
Abstract
Mycoplankton are widespread components of marine ecosystems, yet the full extent of their functional role remains poorly known. Marine mycoplankton are likely functionally analogous to their terrestrial counterparts, including performing saprotrophy and degrading high-molecular weight organic substrates using carbohydrate-active enzymes (CAZymes). We investigated the prevalence of transcribed oceanic fungal CAZyme genes using the Marine Atlas of Tara Ocean Unigenes database. We revealed an abundance of unique transcribed fungal glycoside hydrolases in the open ocean, including a particularly high number that act upon cellulose in surface waters and the deep chlorophyll maximum (DCM). A variety of other glycoside hydrolases acting on a range of biogeochemically important polysaccharides including β-glucans and chitin were also found. This analysis demonstrates that mycoplankton are active saprotrophs in the open ocean and paves the way for future research into the depth-dependent roles of marine fungi in oceanic carbon cycling, including the biological carbon pump.
Similar content being viewed by others


Even though our understanding of marine fungal diversity is increasing [1, 2], a comprehensive knowledge of their active functional ecology remains limited, especially in the open ocean [1]. Fungal activity has been detected in corals, deep sea and coastal sediments, and associated with phytoplankton blooms, including parasites [1, 2], but the full extent that fungi are functionally active throughout the open ocean water column is yet to be established.
Many terrestrial fungi occupy key roles as saprotrophs by decomposing and recycling biogenic matter, making them intrinsic components of healthy functioning ecosystems [3]. In coastal waters there is evidence that planktonic fungi (mycoplankton) degrade and utilise phytoplankton-derived carbohydrate-rich matter in broadly analogous functional modes [4]. However, the extent that carbohydrate-based fungal saprotrophy occurs in the open ocean remains largely speculated [1].
Glycoside hydrolases (GHs) are a widespread group of carbohydrate-active enzymes (CAZymes) [5] that degrade complex polysaccharides and are categorised into substrate-specific families. In terrestrial fungi, secreted CAZymes are key to the functional potential of saprotrophs and are the primary mode of degradation of high-molecular weight (HMW) polysaccharides (e.g. cellulose). Coastal saprotrophic mycoplankton also employ secreted GHs to degrade phytoplankton-derived HMW carbohydrate-based substrates [4], but the prevalence and identity of the specific GH families of active open ocean mycoplankton are unknown.
Metagenomes from the Tara Oceans project have been used to assess mycoplankton diversity [6, 7], but the associated metatranscriptomes are yet to be fully explored from a fungal perspective. We interrogated the Marine Atlas of Tara Ocean Unigenes (MATOU) metatranscriptomic occurrences database [8] for transcribed fungal GH genes to explore broad-scale depth-dependent structuring in the oceans. The MATOU database consists of all unique eukaryotic genes assembled from the Tara Oceans metatranscriptomes (unigenes), their associated taxonomy and occurrence within samples (full methods described in [8]).
To identify GHs within the MATOU database, reference libraries were created for 61 fungal GH families and clustered using CD-Hit [9]. The MATOU unigenes were searched against these libraries using Diamond v.0.9.22 [10], yielding a database where each positive unigene match represents a unique GH (similar genes clustered when similarity < 95% over 90% of the smallest sequence [8]). The database was screened for non-fungal unigenes using the MATOU taxonomy (Fig. 1a, Supplementary Fig. 1). After removal of redundant matches (i.e. where multiple GHs matched to a single unigene), 1,326 unique fungal GH unigenes were found (~0.001% of the entire unigene catalogue) that occurred 44,386 times in all Tara Oceans samples.

(a) Pipeline describing the steps involved in identifying fungal CAZymes within the Tara Oceans MATOU database. A fungal GH protein sequence reference database was created from all the 61 characterised GH subfamilies found in fungi. The database was consolidated by clustering sequences at 95 % identity using CD-Hit before Diamond BLAST databases were generated for each subfamily. Unigenes were searched against each of these 61 databases using the following thresholds: e value > 1e−30, score > 1, subject Cov > 75%, keeping only the best alignments. Positive matches were then screened using the MATOU taxonomy to discriminate between fungal and non-fungal unigenes. Occurrences of each unigene within the Tara Oceans transcriptomes were returned. (b) Fungal GH groups found in the MATOU database ranked by abundance over all Tara Oceans samples. (c) Total numbers and taxonomy (including Ascomycota (green), Basdiomycota (orange), and Unassigned (yellow)) of unique fungal unigenes from the ten most abundant GH groups.
