Article Text

Abstract
The cytokine interleukin (IL)−6 performs a diverse portfolio of functions in normal physiology and disease. These functions extend beyond the typical role for an inflammatory cytokine, and IL-6 often displays hormone-like properties that affect metabolic processes associated with lipid metabolism, insulin resistance, and the neuroendocrine system. Consequently, the biology of IL-6 is complex. Recent advances in the field have led to novel interpretations of how IL-6 delivers immune homeostasis in health and yet drives disease pathology during infection, autoimmunity, and cancer. Various biological drugs that target IL-6 are in clinical practice or emerging in clinical trials and pre-clinical development programmes. The challenge is knowing how and when to apply these therapies. In this review, we will explore the biology behind IL-6 directed therapies and identify some key hurdles for future investigation.
Statistics from Altmetric.com
A panel of international experts in the field of rheumatology recently came together to consider IL-6 biology and the therapeutic targeting of this cytokine in disease.
Considering the biological properties of IL-6
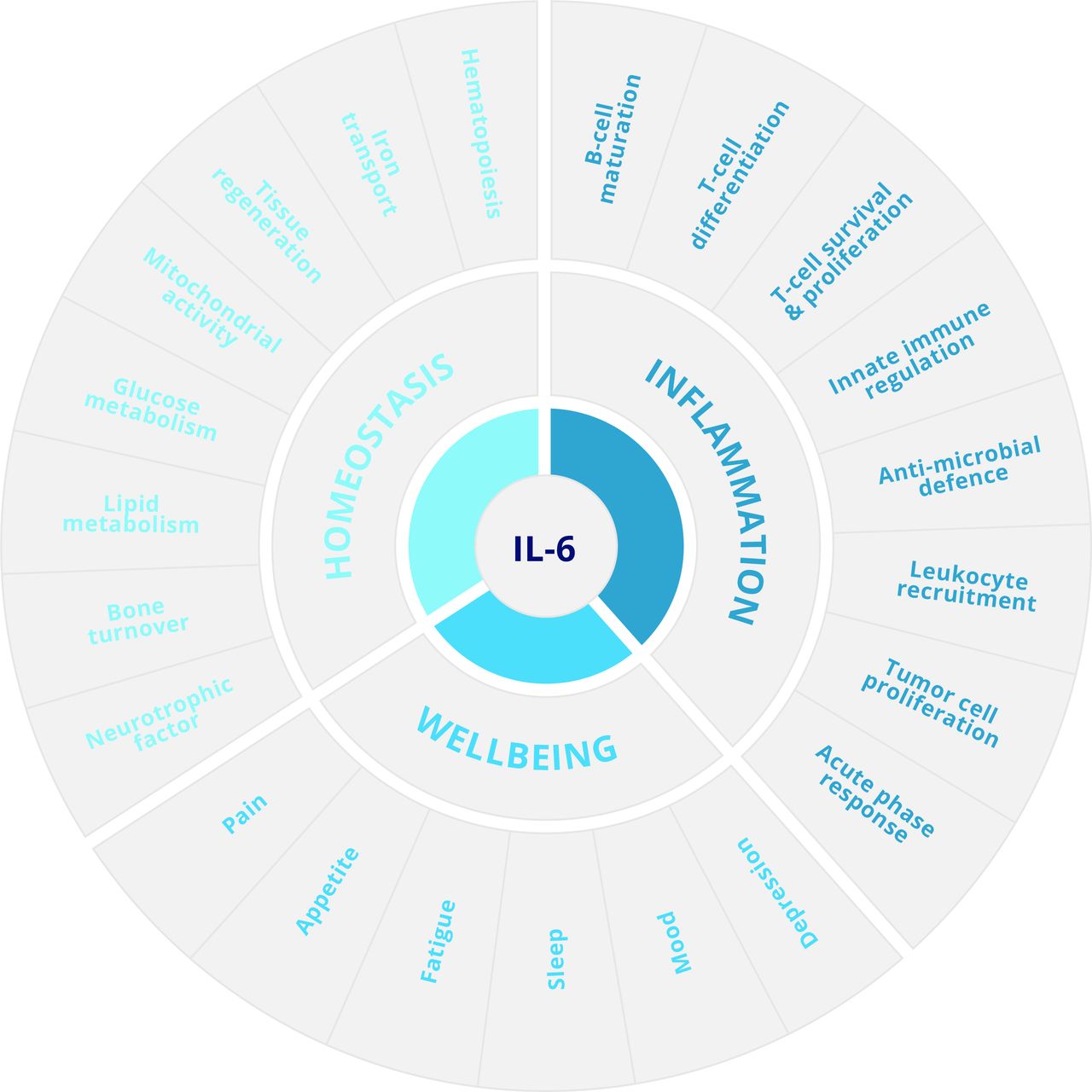
IL-6 is a broad acting cytokine and an important therapeutic target for the treatment of diseases such as inflammatory rheumatic diseases and some cancers. In this regard, IL-6 is characterised by an ability to steer the survival, proliferation, and differentiation of cells and can direct both pro-inflammatory and anti-inflammatory outcomes in infection, autoimmunity, and cancer (figure 1).1–3 For example, studies of acute infection show that IL-6 controls antimicrobial host defence and prevention of excessive tissue injury. However, these same activities also promote the progression and maintenance of chronic inflammation; IL-6 deficient mice are often protected from disease in experimental models of arthritis, lupus, encephalomyelitis, and plasmacytomas. For example, IL-6-deficient mice show limited hallmarks of joint pathology in both antigen-induced and collagen-induced arthritis.4 5 In these inflammatory settings, IL-6 controls leucocyte recruitment, determines the activity and maintenance of the inflammatory infiltrate, and drives various innate and adaptive immune responses.1–3 6–8

