
Abstract
Loop-mediated amplification (LAMP) is an isothermal amplification technique favored in diagnostics and point-of-care work due to its high sensitivity and ability to run in isothermal conditions. In addition, a visual readout by lateral flow strips (LFS) can be used in conjunction with LAMP, making the assay accessible at the point-of-care. However, the amplicons resulting from a LAMP reaction varied in length and shape, making them undiscernible on a double-stranded DNA intercalating dye stained gel. Standard characterization techniques also do not identify which amplicons specifically bind to the LFS, which generate the visual readout. We aimed to standardize our characterization of LAMP products during assay development by using fluorescein amidite (FAM) and biotin-tagged loop forward and backward primers during assay development. A pvuII restriction enzyme digest is applied to the LAMP products. FAM-tagged bands are directly correlated with the LFS visual readout. We applied this assay development workflow for an HPV 16 assay using both plasmid DNA and clinical samples to demonstrate proof of concept for generalized assay development work.
Similar content being viewed by others

Introduction
Loop-mediated amplification (LAMP) is an isothermal nucleic acid amplification method that produces a range of amplicons of different sizes and shapes, unlike the gold standard polymerase chain reaction (PCR) [1], which produces one amplicon of a predictable length. Although the primary focus here is on LAMP, there are other isothermal amplification techniques for point-of-care settings, including but not limited to helicase-dependent amplification (HDA) and recombinase polymerase amplification (RPA) [2,3,4]. LAMP uses Bst polymerase for strand displacement and four base primers—a forward primer (F3), backward primer (B3), forward inner primer (FIP), and backward inner primer (BIP)—to amplify targeted DNA sequences. The base primers produce stem-loop dumbbell structures that become the starting template for exponential amplification. Two additional primers, called the loop forward (LF) and loop backward (LB), are added to the base primer set to further accelerate LAMP reactions and increase sensitivity [5]. LAMP’s combined set of properties—isothermal amplification, the increased number of amplicon products, and accelerated amplification—minimize the need for instrumentation and make LAMP attractive for point-of-care (POC) diagnostic applications.
Furthermore, combining a LAMP assay with a simple readout method like lateral flow strips (LFS) can result in an accessible POC test. LFS allow for inexpensive qualitative colorimetric assays with appropriate flow and amplification controls that lead to rapid and intuitive visual readouts [6,7,8]. With a full LAMP primer set, minor modifications to tag the primers can accommodate commercially available LFS readouts.
Although there are numerous reports of paper microfluidic assays that address aspects of sample preparation and reagent storage, isothermal amplification of DNA, and antibody-based lateral flow detection—including devices and methods that incorporate LAMP and LFS—most of these have not included efforts to determine which of the many LAMP amplicon products correlate with a positive test line [9,10,11,12,13,14]. The lack of this characterization step leads to two problems. First, if it is unknown which products lead to the positive readout on the test strip, it is impossible to optimize the assay to produce more of these products during assay development. Second, without this information, it is difficult to figure out the source of false positive tests. Elucidating the specific DNA amplicons that bind to the test strip will make for a more robust LAMP-LFS assay development workflow.
We aim to distinguish LAMP-specific products from non-specific amplification. Traditionally, positive amplification for LAMP is detected via agarose gel electrophoresis using a double-stranded DNA intercalating dye like SYBR Green or ethidium bromide. The resulting gel does not have the resolution to distinguish between non-specific primer amplification and target-specific amplification. In some POC applications, analysis involves using intercalating dye in situ, with the real-time fluorescence increase observed by eye or using a portable reader [15]. In this embodiment, the user does not have an internal control to detect the presence and amount of non-specific amplification. Leveraging our understanding of primer design and associated amplicon readouts on LFS, we are able to investigate and develop a characterization workflow for use during LAMP assay development to identify and validate target-specific amplicons. This workflow is meant to be used only during the assay development and validation processes, since the resulting increase in steps would be unnecessary and cumbersome when the test is cleared for deployment.