The top ten GH families containing the greatest number of unique genes were determined by ranking the sum of all unigene occurrences from all samples for each family (Fig. 1b). Overall, the greatest number of unique GHs were involved in cellulose/hemicellulose degradation (GH7). Other substrates of the most abundant GH families included β-glucans (GH17 and GH72), β-glycans (GH5, GH16, GH3), α-glucans (GH13), chitin (GH18), N-/O-glycans (GH47) and xylan (GH43). Fungal GHs were dominated by genes originating from the Ascomycota, except for the abundant GH7s, many of which were unclassified fungal genes (Fig. 1c).
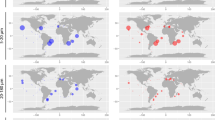
Transcribed fungal GH genes were recovered from 66 stations (Fig. 2). The number of unique GH7s was greatest in the surface and deep chlorophyll maximum (DCM), especially in high productivity areas such as the Mediterranean Sea and the Indian Ocean (Fig. 2b). At stations where concomitant samples were available from the surface, DCM and mesopelagic, a distinct drop in the number of unique GH7s was seen in the mesopelagic. The number of unique unigenes in other abundant GH groups was more heterogeneous between sites (Fig. 2c).

(a) Global map indicating Tara Oceans stations searched for fungal GH unigenes in the surface, deep chlorophyll maximum (DCM) and mesopelagic. (b) Mean unique unigenes/station for each of the major oceanic regions sampled. (c) Depth-dependent partitioning of fungal GH unigenes in the surface (SUR), DCM and mesopelagic (MES).
The high prevalence of unique GH7 transcribed genes in surface waters is likely a response to increased ‘fresh’ phytoplankton-derived matter in the photic zone. Cellulose is a key structural component of many phytoplankton cells [11]. Polysaccharides, such as cellulose, also represent a primary source of particulate organic carbon (POC) [12]. Of the enzymes responsible for cellulose degradation, those within the GH7 family are typically the most active, and are important in biomass degradation by terrestrial fungi [13]. The decline in the number of unique GH7 genes below the DCM in the mesopelagic zone in the six sites sampled suggests a shift in mycoplankton functionality due to depletion of readily available phytoplankton-derived carbon sources, and corresponds with the decrease in overall polysaccharide concentrations between surface waters and the mesopelagic zone [14].
Amongst the other major GH groups, the greatest number of unique genes was in the GH17 family, a group of CAZymes that degrade β-glucans. Cladosporium mycoplankton isolated from coastal waters have been shown to secrete GH17 β-1,3-glucosidase when utilising laminarin [4], which is an algal-derived β-glucan that is a major POC component [15]. Also prevalent were GH18 genes, responsible for degrading chitin, another major polysaccharide and important component of zooplankton and some phytoplankton, suggesting that chitin degradation is a functional role of marine mycoplankton, as with fungi in freshwater lake ecosystems [16].
While the number of transcribed GHs is a strong indicator of active carbohydrate metabolism in mycoplankton communities, the identity of these fungi remains uncertain. The extent of GH7 genes with unresolved fungal taxonomy opens interesting questions about the phylogeny of these taxa. The majority of the other GHs were affiliated to the Ascomycota, in line with phylogenetic studies that show the phylum dominates open ocean mycoplankton diversity [6]. However, there is a lack of early-diverging taxa (e.g. Chytridiomycota) within the MATOU database. Since Chytridiomycota parasitism of phytoplankton takes place in the open ocean [17] their absence highlights outstanding gaps in our understanding of the functional ecology of marine fungi.
The vertical flux of POC in the open ocean is an essential feature of the biological carbon pump (BCP), sustaining the oceans capability to sequester carbon [18]. Biogeochemical models of the BCP do not currently consider fungi (e.g. [19]). Given that marine snow is an apparent hotspot for fungi [20], and that here we show differences in fungal GH expression suggesting depth-dependent resource partitioning in relation to POC-associated substrates, the recently proposed ‘mycoflux’ [2] should be considered within a contemporary view of the BCP.