The IL-6 portfolio of functions in normal physiology and disease.
IL-6 is recognised as a prominent lymphokine that determines the proliferative survival and effector characteristics of T cells and B cells, but it also controls both adaptive and innate immunity; in fact, IL-6 plays a role in almost every aspect of the innate immune system.1 6 However, perhaps more striking is that IL-6 can elicit functions reminiscent of a hormone, including effects on the normal homeostatic control of vascular function, lipid metabolism, insulin resistance, iron transport, mitochondrial activities, the neuroendocrine system, and neuropsychological behaviour (figure 1).1 9–13 The involvement of IL-6 in these processes is reflected by the impact of IL-6-directed therapies on lipid biosynthesis and anaemia, as well as effects on patients’ well-being including pain, mood, depression, fatigue, and sleep (figure 1).8 11 14–17 IL-6 also plays an important role in maintaining the functional integrity of tissues and organs (such as the liver and mucosal barrier surfaces).18 19 For example, IL-6 is involved in maintaining the epithelial barrier within the gut, and disruption or blockade of IL-6 receptor signalling causes a loss of immune homeostasis and mucosal integrity.19–21 These findings show the importance of IL-6 biology at the barrier surface, and they also help to explain why a previous history of gastric perforations or diverticulitis is considered a contraindication for therapy with tocilizumab. Thus, when dealing with the application of IL-6 targeted interventions it is important to reflect on both the contribution of IL-6 to normal physiology and its involvement in the underlying pathology.
IL-6 signalling
IL-6 biology is highly complex, and understanding how IL-6 receptor signalling can bring about a diverse set of biological outcomes is challenging. In this regard, IL-6 has been reported to use three distinct forms of receptor signalling—classical IL-6 receptor signalling, IL-6 trans-signalling, and a third recently reported mechanism described as IL-6 trans-presentation.1 10 17 22 In the context of this review, we will compare the activities associated with classical IL-6 receptor signalling and IL-6 trans-signalling.