To implement the characterization workflow, we tag loop primers with a fluorescent tag, 6-FAM (fluorescein), to enable downstream detection via two modalities: (1) amplicon binding to LFS containing an anti-FAM test line and (2) fluorescent detection of LAMP amplicons on a gel without additional staining. For the first modality, the resulting amplicons incorporating the FAM-tagged primers will bind to the test line composed of anti-FAM antibodies. Second, we include a restriction enzyme digest of the LAMP product, which has been previously shown to determine the specificity of positive LAMP amplification [16, 17]. We chose a restriction enzyme specific to our target region to identify unique bands later visible using gel electrophoresis. They enzyme selection is determined by the sequence of the target region and the number of restriction sites present. The fluorescently tagged amplicons are also visible on an unstained agarose or acrylamide gel after imaging with appropriate filters specific to the fluorescent tag to screen out non-FAM-tagged DNA amplicons, identifying the assay’s target-specific amplicons. This characterization strategy allows us to determine which FAM-tagged LAMP target-specific products bind to the test lines on the LFS.
As a proof of concept to demonstrate that this assay development workflow will work for validation with clinical samples, we designed a LAMP assay for the high-risk human papillomavirus, HPV 16 [12]. The target is the E7 oncogene of HPV 16, which is present in cervical lesions caused by HPV in both the dermis and epidermis of infected individuals [18, 19]. The E7 oncogene is present in cells supporting viral genomic amplification and produces the E7 protein responsible for mediating cell proliferation during mitosis [19]. We tested discarded cervical swab samples acquired from the BUMC Pathology Lab under an approved Institutional Review Board Protocol. These samples are provided to us with standard of care cobas 4800 laboratory test results. The cobas 4800 HPV Test (Roche Diagnostics) uses an automated real-time TaqMan PCR-based qualitative molecular HPV screening test to test cervical swab samples. The cobas test targets the L1 oncogene that produces a capsid protein that is the structural element of HPV virions during assembly and release [19]. Although the targeted genes for our assay and the cobas test are different, L1 and E7 are both one copy genes, and both proteins are important in HPV infection of the epithelium.
This assay development workflow allows for more comprehensive characterization than current protocols for the design and validation of isothermal nucleic acid amplification tests with LFS readouts. Enhanced characterization during assay development will lead to tests with lower false positive gel electrophoresis results during characterization and lower false negative rates in final test design. This method also provides a clear protocol to follow for troubleshooting.
Materials and methods
Loop-mediated amplification with fluorescent loop primers for HPV 16
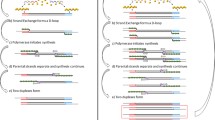
A 6-primer set was designed and optimized for HPV 16 loop-mediated amplification (LAMP) (Fig. 1a, Table 1). All primers were synthesized by IDT (Integrated DNA Technologies [IDT], Coralville, IA, USA). Starting from a 4-primer baseline set, two additional loop primers—5′-FAM-labeled loop forward (LF) and a 5′-biotin-labeled loop backward (LB)—were designed and added to the reaction (Fig. 1b). The FAM fluorescent tag enables downstream detection of the amplicons via LFS and fluorescent gel imaging (Fig. 1c). The 5′ biotin tag enables visual detection of positive amplification via streptavidin-tagged gold nanoparticles.

Schematic of FAM-tagged loop primers for LAMP and analysis workflow. a The schematics show the locations of the parts of primers used in the LAMP reaction. The table further demonstrates how to design each primer for a LAMP assay; specifically, we are targeting the E7 gene in HPV 16. The simplest products are shown with the corresponding primer regions highlighted. b The two loop primers are tagged on the 5′ end, which allows for amplification to occur without the tags inhibiting it. The stylized example amplicon shows how the tags would be integrated into the products. c The FAM tag is specific to amplicon products that attach to the anti-FAM test line. A gel run on post-digested amplicons imaged for FAM will display bands specific to HPV 16. Using the combination of fluorescent gels and LFS, LAMP products are characterized
Linearized HPV 16 E7 gene pUC-57 plasmid DNA stocks were used as quantitation standards for LAMP assay development; plasmids were obtained from either gene synthesis services (GENEWIZ, Cambridge, MA) or cloned in-house as described previously [12]. The LAMP reaction contained 1× Isothermal Amplification Buffer (New England Biolabs, Ipswich, MA), 1 mM dNTP (Invitrogen, Carlsbad, CA), 0.3 M betaine (Sigma-Aldrich, St. Louis, MO), 8 U Bst 2.0 DNA polymerase (New England Biolabs, Ipswich, MA) with final primer and probe concentrations at 0.2 μM F3 and B3, 1.6 μM FIP and BIP, 0.8 μM LF and LB. The reaction was performed at 63 °C for 60 min. All reactions were run using either the QuantStudio 5 (Applied Biosystems, Foster City, CA) or 2720 Thermal Cycler (Applied Biosystems, Foster City, CA). Endpoint amplification was characterized with either TBE agarose gels, non-denaturing 12% TBE acrylamide gels, or 10% denaturing urea gel electrophoresis. Results are classified as LAMP-LFS positive or LAMP-LFS negative based on gel quantification.
