Theoretical Insights into the Structure of the Aminotris(Methylenephosphonic Acid) (ATMP) Anion: A Possible Partner for Conducting Ionic Media



1
Chemistry Department, University of Rome “La Sapienza”, Piazzale Aldo Moro 5, 00185 Rome, Italy
2
School of Biological and Chemical Science, Queen Mary, University of London, London E1 4NS, UK
3
Department of Chemistry, McMaster University, Hamilton, ON L8S 4M1, Canada
4
Department of Chemistry, The University of Hong Kong, Hong Kong, China
*
Author to whom correspondence should be addressed.
Symmetry 2020, 12(6), 920; https://doi.org/10.3390/sym12060920
Submission received: 5 May 2020
/
Revised: 21 May 2020
/
Accepted: 27 May 2020
/
Published: 2 June 2020
(This article belongs to the Special Issue Materials Science: Synthesis, Structure, Properties)
Abstract
:We present a computational characterisation of Aminotris(methylenephosphonic acid) (ATMP) and its potential use as an anionic partner for conductive ionic liquids (ILs). We argue that for an IL to be a good candidate for a conducting medium, two conditions must be fulfilled: (i) the charge must be transported by light carriers; and (ii) the system must maintain a high degree of ionisation. The result trends presented herein show that there are molecular ion combinations that do comply with these two criteria, regardless of the specific system used. ATMP is a symmetric molecule with a total of six protons. In the bulk phase, breaking the symmetry of the fully protonated state and creating singly and doubly charged anions induces proton transfer mechanisms. To demonstrate this, we used molecular dynamics (MD) simulations employing a variable topology approach based on the reasonably reliable semiempirical density functional tight binding (DFTB) evaluation of the atomic forces. We show that, by choosing common and economical starting compounds, we can devise a viable prototype for a highly conductive medium where charge transfer is achieved by proton motion.
1. Introduction
Ionic liquids (ILs) can be defined as salts with low melting or glass transition points which display some or all of the following functional physicochemical properties: solvation capabilities, ionic conductivity, low vapour pressure and high thermal stability [1,2]. For a more in-depth discussion of which properties actually define an ionic liquid see ref. [2]. The wide applicability of these liquids in chemistry, industry and technology has generated expectations in technological developments [3,4,5]. Of particular interest is the chemical “tunability” of the component cationic and anionic moieties which permit the task-specific rational modulation of ILs properties for specific applications.
Despite being pure electrolytes, ILs are not as good conductors as expected. Charge transport is mainly achieved by ion drift, whilst conductivity is limited by the high viscosities which, in turn, depend on the strong cohesive forces that characterise a molten salt. To promote efficient charge transport, light charge carriers such as protons should be embedded in the liquid.
In this context, the special class of materials known as protic ionic liquids (PILs) are particularly relevant as, differing from other ILs, they exist in ionised form due to extant proton transfer between the acid and base components. It is precisely the presence of proton transfer equilibria that makes these substances viable candidates to optimise charge transfer phenomena towards the delivery of novel paradigms for highly conductive mediums. While PILs lack the extreme chemical and thermal stability of some of the traditional ILs, they have attracted attention because of their peculiar solvation abilities that make them ideally suited to substitute water and other protic solvents, particularly in biological applications [6]; including in the war on antibiotic resistance [7]. PILs are a very complex chemical species and depending on the structures of the constituent molecular ions, they can show a wide range of peculiar structural patterns from aggregation phenomena [8,9] to intricate hydrogen bonding (H-bonding) networks [10,11] which, in turn, can guide surface effects [12] and phase separations [13]. It therefore follows that the study of their molecular structures is essential to understand, at least in part, their bulk properties. Among the latter the aforementioned ionic diffusion/conduction is particularly interesting for applications in electrochemistry where these substances could potentially act as dry proton conducting prototypes [14,15,16,17,18,19,20].