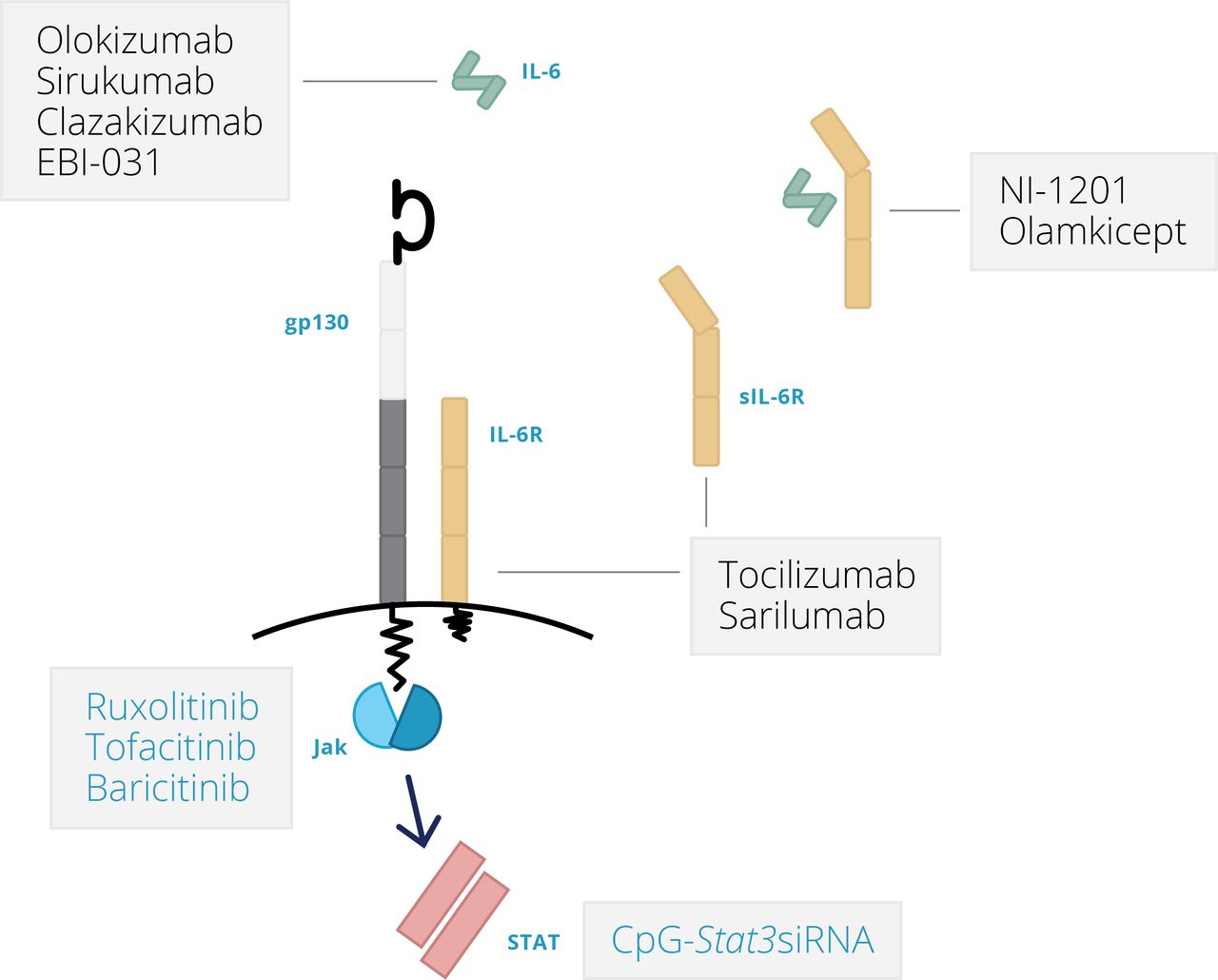
In the classical signalling pathway, IL-6 activates cells by first binding a non-signalling α-receptor (IL-6R; CD126), which, after dimerisation with β-receptor subunit glycoprotein (gp130; also known as CD130), leads to activation of receptor associated kinases within the cell, including the Janus kinases (Jak1, Jak2) and tyrosine kinase 2 (Tyk2) (figure 2) as well as the transcription factors signal transducer and activator of transcription 1 (STAT1) and STAT3 that ultimately bind to promoter regions of responsive genes.9 All cells in the body express gp130. However, membrane-associated IL-6R is only present in a very small number of cell types—namely, leucocyte subsets, hepatocytes, and specialised epithelia, particularly in the gut.1 10 17 23 While classical IL-6 receptor signalling controls some important inflammatory outcomes (for example, the acute phase response and the generation of certain CD4 effector T cells), this mode of IL-6 signalling plays a prominent role in the maintenance of immune homeostasis.