Initial studies were completed to determine the limit of detection of HPV 16 using plasmid DNA to identify appropriate working concentrations (S1). Further details on limit of detection procedures and analysis are in the Electronic Supplementary Material (ESM).
FAM fluorescent DNA ladders
A fluorescent FAM ladder (FL) was synthesized in-house by adding Klenow polymerase (New England Biolabs, Ipswich, MA) and fluorescent dCTPs (Jena Bioscience, Jena, Germany) to mspI-digested pBR ladder (New England Biolabs, Ipswich, MA). The reaction was performed at room temperature for 12 min. The DNA was purified using phenol-chloroform extraction and ethanol precipitation.
Gel imaging
To identify 5′-FAM-specific LAMP products, unstained agarose and acrylamide gels were imaged under 488-nm laser excitation using the Sapphire Biomolecular Imager (Azure Biosystems, Dublin, CA). Images were processed with Sapphire Capture software (Azure Biosystems, Dublin, CA). 1× SYBR Gold (Life Technologies, Carlsbad, CA) or 1× SYBR Green (Life Technologies, Carlsbad, CA) stained gels were imaged using both the VersaDoc (Bio-Rad, California, USA) and the Sapphire Biomolecular Imagers.
Lateral flow strip operation and imaging
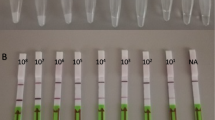
Lateral flow strips were purchased from Bio-UStar Technologies (Hangzhou, China). Each LFS has a single test line (anti-FAM) and a control line (anti-Streptavidin). One microliter of the post-LAMP-amplified reaction was added to the sample pad, followed by a 60 μl aliquot of Hyclone HyPure Water (GE Healthcare Life Sciences, Marlborough, MA) as running buffer. Results were read at 5 min. The strips were imaged at 10 min using a Nikon SMZ18 stereomicroscope (Nikon, Tokyo, Japan), under white LED illumination at ×0.75 magnification.
pvuII digest characterization of LAMP
The HPV 16 LAMP amplicon contains a restriction enzyme site for pvuII. pvuII (New England Biolabs, Ipswich, MA) digests were performed at 37 °C overnight. The digest products were run on a 12% non-denaturing acrylamide gel. The 5′-FAM-labeled products were identified by imaging the unstained gel under 488 nm illumination. Afterwards, all other LAMP amplicons were visualized by staining the gel with 3 μl 10,000× SYBR Green (Life Technologies, Carlsbad, CA) and 30 ml 1× Tris/Borate/EDTA (TBE) buffer for 30 min on a rocking platform. The stained gel was imaged using the Sapphire Biomolecular Imager.
Sequencing pvuII digest bands
pvuII-digested bands were extracted from acrylamide gels with the QIAGEN Gel Extraction Kit (QIAGEN, Valencia, CA) and cloned into the zraI-digested pGEM vector (Promega, Madison, WI). The plasmid backbone and purified digest bands were ligated overnight and transformed into Top10 cells (Invitrogen, Waltham, MA, USA). Colony PCR was performed to select colonies with positive transformations. Liquid cultures of these colonies were subsequently processed with a MiniPrep Kit (QIAGEN, Valencia, CA) and sequenced (Genewiz Sequencing, Cambridge, MA).
Denaturing gel electrophoresis
To eliminate secondary structure from LAMP amplicons, pvuII-digested samples were loaded on a 10% denaturing urea-polyacrylamide gel and run at 70 W for 1 h at 54 °C. The unstained gel was imaged for FAM first (without any additional labeling) to show products specifically containing the labeled primers. It was then subsequently stained to show all DNA on the gel with 15 μl 1× SYBR Gold (Life Technologies, Carlsbad, CA) and 250 ml 1× TBE buffer for 30 min on a rocking platform.