One of the principal challenges in delivering efficiently functionalised PILs is that the degree of ionisation is largely dependent on the ΔpKa between the acid and the conjugate acid of the base. Large ΔpKa values, substantiating complete ionisation, lead to poorly conducting materials (as there would effectively be no ‘free’ protons), whereas low ΔpKa and hence partial ionisation, may result in higher conductivities, yet concomitant with the partial loss of the IL properties due to the existence of a neutral component that can lead to phase separation and evaporation [21,22].
In the pursuit of prototype materials that may overcome these problems and therefore present (i) a high protonic mobility while (ii) retaining a fully ionised state, we have extensively examined a homologous series of compounds made up of a single deprotonated amino acid anion and a choline cation [23,24,25]. We indeed found that the amino acid anions with a protic side chain are able to provide a source of mobile protons, while remaining ionised. The “trick” is due to the fact that proton transfer takes place exclusively between anions and that cations do not directly participate in these protic relocations, thus ensuring the liquid remains (at least predominantly) ionised due to the lack of mutual neutralisation reactions. The anions, in this case, must possess structural features such as to allow themselves to interact via H-bonds strongly enough to overcome the Coulombic repulsion [26]. Unfortunately, PILs based on amino acid anions are rich in amino groups that act as proton scavengers, therefore inducing the transformation of the anions into zwitterionic structures which have been found to be stable enough to quench proton mobility. In other words, the natural tendency of the amino acid anions to form zwitterionic species limits the practical applicability of these substances as proton conductors with, perhaps, the notable exception of phospho-amino acids [27].
As a result of these considerations, in our quest for having a highly conductive ionic liquid, we identified a novel system which suits the two above criteria. The aim of this paper was to present insights of a prototype material based on singly and doubly deprotonated Aminotris(methylenephosphonic acid) (ATMP) and the 1-ethyl-3-methylimidazole (EMIM) cation (Figure 1), employing ab initio electronic structure computations and semiempirical molecular dynamics (MD) to resolve their structural and dynamic characteristics.
Neat liquid phosphoric acid (H3PO4) has the highest intrinsic proton conductivity of any known substance and we believe this behaviour could be replicated using ATMP [28]. The presence of three phosphonic acid chains in ATMP makes this a unique system which possesses a large number of diverse hydrogen bonding (H-bonding) acceptor and donor terminals, which can drive proton motion. Moreover, it lacks any basic group, hence the negligible quenching of proton mobility is expected, as manifested in amino acid-based PILs.
The choice of EMIM as the pairing cation originated from the fact that imidazolium-based ILs have been comprehensively characterised in the literature with favourable properties such as stability and a lack of basicity [29,30,31]. The singly deprotonated ATMP represents, as far as we know, a novel anion component for an IL. ATMP in its completely deprotonated form has been utilised on only one occasion as an IL component of a hybrid catalytic nanomaterial to reduce nitro-anilines [32]. ATMP has also been employed as part of a protic ionic solid to capture protons in an anhydrous proton conducting material [33]. Lastly, ATMP was incorporated in an innovative hybrid ion-exchange material as a cation exchanger [34]. The fact that ATMP has had a role in ion capture and exchange suggests there is potential for this system to serve as an anhydrous fast charge carrier.
In this work, we examined the structure of [ATMP][EMIM] with emphasis on the short-range order using newly computed ab initio data. We also present novel MD simulations of clusters of growing sizes as well as of the bulk phase through an efficient quantum chemical semiempirical method: density functional tight binding (DFTB).
The results demonstrate the favourable formation of stable ATMP anionic clusters, promoted by multiple stabilising and directing H-bonds manifested by the three phosphonic acid side chains. This H-bond network facilitates both intra- and intermolecular proton transfers. The ATMP anions tend to aggregate and our findings support the increasing evidence that some ILs show ‘like charge’ aggregation phenomena [35,36,37]. Herein we attempt to unravel the structural behaviour of this prototypical system with focus on determining the reversibility of the proton ‘jumps’ and to establish the potential of analogous (real) materials as alternative means to achieviing dry proton conduction in highly ionised mediums.