Classical IL-6 receptor signalling and IL-6 trans-signalling pathways.
In contrast, IL-6 trans-signalling is rate limited by the presence of a soluble IL-6 receptor (sIL-6R), which forms a complex with IL-6 that structurally resembles heterodimeric cytokines such as IL-12, IL-23 and IL-27 (figure 2). For example, sIL-6R shares 60% identity with the IL-12p40 subunit.1 In healthy individuals, the concentration of circulating sIL-6R is 25–35 ng/mL.24 25 However, during disease, sIL-6R concentrations may increase by two to threefold, and various inflammatory mediators have been shown to promote the active release of IL-6R from the surface of neutrophils, monocytic cells, lymphocytes, and hepatocytes.1 17 23 26–29
The significance of IL-6 trans-signalling is emphasised by two important features. First, the heterodimeric IL-6/sIL-6R complex is an agonist for cells expressing gp130 and provides a mechanism to explain how cell types that are typically refractory to IL-6 become responsive to IL-6 stimulation. IL-6 trans-signalling, therefore, broadens the types of cells that may elicit IL-6 activities—for example, stromal tissue cells including fibroblasts, endothelial cells, and smooth muscle cells.1 6 17 23 Second, evidence from experimental models increasingly shows that IL-6 trans-signalling enhances IL-6 responsiveness and drives inflammatory events.1 10 17 23 IL-6 trans-signalling is, therefore, considered a primary regulator of IL-6 biology in local pathology. Characterisation of these activities has significantly benefitted from the identification of the soluble gp130 molecule, which circulates at high concentrations in healthy individuals (200–400 ng/mL). This protein antagonises IL-6 activities driven by sIL-6R, leading to competitive inhibition of the IL-6/sIL-6R response, and selectively inhibits IL-6 trans-signalling in both in vitro and in vivo models.1 17 23 An engineered chimeric sgp130 modality is now in clinical trials as olamkicept.
Considering the contribution of IL-6 in inflammation
Physiological concentrations of IL-6 are relatively low (1–5 pg/mL) in human serum. These concentrations are rapidly elevated in response to infection, trauma or injury, and in extreme situations (for example, meningococcal septic shock) may reach quantities in the mg/mL range.30 IL-6 measurements are often better predictors of disease activity than assessments of C reactive protein (CRP).1 Elevated concentrations of IL-6 are, therefore, an early indicator of infection or inflammation and are integral to immunological host defence. Studies in IL-6 deficient mice emphasise that IL-6 is required for antimicrobial host immunity.31 These outcomes occur frequently in humans. For example, children with inhibitory autoantibodies to IL-6 often present with recurrent staphylococcal cellulitis and subcutaneous abscesses.32 However, these same activities often contribute to the maintenance of chronic inflammation, and IL-6 deficient mice show limited hallmarks of pathology in models of arthritis, autoimmune encephalomyelitis, lupus, and other inflammatory disorders.1 IL-6 is, therefore, a key orchestrator of pathology, and these mouse studies have directly contributed to the successful clinical introduction of tocilizumab and the development of other IL-6 directed biological drugs.