Extraction of captured products from lateral flow strips
For each LAMP reaction (no template control, HPV 16 plasmid, and patient samples), 7 LFS were run for both the undigested and pvuII-digested products. Figure 2 provides a condensed schematic of the LFS extraction and elution process. For the undigested LAMP reaction products, a total of 3 μl was placed on each of the 7 LFS strips per reaction. For digested LAMP amplicons, a total of 10 μl was pipetted onto each LFS. LFS were dried at room temperature for 10 min before imaging as previously described. All equipment was cleaned with DNA AWAY (Thermo Fisher Scientific, Waltham, MA) and 70% ethanol between strips to mitigate concerns about strip contamination when working with open devices. (Note the process above is for assay development only and that in the final embodiment of these tests, the reaction volume and LFS will be completely contained in a plastic cartridge that will close before the reaction is run and never need to be opened again, thus containing all the amplicons.) To extract the reaction products from the LFS test line for further analysis, each test line was cut out of the LFS (a total of 7 strips for each reaction) and placed into 50 ml of 1× phosphate-buffered saline (PBS) and dialyzed for 1 h at 4 °C on a rotating apparatus. The PBS was removed, and fresh PBS was added for another hour of washing to remove non-specifically bound products. After washing, all 7 cut out test lines for a single reaction was pooled and added to a 1.7-ml tube with 75 μl of elution buffer (QIAGEN, Valencia, CA) and heated for 10 min at 95 °C to elute the bound DNA. After removing the paper, the eluted DNA was recovered using isopropanol precipitation. DNA concentration and purity were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA).

Schematic for lateral flow strip extraction performed on both LFS-negative and LFS-positive strips
BD SurePath cervical clinical samples
A total of 110 discarded, de-identified BD SurePath (BD, Franklin Lakes, NJ) cervical swabs were obtained from the Biospecimen Archive Research Core (BARC) at Boston University Medical Center (Boston, MA). The clinical samples were previously tested with the cobas 4800 HPV Test (Roche Diagnostics, Basel, Switzerland) and categorized as positive HPV 16, positive HPV18, or “Other HPV” by the pathology lab. The BD SurePath is a liquid-based pap test that consists of a cervical swab submerged into an alcohol-based SurePath Preservative Fluid (SPPF). Since SPPF contains < 0.2% formaldehyde, a PBS wash is required to remove the formaldehyde prior to DNA extraction [20, 21]. All clinical samples were received by our lab and aliquoted from the original container into 1.7-ml tubes. All tubes were spun down at 13,000 RPM to pellet the sample and the supernatant was discarded. The pellets were then washed in 1 ml of 1× PBS two times, requiring a 15-min spin down at 13,000 RPM after each wash; the supernatant was discarded each time. Lastly, the tubes were numbered and kept in the − 80 freezer until further testing. DNA extractions were performed on prepared tissue samples using a DNeasy Blood and Tissue kit (QIAGEN, Valencia, CA). Sixty-five samples were cobas-negative and 45 were cobas-positive for the L1 HPV 16 loci (see ESM, separate Excel file).
PCR to quantify clinical samples
All extracted clinical samples were tested by quantitative PCR using QuantStudio 5 (Applied Biosystems, Foster City, CA) with a multiplexed HPV 16 and RNase P assay (RNase P as an internal control for human DNA) to quantify the total concentration in copies/μl. Our lab previously designed a primer and probe set for the HPV 16 E7 Gene and RNase P (ESM Table S1) [12]. The RNase P reference plasmid was cloned in-house while initial plasmids were synthesized by GENEWIZ (Cambridge, MA). Each PCR reaction was 25 μl with 1× TaqMan Buffer, 3.5 mM MgCl2, 200 μM dNTPS, 0.1× ROX Reference Dye, 0.025 U/μl Taq DNA polymerase, 5 μl sample or plasmid DNA, and 50 nM primers and 50 nM probes for both HPV 16 and RNase P (ESM Table S1). The following conditions were used for each experiment: initial denaturing at 95 °C for 10 min, 40 cycles of 95 °C for 30 s, 55 °C for 30 s, 60 °C for 1.5 min, ending with a final extension of 60 °C for 5 min. The standard curves were determined by titrated HPV 16 E7 gene and RNase P plasmid DNA stocks using the QuantStudio Design and Analysis Software (Applied Biosystems, Foster City, CA). Triplicates were run for each of the clinical samples. qPCR results and analysis are in the ESM.