While it is very likely that this compound can be easily synthetised by a salt exchange reaction, the actual bulk properties of the real material are to date unknown. It is known that the anion can act as an ionic partner of a room temperature ionic liquid (RTIL) [32]. In principle, a suitable choice of the cation (or a mixture of cations) would ultimately lead to an ionic medium with the desired melting point. As we show herein, the possible conductive properties of the system are entirely due to the anions, with the cations playing a spectator role. The aim of this study was to show that by a suitable choice of the anion we can produce a proton conducting, highly ionised medium without the onset of neutral components. Even in the unlikely case that the system was not liquid under all circumstances, the results herein presented remain relevant, as they reveal the molecular mechanisms which are the basis of the conductive properties of well known ionic solid phases such as those previously reported [33].
2. Computational Methods
Initial ab initio electronic structure calculations were performed on the separate EMIM cation and ATMP anion to determine the stable geometric structures of the ions. This was followed by calculations on the isolated dimers consisting of cation–anion and anion–anion combinations with the ATMP anions in differing deprotonation states, to characterise the following dimeric combinations: [EMIM]+[ATMP]−, [EMIM]+[ATMP]2−, [ATMP]−[ATMP]−, [ATMP]2− [ATMP]2−, [ATMP]−[ATMP]2− and [ATMP][ATMP]2− (i.e., cation–anion, cation–dianion, anion–anion, dianion–dianion, anion–dianion and neutral–dianion). The computations were carried out both in the gas phase and in a self consistent reaction field (SCRF) to mimic the dielectric effects of the solvent, using the polarizable continuum solvent model (PCM) [38]. The latter was employed to introduce an environmental dielectric screening to account for the fact that the dimers with large charge separations were unstable in vacuum. As the dielectric properties of the material were unknown at that moment and that, even for similar well known ILs the measurement of dielectric properties was still missing [2], the model solvent used was acetonitrile (ε = 35.7) as its dielectric constant has the typical values of many PILs [39,40].
Four different conformers for each of the dimers above were optimised and the lowest energy one was chosen as representative of the minima structure (see Section S1 of the supporting information for further details).
A full unconstrained optimisation was performed for the monomeric and the lowest energy dimeric structures followed by the evaluation of the harmonic frequencies to determine the approximate thermodynamics. We used the D3-B3LYP [41] functional with the 6-311+G(d,p) basis set [42] which was suitable for the study of PILs based on phosphorylated species. The Gaussian16 programme package [43] was utilised for all the ab initio calculations.
The study of larger ionic aggregates and of the bulk phase was achieved by MD based on DFTB, a semiempirical, quantum chemical method with an accuracy comparable to density functional theory (DFT), albeit with the added advantage of being less computationally demanding. DFTB has been previously used with success to simulate the structure and properties of ILs, as well as proton transfer capabilities [44,45,46,47]. A validation of the DFTB approach is presented in Section S1 of the supporting information, where we show how it was able to reproduce the dimer association energies computed with the DFT (B3LYP) method with sufficient accuracy.
We made use of the full DFTB3 method which was the latest extension that included the third-order effects [48] in the exchange–correlation potential expansion, facilitating an accurate treatment of H-bonding. Furthermore, this model improved the description of ions pairs as the Coulomb interaction parameter on the surrounding charges was accounted for. DFTB simulations were performed using the 3-ob Slater–Koster parameter set, whilst concurrently estimating dispersion forces through implementation in the Slater–Kirkwood polarizable atom model. The DFTB simulations were carried out on isolated clusters of the following differing sizes: 4 ionic pairs for 250 ps and 6 ionic pairs for 100 ps. Another DFTB simulation was performed with NVT periodic boundary conditions with density set to 1.20 g/cm3 consisting of 9 ionic pairs for ~50 ps with a 1 fs timestep. A Nosé–Hoover thermostat was employed with a constant temperature set at 300 K for all simulations.
In all the above simulations, initially, the ATMP anions were singly deprotonated resulting in a proton to ATMP ratio of 5:1. In principle ATMP can also act as a component of an ionic medium in its doubly and triply deprotonated forms giving rise to a proton:ATMP ratio of 4:1 and 3:1. The latter however is an anion unlikely to lose its remaining protons given that the second pKa of the phosphoric group is 7.2. Hence, the triply deprotonated form can hardly be the component of a conductive medium. The doubly deprotonated form, instead, still has a mobile proton on one of the phosphonic groups and therefore we set up an additional simulation of a neutral cluster containing three ATMP dianions coupled with 6 cations. The simulation was carried out as detailed above for 300 ps.