How does IL-6 contribute to both local and systemic disease processes? First and foremost, IL-6 is characterised as a lymphokine—a cytokine that affects the survival, proliferation, differentiation, and effector properties of T cells and B cells. These properties often help to distinguish the biological activity of IL-6 from that of tumour necrosis factor (TNF)-α. However, IL-6 is also a potent regulator of innate immune cell activation and a primary activator of the hepatic acute phase response (for example, CRP, serum amyloid A, fibrinogen, and thrombopoietin).11 18 33 Indeed, IL-6 is also considered an adipokine and myokine with influences on both adipose tissues and muscle. Structural cells within the inflamed synovium of patients with rheumatoid arthritis typically lack IL-6R expression. These cells express gp130, and regulation of IL-6 trans-signalling within the inflamed joint contributes to chemokine directed leucocyte recruitment, the retention and activation of the inflammatory infiltrate, the transcriptional activation of matrix metalloproteases linked to cartilage damage, and effects in bone resorption.17 33 For example, analysis of synovial fluid from patients with rheumatoid arthritis shows an increase in sIL-6R that correlates with leucocyte infiltration and more advanced stages of disease activity.34 Mouse models have shown that the addition of sgp130 into an inflamed joint significantly reduces the degree of inflammation and associated pain, suggesting that IL-6 trans-signalling is responsible for driving local disease outcome.34–37
IL-6 receptor signalling promotes activation of the Jak-STAT pathway.9 38 This pathway has evolved to sense and interpret environmental signals that promote cellular growth and functional identity.39 Many of the responses attributed to IL-6 are controlled by the latent transcription factors STAT1 and STAT3.1 6 17 23 For example, genetic studies in mice show that STAT1 and STAT3 activities determine the severity and duration of experimental arthritis.17 36 40 The relevance of the Jak-STAT pathway (and potentially IL-6 biology) to rheumatoid arthritis is perhaps further illustrated by genome-wide association studies.41–43 For example, around 20% of gene variants predisposing a genetic susceptibility to rheumatoid arthritis are linked to IL-6 or the Jak-STAT pathway, either directly in the signalling cascade (for example, Tyk2, IL-6ST, IL-6R), through genes transcriptionally controlled by IL-6 (such as IL-21 and IL-2RB) or responsible for IL-6 expression (such as IRF5 and IRF8). Computational network analysis of genes with genetic risk alleles linked with rheumatoid arthritis identifies STAT1 activity as an important feature of disease development.42 This information offers us opportunities to think about the way IL-6 controls the pattern of inflammation and its potential contribution to the heterogeneity of joint pathology seen clinically.33 A more detailed understanding of disease activity will ultimately improve the diagnosis and stratification of patients and inform the most appropriate course of therapy.
Although the clinical introduction of tocilizumab as a frontline biological drug for the treatment of rheumatoid arthritis illustrates the significance of IL-6 in this disease, this therapy has been less impressive in most other prevalent autoimmune diseases.1 44 This does not mean that IL-6 does not contribute to these diseases. Instead, the failure of IL-6 receptor inhibition to improve clinical outcomes in these conditions often reflects the contribution of IL-6 to immune homeostasis and normal tissue physiology.1 33 44 These clinical observations emphasise the heterogeneity of the inflammatory response and imply that the activity of some other cytokines (for example, IL-1, IL-17, IL-23, and TNFα) maintains the cytokine network at these inflammatory sites.44 Understanding how IL-6 contributes to the maintenance of immune homeostasis, yet promotes chronic disease progression, will ultimately help to identify how and when IL-6 directed interventions may best be applied.