LAMP on clinical samples
LAMP was performed on 24 clinical samples in technical quintuplicates with the previously optimized conditions. These 24 clinical samples were selected from 4 pools of post-qPCR-classified samples (true negative, true positive, false negative, false positive groups denoted in ESM Fig. S2). Upon applying the LAMP reaction products onto lateral flow strips, each clinical sample replicate was tagged as “lateral flow strip positive (LP)” or “lateral flow strip negative (LN)” depending on whether both the control and test lines are visible on the strips. Subsequently, 5 μl aliquots of each LAMP technical replicate was loaded onto non-denaturing 12% acrylamide gels. The results were analyzed using a modified binomial logistic regression model in MATLAB. Each sample was categorized based on the number of LP replicates—values ranged from 0 to 5. The detailed data for these samples are located in ESM Fig. S3.
Results and discussion
Post-digest LAMP HPV 16 products are validated by characterization methods that correlate with LFS results
Conventional LAMP characterization during assay validation usually consists of running the undigested dsDNA products on an agarose gel; positive amplification for LAMP is defined as the presence of a smear of banded products as shown by the positive gel lanes. Furthermore, investigators usually include a gene-specific restriction enzyme digest in the characterization process to decrease the false positive rate of the assay. In this work, we include FAM-tagged loop primers in the complete LAMP primer set and use them as location trackers in our gel analysis. In combination with pvuII digestion, the FAM-tagged loop primers allow us to identify the gel bands that are directly associated with the LAMP products that are captured by the anti-FAM test line on the LFS. The analysis of these bands in addition to the elution and analysis of the bound products from the LFS test lines allow confirmation that specific amplification products rather than spurious products are resulting in positive LFS results.
Sequencing post-digest LAMP bands indicates HPV 16-specific products
To validate the specificity of the pvuII digest assay alone, we gel-purified and sequenced 2 selected bands (A and B from Fig. 3a) from a pvuII-digested LAMP reaction. Our sequence analysis suggests that post-digest segments at sizes 146 bp and 92 bp correspond to the two predicted HPV 16-specific LAMP products shown in Fig. 3b. Bands A and B are pvuII digest-specific bands visible in a stained gel.

Sequencing pvuII-digested LAMP amplicons. a PvuII-digested products were run on a non-denaturing gel and bands A and B were extracted. b The schematic shows the corresponding LAMP amplicon diagram for the sequenced bands A and B
The FAM loop primers produce distinct, HPV 16-specific FAM bands that correlates with positive LFS visual readouts
Upon establishing baseline results from the pvuII digest assay, we investigated how the FAM-tagged LF primers are integrated into the HPV 16 LAMP products and whether these FAM-labeled moieties were indeed captured by the anti-FAM antibodies on the LFS test line. To start, we took FAM-labeled pvuII-digested LAMP products and ran a denaturing urea acrylamide gel to dissociate any confounding secondary structures like LAMP-specific dumbbells or extraneous DNA hybridizations associated with unused primer strands. The unstained (no intercalating dyes) gel in Fig. 4, panels a and f, shows two prominent FAM-specific bands in 94–114 bp (band C) and 71–80 bp (band D) after the inclusion of a pvuII restriction digest.

FAM band comparisons of pvuII-digested and undigested lateral flow strip elutions. Three different representations of the LAMP amplicon products are shown. Gel a and b are LAMP amplicons digested with pvuII (independent of LFS). Gels c, d, f, and g are eluted products from the LFS itself. Furthermore, gels c and d contain uncleaved LFS-LAMP products (pvuII−), whereas gels f and g are cleaved LFS-LAMP products (pvuII+). The FAM bands 94–114 (band C) bp and 71–80 bp (b and D) are highlighted as the HPV 16 FAM amplicons. Gel b also shows the previously sequenced bands of 146 (band A) and 92 (band B). The LFS strips that DNA was eluted from are shown in LFS images e and h of Fig. 4. The gels in panels a and b are pvuII-digested LAMP products; the lateral flow strips results are equivalent to the LFS shown in panel h
The data suggest two things: First, our results show that these two FAM bands are correlated with HPV 16-positive samples. Moreover, in panel f, the existence of the two FAM bands is also correlated with a positive lateral flow readout on the anti-FAM test line. Additionally, the LFS are positive regardless of whether or not the LAMP products were cleaved with pvuII (Fig. 4, panel c) and the LFS results are the same (Fig. 4, panels e and h). Combining these two observations, we believe that the FAM bands C and D are a direct visual confirmation that an HPV 16-positive sample leads to a positive test line on the LFS. When we applied the same procedures to BD SurePath samples that were previously ruled qPCR- and LAMP-positive (samples #160, #163, and #167), we observed the two FAM bands C and D. Conversely, for qPCR- and LAMP-negative samples (patients #122 and #126), the FAM bands C and D were absent; instead, the FAM loop primers appear to integrate into the LAMP products at various unpredictable loci. These results highlight the value of using the FAM loop primers as “tracer” molecules that can dramatically enhance the interpretation accuracy of LAMP-dependent lateral flow assays.
