA that exacerbates asthma by regulating β-catenin. PLoS ONE 11 e0149257
Gonzalez-Reiche AS, Hernandez MM, Sullivan M, Ciferri B, Alshammary H, et al. 2020 Introductions and early spread of SARS-CoV-2 in the New York City area. medRxiv https://doi.org/10.1101/2020.04.08.20056929
Harvala H, Frampton D, Grant P, Raffle J, Ferns RB, et al. 2017 Emergence of a novel subclade of influenza A(H3N2) virus in London, December 2016 to January 2017. Eurosurveillance 22 30466
He Y, Zhou Y, Wu H, Luo B, Chen J, et al. 2004 Identification of immunodominant sites on the Spike protein of severe acute respiratory syndrome (SARS) coronavirus: Implication for developing SARS diagnostics and vaccines. J. Immunol. 173 4050–4057
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, et al. 2020 SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181 271–280
Huang Q, You W, Li Y, Sun Y, Zhou Y et al. 2018 Glucolipotoxicity-inhibited miR-299-5p regulates pancreatic β-cell function and survival. Diabetes 67 2280–2292
Jia Y, Shen G, Zhang Y, Huang K-S, Ho H-Y, et al. 2020 Analysis of the mutation dynamics of SARS-CoV-2 reveals the spread history and emergence of RBD mutant with lower ACE2 binding affinity. bioRxiv https://doi.org/10.1101/2020.04.09.034942
Jones S, Nelson-Sathi S, Wang Y, Prasad R, Rayen S, et al. 2019 Evolutionary, genetic, structural characterization and its functional implications for the influenza A (H1N1) infection outbreak in India from 2009 to 2017. Sci. Rep. 9 14690
Kitchen J, Saunders RE and Warwicker J 2008 Charge environments around phosphorylation sites in proteins. BMC Struct. Biol. 8 19
Krupa A, Preethi G and Srinivasan N 2004 Structural modes of stabilization of permissive phosphorylation sites in protein kinases: distinct strategies in Ser/Thr and Tyr kinases. J. Mol. Biol. 339 1025–1039
Lai MMC and Cavanagh D 1997 The molecular biology of coronaviruses; in Advances in Virus Research Eds. Maramorosch K, Murphy FA and Shatkin AJ (Academic Press, New York) pp 1–100
Li F 2016 Structure, function, and evolution of coronavirus Spike proteins. Annu. Rev. Virol. 3 237–261
Lu R, Zhao X, Li J, Niu P, Yang B et al. 2020 Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395 565–574
Madeira F, Park YM, Lee J, Buso N, Gur T, et al. 2019 The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47 W636–W641
Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E et al. 2020 Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 18 179
Ramakrishnan C and Ramachandran GN 1965 Stereochemical criteria for polypeptide and protein chain conformations: II. Allowed conformations for a pair of peptide units. Biophys. J. 5 909–933
Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A and Mesirov JP 2017 Variant review with the integrative genomics viewer. Cancer Res. 77 e31–e34
Surjit M, Kumar R, Mishra RN, Reddy MK, Chow VTK et al. 2005 The severe acute respiratory syndrome coronavirus nucleocapsid protein is phosphorylated and localizes in the cytoplasm by 14-3-3-mediated translocation. J. Virol. 79 11476–11486
Surjit M and Lal SK 2008 The SARS-CoV nucleocapsid protein: A protein with multifarious activities. Infect. Genet. Evol. 8 397–405
Surjit M, Liu B, Chow VTK and Lal SK 2006 The nucleocapsid protein of severe acute respiratory syndrome-coronavirus inhibits the activity of cyclin-cyclin-dependent kinase complex and blocks S phase progression in mammalian cells. J. Biol. Chem. 281 10669–10681
Trobaugh DW and Klimstra WB 2017 MicroRNA regulation of RNA virus replication and pathogenesis. Trends Mol. Med. 23 80–93
Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, et al. 2020 Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181 281–292
Walls AC, Tortorici MA, Bosch B-J, Frenz B, Rottier PJM, et al. 2016 Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 531 114–117
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, et al. 2020 Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367 1260–1263
Wu F, Zhao S, Yu B, Chen Y-M, Wang W, et al. 2020 A new coronavirus associated with human respiratory disease in China. Nature 579 265–269
Yadav P, Potdar V, Choudhary M, Nyayanit D, Agrawal M, et al. 2020 Full-genome sequences of the first two SARS-CoV-2 viruses from India. Indian J. Med. Res. 151 200–209
Zhang T, Wu Q and Zhang Z 2020 Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr. Biol. 30 1346–1351
Zhu N, Zhang D, Wang W, Li X, Yang B et al. 2020 A Novel Coronavirus from Patients with Pneumonia in China, 2019. New Eng. J. Med. 382 727–733
Acknowledgements
We acknowledge the financial and overall support provided by the Department of Biotechnology, Ministry of Science and Technology, India, and Indian Council of Medical Research and all laboratory staff of the NICED- VRDL network for laboratory support during the ongoing COVID-19 pandemic. We also acknowledge the assistance provided by Dr. Sillarine Kurkalang (NIBMG), Mr. Sumitava Roy (NIBMG) in reviewing the sequence data, Ms. Soumi Sarkar (NIBMG) for assistance in statistical analysis, and Mr. Anand Bhushan and Ms. Meghna Chowdhury for providing assistance in laboratory support and logistics. SD and NS would like to acknowledge support from J C Bose fellowship. We also thank DBT-IISc partnership programme at IISc, Bengaluru, and the National Genomics Core at NIBMG. HR and SC would like to acknowledge support from CSIR-SPM fellowship and DST-INSPIRE fellowship, respectively. TG would like to acknowledge DBT-RA fellowship.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Corresponding editor: Sudha Bhattacharya
This artice is part of the Topical Collection: COVID-19: Disease Biology & Intervention.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Maitra, A., Sarkar, M.C., Raheja, H. et al. Mutations in SARS-CoV-2 viral RNA identified in Eastern India: Possible implications for the ongoing outbreak in India and impact on viral structure and host susceptibility. J Biosci 45, 76 (2020). https://doi.org/10.1007/s12038-020-00046-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12038-020-00046-1