The advantage of using a semiempirical method was twofold in this context: (i) computational efficiency is very high thus we can explore timescales much longer than single proton transfer events and (ii) variable chemical topology (i.e., bond breaking and forming) is naturally accounted by the evaluation of the gradient of the electronic energy (the interatomic forces). Another option for tackling the simulation of the systems where chemical bonds are free to break/form is ab initio molecular dynamics (AIMD) which provide the advantage of computing the ‘exact’ nuclear forces from the electronic structure. However, this ‘rigorous’ approach is extremely costly. Our intention here was to reduce the computational cost thus extending the current limits of the simulation to timescales that can adequately sample proton transfer events.
3. Results and Discussion
3.1. Ab Initio Dimer Computations
It is understood from earlier studies [24] that some of the bulk properties of PILs share a correlation with the ability of the molecular ions to form H-bonds. The presence of both phosphonic and phosphate groups on a singly deprotonated ATMP ion suggests that the five H-bonding donors can contribute to a stable cooperative network of anionic dimers and oligomers, along with the deprotonated oxygen which acts as a principal H-bond acceptor. It is expected that tautomeric reactions can arise within the same anion as well as the inter-anion proton transfer processes due to the acidic functionalities.
We initiated our computational assessment by determining the enthalpies associated with differing ionic pairs that can be formed in the bulk system in both gas- and solvated-phases. Such resolution of the structural features in isolated environs assists in the understanding of the atomistic and dynamical behaviours of these systems. A conformational analysis for the various dimer variants was performed and its results are presented in Section S1 of the supporting information. Four different possible minimum structures were found for each dimer variant. Here we shall present only the results for the lowest energy minima.
Relevant energetic trends are listed in Table 1 which contains the association enthalpies of the following dimers: cation–anion, cation–dianion, anion–anion, dianion–dianion, anion–dianion and neutral–dianion. From the frequency calculations, each structure reported here was confirmed as a minimum; even those referring to a like charged dimer in the absence of a solvent model. This fact tells us that the comprising H-bonds are strong enough to bind these structures in a metastable local minimum with a positive energy with respect to the bimolecular dissociation limit.
The optimised geometries of the oppositely charged ionic pairs [EMIM]+[ATMP]− and [EMIM]+[ATMP]2− are shown in Figure 2. They display a relatively weak H-bond that tethers one of the imidazole hydrogens to the deprotonated oxygen of ATMP. The acceptor/donor H—bond angles are moderately obtuse at 112° and 130°, as typical of weak H-bonds, for [EMIM]+[ATMP]− and [EMIM]+[ATMP]2− respectively. As expected, we observe that [EMIM]+[ATMP]−, in a model solvent, has a smaller binding energy (~23 kcal/mol) than does its cation-dianion complement [EMIM]+[ATMP]2− (~30 kcal/mol).
The stable geometries of the anionic dimers of ATMP are reported in Figure 3. The like charged complexes have association energies which are larger than their respective oppositely charged pairs, even the [ATMP]2−[ATMP]2− structure. The presence of strong inter-anion H-bonds stabilises like charge complexes to such an extent that they overcome the Coulombic-like charge repulsion. The main H-bonding motif, as expected, is characterised by the appearance of PO–H–OP interactions in various charged states as shown in Figure 3. These H-bonds are much more robust than those between anions and cations and show angles very near 180°, specifically 171°, 161°, 172° & 175° for [ATMP]−[ATMP]−, [ATMP]2−[ATMP]2− and [ATMP]−[ATMP]2−, respectively.