To further underline the value of using FAM loop primers during process development for LAMP-LFS assays, we looked at the LAMP products on a denaturing gel using SYBR Gold stain (or equivalently, with ethidium bromide). As expected, although the FAM bands C and D are still visible after SYBR Gold staining, the FAM-specific information is lost (Fig. 4, panels b, d, g); instead, the stain reveals a wider range of different band sizes that tend to occlude the HPV-specific bands that we wish to isolate. In addition, sequenced bands A and B are identifiable on a stained gel which confirms that the pvuII digest bands are unique from the FAM-specific bands C and D and the combination of these bands are present in a positive HPV 16 result. More information on the efficacy of using FAM-tagged loop primers in assays involving clinical samples are discussed in further detail in the ESM.
LFS test line elution confirms FAM-specific LFS-positive results
On our lateral flow strips, the anti-FAM line captures HPV 16-specific amplicons doubly labeled with FAM and biotin, which can bind to streptavidin-coated gold nanoparticles. In a positive test, the test line collects enough streptavidin-labeled gold nanoparticles to produce a concentrated, visible red line. By excising the anti-FAM antibody test line from the strip after running the test (samples were either pre-digested with pvuII or left as is), we were able to elute the FAM-labeled LAMP products from the strip, and look for the signature bands. In Fig. 4, panels f and h, we observed that pvuII-digested LAMP products eluted from the LFS test line possess the same two FAM bands C and D as the pvuII-digested LAMP products that were never run on an LFS (Fig. 4, panel a). Conversely, if the LFS-eluted LAMP products were not treated with pvuII, it is not possible to elucidate both bands C and D to definitively identify a LAMP HPV 16-positive sample via gel (Fig. 4, panels c and d). In the context of clinical samples, these results suggest that a FAM-tagged loop primer can be used as a vehicle to prove, on a molecular level, whether a positive visual readout on the LFS corresponds to a positive HPV 16 sample.
Confirmed proof-of-concept for FAM-specific LFS readout on clinical samples with high sensitivity
To perform a complete analysis of this new characterization method for LAMP, we tested a select set of HPV 16 qPCR-positive and qPCR-negative clinical samples for proof of concept. Each sample was tested in quintuplicate to determine if the sample would be LAMP-LFS negative or LAMP-LFS positive depending on the total number of positive test strips. In our metric, a sample is deemed to be LAMP-LFS positive if the 5 quintuplicate lateral flow strips are positive and every technical replicate has the HPV 16-specific FAM bands C and D on either a non-denaturing acrylamide gel or a denaturing urea acrylamide gel (ESM Fig. S3). When comparing clinical samples (n = 24) assayed with experimental LAMP-LFS versus the gold standard qPCR, the sensitivity is 60% and the specificity is 89% for the LAMP-LFS assay (Table 2). The positive predictive value (PPV) is 90% and the negative predictive value is 57% (NPV). Our characterization method is able to cross-reference FAM gel bands as well as LFS strip results, confirming that a lateral flow strip negative sample has no FAM bands present and vice versa. For instance, although patient samples #122 and #126 (Fig. 4, panel a) both have fluorescent bands present on a urea gel, we are able to confirm that both samples are HPV 16-negative by virtue of qPCR, by the absence of the FAM bands C and D, and by the fact that the LFS test line is negative.
High sensitivity is valued in a point-of-care screening tests for HPV 16, when more specific follow-up tests can be performed to confirm infection. However, specificity is valued to avoid incorrectly administered treatments or procedures, in settings where testing and treatment for suspect cervical lesions are in place [22]. In our small and limited sample set, the LAMP-LFS assay was more specific than sensitive. More robust statistical analysis will require a larger sample set. This small proof-of-concept test on clinical samples reinforces our hypothesis that including a fluorescent primer can aid researchers in developing a robust LAMP assay for HPV 16 by limiting false positive and false negative gel results (compared with a conventional, non-denaturing agarose gel). Assay developers can also leverage the FAM loop primer to expand the understanding of how changes in the loop primer sequence can improve the assay sensitivity and FAM-tagged amplicons that bind to the lateral flow strip.