The role of IL-6 in chronic disease progression
Initial studies of cytokine targeted therapies suggested that diseases sharing common mechanisms of pathophysiology might respond to a variety of biological interventions, but with varying efficacy. Clinical experience with TNF blockers shows that TNFα inhibition is effective in many different diseases including ankylosing spondylitis, juvenile idiopathic arthritis, psoriasis, and psoriatic arthritis. However, recent trials show that blockade of IL-23 (for ankylosing spondylitis), IL-6 (for juvenile idiopathic arthritis), and IL-17 (for psoriasis) may represent viable alternate intervention strategies for the treatment of these disorders.3 33 These findings illustrate the context dependent nature of inflammatory reactions and support the presence of a cytokine hierarchy that is specific to a defined disease process.1 3 33 44 In this regard, IL-6 receptor inhibition has shown excellent efficacy in rheumatoid arthritis but has had limited success in diseases in which IL-6 contributes to epithelial homeostasis or the maintenance of barrier integrity.1 20 This is reflected by the adverse incidence of gastric perforations and associated diverticulitis as a response to IL-6 inhibition. The involvement of IL-6 at epithelial barriers is further illustrated by its role in antimicrobial host defence.1 17 32 45 Here, clinical trials of tocilizumab in atopic dermatitis showed that disruption of IL-6 signalling improved the development of skin lesions but caused an adverse incidence of bacterial superinfections.45 These outcomes were not unexpected and could be predicted on the basis of the biology of IL-6 in viral, parasitic, and bacterial infection.1 In this regard, it should be noted that an ancestral IL-6-like system in fruit flies promotes innate immunity in response to bacterial infection.1 This illustrates the importance of understanding fundamental cytokine biology before starting a clinical study.
The current status of IL-6 blocking therapies
Structure-function studies have provided valuable insight into amino acid residues and sequences responsible for IL-6 docking to its cognate IL-6R and signal transducing receptor gp130.9 46 47 These interactions are complex, and a fully functioning IL-6 receptor consists of a hexameric structure involving two molecules of IL-6, two molecules of IL-6R, and two molecules of gp130. Ultimately, this has led to the pharmacological design of IL-6 directed therapies that prevent IL-6 binding to IL-6R (tocilizumab and sarilumab, targeting IL-6R; sirukumab and clazakizumab, targeting IL-6), IL-6 engagement with gp130 (olokizumab and EBI-029, targeting IL-6), and others that bind unique motifs potentially linked to the regulation of IL-6 trans-signalling (olamkicept—a chimeric engineered sgp130Fc fusion protein; NI-1201, targeting IL-6R) (figure 3).1 17 Following a negative review from the FDA, sirukimab development and registration has been stopped. It should also be noted that Jak inhibitors (such as tofacitinib, ruxolitinib, and baricitinib) also inhibit aspects of IL-6 signalling, but these are less specific for IL-6 and target other cytokines that use the Jak-STAT pathway (figure 3).1 48 49 With the advent of other biological drugs (for example, tildrakizumab against IL-23, NNC01140006 against IL-21) and small molecule inhibitors (such as Jak inhibitors) that target similar downstream signalling pathways to IL-6, differentiating between the modes of action of these drugs has become essential. To do this, we must answer the following questions: which clinical indications are likely to benefit from IL-6 inhibition; is there a therapeutic advantage to blocking IL-6 versus IL-6R in certain conditions; and can IL-6 trans-signalling be targeted therapeutically in human disease, and does this strategy reduce the incidence of contraindications potentially associated with classical IL-6 receptor signalling? These questions deserve to be considered in two ways. First, the efficacy of responses to these drugs needs to be tested across various inflammatory settings using ‘umbrella’ or ‘bucket’ trial designs. Second, similar investigations need to be conducted in defined patient groups in which an individual disease process may be subdivided into distinct pathotypes that reflect differences in local inflammation and responses to therapy. In both instances, we need to predict the course of pathology in patients whose disease is undifferentiated at presentation and to identify the major contributing pathways that drive disease progression in individual patients.
{kind=link}
{kind=link}
{kind=link}
Therapies directed against IL-6 or IL-6R.