References
Amend A, Burgaud G, Cunliffe M, Edgcomb VP, Ettinger CL, Gutiérrez MH, et al. Fungi in the marine environment: open questions and unsolved problems. mBio. 2019;10:e01189–18.
Grossart H-P, Wyngaert SV den, Kagami M, Wurzbacher C, Cunliffe M, Rojas-Jimenez K. Fungi in aquatic ecosystems. Nat Rev Microbiol. 2019;1:339–354.
Frąc M, Hannula SE, Bełka M, Jędryczka M. Fungal biodiversity and their role in soil health. Front Microbiol. 2018;9:707.
Cunliffe M, Hollingsworth A, Bain C, Sharma V, Taylor JD. Algal polysaccharide utilisation by saprotrophic planktonic marine fungi. Fungal Ecol. 2017;30:135–8.
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–5.
Hassett BT, Vonnahme TR, Peng X, Jones EBG, Heuzé C. Global diversity and geography of planktonic marine fungi. Bot Mar. 2019.
Morales SE, Biswas A, Herndl GJ, Baltar F. Global structuring of phylogenetic and functional diversity of pelagic fungi by depth and temperature. Front Mar Sci. 2019;6:131.
Carradec Q, Pelletier E, Silva CD, Alberti A, Seeleuthner Y, Blanc-Mathieu R, et al. A global ocean atlas of eukaryotic genes. Nat Commun. 2018;9:373.
Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinforma Oxf Engl. 2012;28:3150–2.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60.
Domozych D. Algal cell walls. In: eLS. Wiley, Hoboken; 2019. p. 1–11.
Smetacek V, Hendrikson P. Composition of particulate organic-matter in kiel bight in relation to phytoplankton succession. Oceano Acta. 1979;2:287–98.
Payne CM, Knott BC, Mayes HB, Hansson H, Himmel ME, Sandgren M, et al. Fungal cellulases. Chem Rev. 2015;115:1308–448.
Pakulski JD, Benner R. Abundance and distribution of carbohydrates in the ocean. Limnol Oceanogr. 1994;39:930–40.
Becker S, Tebben J, Coffinet S, Wiltshire K, Iversen MH, Harder T, et al. Laminarin is a major molecule in the marine carbon cycle. Proc Natl Acad Sci USA. 2020;117:6599–607.
Wurzbacher CM, Bärlocher F, Grossart H-P. Fungi in lake ecosystems. Aquat Micro Ecol. 2010;59:125–49.
Gutiérrez MH, Jara AM, Pantoja S. Fungal parasites infect marine diatoms in the upwelling ecosystem of the Humboldt current system off central Chile. Environ Microbiol. 2016;18:1646–53.
Boyd PW, Claustre H, Levy M, Siegel DA, Weber T. Multi-faceted particle pumps drive carbon sequestration in the ocean. Nature. 2019;568:327–35.
Giering SLC, Sanders R, Lampitt RS, Anderson TR, Tamburini C, Boutrif M, et al. Reconciliation of the carbon budget in the ocean’s twilight zone. Nature. 2014;507:480–3.
Bochdansky AB, Clouse MA, Herndl GJ. Eukaryotic microbes, principally fungi and labyrinthulomycetes, dominate biomass on bathypelagic marine snow. ISME J. 2017;11:362–73.
Acknowledgements
NAMC is supported by the NERC MARINe-DNA project grant NE/N006151/1. MC is also supported by the NERC grant NE/N006151/1 and ERC MYCO-CARB project grant 772584. The authors wish to acknowledge the constructive comments of the reviewers, whose input led to a much improved manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Chrismas, N., Cunliffe, M. Depth-dependent mycoplankton glycoside hydrolase gene activity in the open ocean—evidence from the Tara Oceans eukaryote metatranscriptomes. ISME J 14, 2361–2365 (2020). https://doi.org/10.1038/s41396-020-0687-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-0687-2
This article is cited by
-
Global contribution of pelagic fungi to protein degradation in the ocean
Microbiome (2022)
-
Harnessing microbial wealth for lignocellulose biomass valorization through secretomics: a review
Biotechnology for Biofuels (2021)
-
Potential and expression of carbohydrate utilization by marine fungi in the global ocean
Microbiome (2021)