Addressing post-amplification contamination
For all experiments performed in both the ESM and main text, LAMP and PCR amplicons were tested in various modes, i.e., gels and LFS. The handling of amplicons was performed in a different lab space from where reagents and test strips were prepared prior to amplification. All surfaces and equipment were cleaned with RNase AWAY (Thermo Scientific, Waltham, MA), DNA AWAY (Thermo Scientific, Waltham, MA), and 70% ethanol to eliminate contamination. For instances where these experiments would be performed in a point of care setting, all steps would be automated and contained within an instrument or device to prevent user error and contamination.
Conclusions
We have demonstrated a characterization method to determine the specificity of LAMP-LFS tests by identifying FAM bands specific to HPV 16 and correlating the bands to LFS results. This technique leverages a restriction enzyme-digested band in combination with gel electrophoresis to first identify FAM bands and correlate them with products eluted from the test lines of LFS. Expanding analysis of the post-digest LAMP products, we showed that the integration of FAM-tagged loop primers produces target-specific amplicons that are distinct to the target DNA. The target-specific FAM-tagged products were quantified for both HPV 16 plasmid DNA and HPV 16-positive clinical samples via gel electrophoresis, eliminating false positive gel results. Furthermore, the FAM-tagged products were proven to be the same products bound to the LFS anti-FAM test line by comparing pre- and post-digest LAMP amplicon products extracted from LFS strips. The combined workflow presented here outlines a straightforward characterization strategy to improve LAMP assay development and optimization.
References
Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28(12):E63. https://doi.org/10.1093/nar/28.12.e63.
Zanoli LM, Spoto G. Isothermal amplification methods for the detection of nucleic acids in microfluidic devices. Biosensors (Basel). 2013;3(1):18–43. https://doi.org/10.3390/bios3010018.
Jiang W, Ren Y, Han X, Xue J, Shan T, Chen Z, et al. Recombinase polymerase amplification-lateral flow (RPA-LF) assay combined with immunomagnetic separation for rapid visual detection of Vibrio parahaemolyticus in raw oysters. Anal Bioanal Chem. 2020. https://doi.org/10.1007/s00216-020-02532-9.
Kolm C, Martzy R, Fuhrer M, Mach RL, Krska R, Baumgartner S, et al. Detection of a microbial source tracking marker by isothermal helicase-dependent amplification and a nucleic acid lateral-flow strip test. Sci Rep. 2019;9(1):393. https://doi.org/10.1038/s41598-018-36749-7.
Nagamine K, Hase T, Notomi T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol Cell Probes. 2002;16(3):223–9. https://doi.org/10.1006/mcpr.2002.0415.
Lee D, Shin Y, Chung S, Hwang KS, Yoon DS, Lee JH. Simple and highly sensitive molecular diagnosis of Zika virus by lateral flow assays. Anal Chem. 2016;88(24):12272–8. https://doi.org/10.1021/acs.analchem.6b03460.
Yongkiettrakul S, Kampeera J, Chareanchim W, Rattanajak R, Pornthanakasem W, Kiatpathomchai W, et al. Simple detection of single nucleotide polymorphism in Plasmodium falciparum by SNP-LAMP assay combined with lateral flow dipstick. Parasitol Int. 2017;66(1):964–71. https://doi.org/10.1016/j.parint.2016.10.024.
Huang HL, Zhu P, Zhou CX, He S, Yan XJ. The development of loop-mediated isothermal amplification combined with lateral flow dipstick for detection of Karlodinium veneficum. Harmful Algae. 2017;62:20–9. https://doi.org/10.1016/j.hal.2016.11.022.
Zhang J, Cao J, Zhu M, Xu M, Shi F. Loop-mediated isothermal amplification-lateral-flow dipstick (LAMP-LFD) to detect Mycoplasma ovipneumoniae. World J Microbiol Biotechnol. 2019;35(2):31. https://doi.org/10.1007/s11274-019-2601-5.