Considering the biology behind IL-6 therapy—A Summary
For a cytokine that was first described more than 30 years ago, it is amazing that we continue to acquire new insights into the workings of IL-6. It signals through a variety of mechanisms, mediates many inflammatory processes and homeostatic events, and is tightly regulated at the transcriptional (for example, microRNAs, RNA binding proteins, circadian control factors) and protein level (for example, IL-6R shedding, sgp130 antagonism) through mechanisms that either inhibit or promote its action.1 17 The success of IL-6 targeted therapies in certain clinical conditions illustrates the prominent role of IL-6 in infection and disease. Imitation is often the sincerest form of flattery, and viruses such as human herpesvirus eight and human cytomegalovirus have both evolved elegant ways to decoy IL-6 responses.50 51 These forms of viral mimicry and adaptation contribute to the persona of IL-6 and emphasise its functional importance in controlling host immunity.
Pharmaceutical companies and researchers have developed an array of new strategies to block this important cytokine. These drugs often display unique modes of IL-6 or IL-6R inhibition, and it is hoped that these therapies will display unique efficacies in different clinical settings. For example, will these alternative interventions limit the development of adverse effects associated with IL-6R blockade in some indications? The ability to apply approaches and technologies from other clinical specialties, such as oncology and genetic screening services, offer opportunities to establish how IL-6 sits within a particular cytokine network specific to a disease process or pathology. In this regard, we will increasingly see the application of systems based methods to obtain a more holistic view of drug action in different disease settings.52 Insight derived from these approaches will provide valuable bioinformatic datasets and biomarkers relevant to the design and interpretation of future clinical trials.
References
Footnotes
Funding This initiative is sponsored by R-Pharm through the provision of an unrestricted educational grant. R-Pharm has had no influence over the content.
Competing interests DA declares no conflicts. EHC reports grants from NovImmune AG, grants and personal fees from Pfizer, grants from UCB, grants and personal fees from Roche, personal fees from Abbvie, Biogen, Bristol Myer Scripps, Chugai Pharma, Eli Lilly, Hospira, Janssen, Novartis, Regeneron, R-Pharm and Sanofi-Aventis; SAJ reports non-financial support from CESAS Medical during the conduct of the study; personal fees from CESAS Medical, Eleven Biotherapeutics, grants and personal fees from Roche Pharmaceuticals, personal fees and other from Genentech, grants and personal fees from Glaxo-Smith-Kline, Chugai Pharmaceuticals, Ferrin Pharmaceuticals, Regeneron/Sanofi, grants, personal fees and non-financial support from NovImmune AG, outside the submitted work; IM reports grants from Roche and Refereron, during the conduct of the study; grants and personal fees from Abbvie, Astra Zeneca, Celgene, GSK, Janssen, Lilly, Novartis, Pfizer, Roche, grants and personal fees from Refereron, outside the submitted work; JS reports grants from Abbvie, Astra-Zeneca, Janssen, Lilly, MSD, Pfizer, Roche, personal fees from Abbvie, Amgen, Astra-Zeneca, Astro, BMS, Celgene, Celltrion, Chugai, Gilead, Glaxo, ILTOO, Janssen, Lilly, Medimmune, MSD, Novartis-Sandoz, Pfizer, Roche, Samsung, Sanofi, UCB during the conduct of the study; TT reports grants, personal fees and other from Astellas Pharma Inc, Abbvie GK, Mitsubishi Tanabe Pharma Co, grants and personal fees from Bristol–Myers KK, Chugai Pharmaceutical Co Ltd, Daiichi Sankyo Co, Ltd, Pfizer Japan Inc, grants from Takeda Pharmaceutical Co, Ltd, Teijin Pharma Ltd, Asahikasei Pharma Corp, Eisai Co Ltd, AYUMI Pharmaceutical Corporation, grants and other from Taisho Toyama Pharmaceutical Co Ltd, Nipponkayaku Co Ltd, other from Astra Zeneca KK., Eli Lilly Japan KK, Novartis Pharma KK, Janssen Pharmaceutical KK, outside the submitted work; The participants/authors received personal fees for their participation in the round table.
Provenance and peer review Commissioned; externally peer reviewed.
Correction notice Minor typographical errors have been corrected since first published.