Wang Y, Li H, Wang Y, Zhang L, Xu J, Ye C. Loop-mediated isothermal amplification label-based gold nanoparticles lateral flow biosensor for detection of enterococcus faecalis and Staphylococcus aureus. Front Microbiol. 2017;8:192. https://doi.org/10.3389/fmicb.2017.00192.
Mamba TS, Mbae CK, Kinyua J, Mulinge E, Mburugu GN, Njiru ZK. Lateral flow loop-mediated isothermal amplification test with stem primers: detection of Cryptosporidium species in Kenyan children presenting with diarrhea. J Trop Med. 2018;2018:7659730. https://doi.org/10.1155/2018/7659730.
Rodriguez NM, Wong WS, Liu L, Dewar R, Klapperich CM. A fully integrated paperfluidic molecular diagnostic chip for the extraction, amplification, and detection of nucleic acids from clinical samples. Lab Chip. 2016;16(4):753–63. https://doi.org/10.1039/c5lc01392e.
Phillips EA, Moehling TJ, Bhadra S, Ellington AD, Linnes JC. Strand displacement probes combined with isothermal nucleic acid amplification for instrument-free detection from complex samples. Anal Chem. 2018;90(11):6580–6. https://doi.org/10.1021/acs.analchem.8b00269.
Tang R, Yang H, Choi JR, Gong Y, Hu J, Wen T, et al. Paper-based device with on-chip reagent storage for rapid extraction of DNA from biological samples. Microchim Acta. 2017;184(7):2141–50. https://doi.org/10.1007/s00604-017-2225-0.
Chen J, Shao N, Hu J, Li R, Zhu Y, Zhang D, et al. Visual detection of multiple nucleic acids in a capillary array. J Vis Exp. 2017;(129). https://doi.org/10.3791/56597.
Nkouawa A, Sako Y, Nakao M, Nakaya K, Ito A. Loop-mediated isothermal amplification method for differentiation and rapid detection of Taenia species. J Clin Microbiol. 2009;47(1):168–74. https://doi.org/10.1128/JCM.01573-08.
Kong X, Qin W, Huang X, Kong F, Schoen CD, Feng J, et al. Development and application of loop-mediated isothermal amplification (LAMP) for detection of Plasmopara viticola. Sci Rep. 2016;6:28935. https://doi.org/10.1038/srep28935.
Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189(1):12–9. https://doi.org/10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F.
Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clin Sci (Lond). 2006;110(5):525–41. https://doi.org/10.1042/CS20050369.
Krevolin MD, Hardy D, Pane J, Aslam S, Behrens CM. Development and validation of a preanalytic procedure for performing the cobas HPV test in SurePath preservative fluid. J Mol Diagn. 2017;19(2):288–94. https://doi.org/10.1016/j.jmoldx.2016.10.003.
Bjonness-Jacobsen EC, Eriksen AK, Hagen VN, Ostbye KM, Witterso A, Pedersen MK, et al. The effect of the small amount of formaldehyde in the SurePath liquid when establishing protocols for immunocytochemistry. Cytojournal. 2016;13:27. https://doi.org/10.4103/1742-6413.194163.
Kuhn L, Denny L. The time is now to implement HPV testing for primary screening in low resource settings. Prev Med. 2017;98:42–4. https://doi.org/10.1016/j.ypmed.2016.12.030.
Acknowledgments
The authors would like to thank the Clinical & Translation Science Institute’s Study Design and Statistical Analysis Consult group who provided additional insight on the applications of data studies on patient samples. This publication was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through BU-CTSI Grant Number 1UL1TR001430. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Funding
This work was funded by NIH U54 EB015403 and NIH U54 EB015408, both parts of the NIH Point of Care Technology Research Network (https://POCTRN.org). The Boston University Precision Diagnostics Center has funded the analysis of the patient samples
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Statement of ethical approval
All patient samples were acquired through the Biospecimen Archive Research Core (BARC) at Boston University Medical Center. These samples are collected from medical procedures performed at the Boston Medical Center (BMC). The use of the discarded human cervical swab samples was approved by the Boston University Institutional Review Board under protocol #3231.
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Published in the topical collection featuring Female Role Models in Analytical Chemistry.
Rights and permissions
About this article

Cite this article
Landaverde, L., Wong, W., Hernandez, G. et al. Method for the elucidation of LAMP products captured on lateral flow strips in a point of care test for HPV 16. Anal Bioanal Chem 412, 6199–6209 (2020). https://doi.org/10.1007/s00216-020-02702-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-020-02702-9