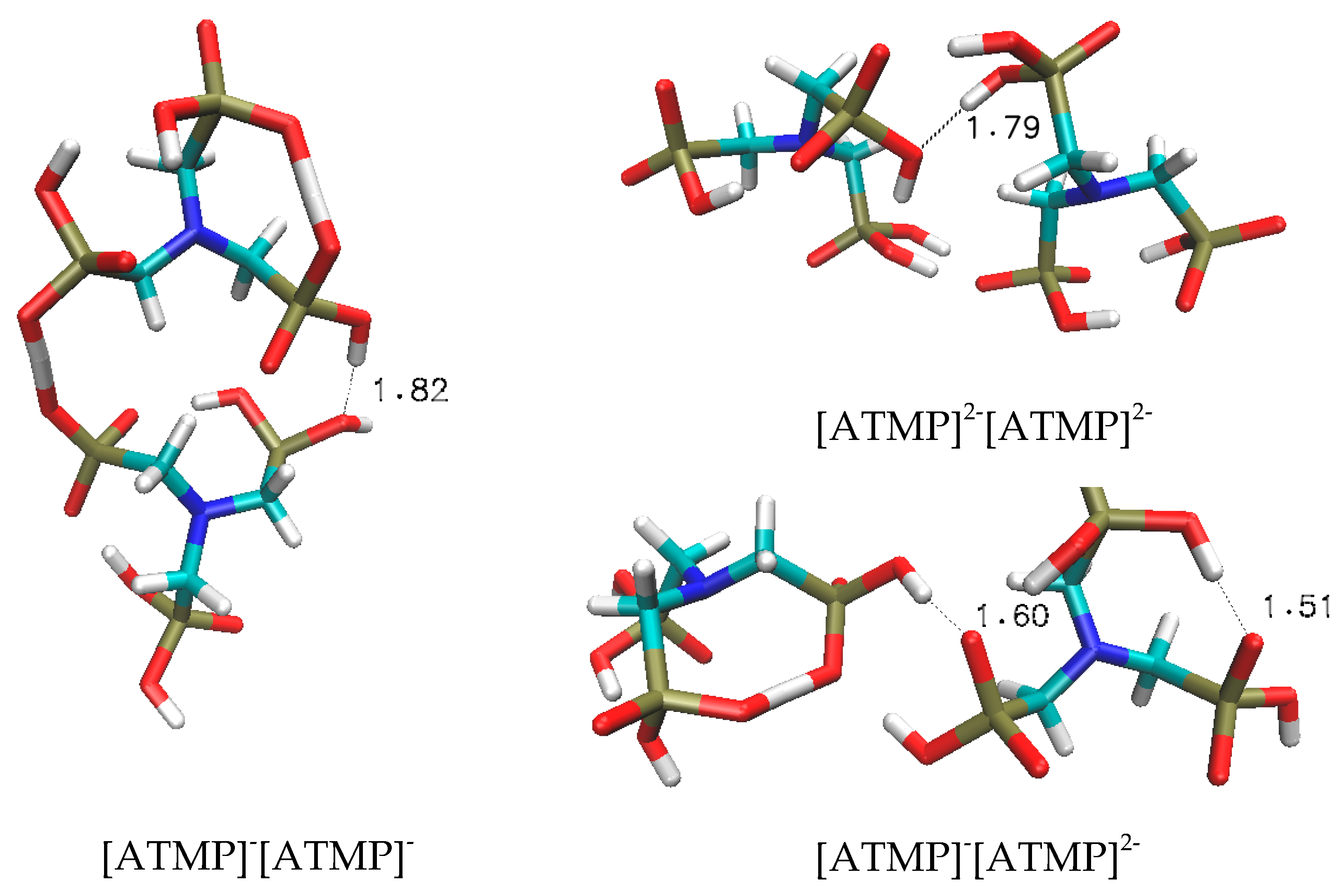
The [ATMP]−[ATMP]− dimer displays an association enthalpy of −37.6 kcal/mol, proving to be the most stable among the analysed complexes; this is exceptional considering that it is a doubly charged species. More impacting on convention is the −36.6 kcal/mol stabilisation of the triply charged [ATMP]−[ATMP]2− dimer and the −34.6 kcal/mol of the quadruply charged variant. Such pronounced associative stabilisation suggests that these dimers may also exist in the bulk phase, with dissociative electrostatic repulsion being overcome by the robust H-bonding.
These relatively simple ab initio electronic structure determinations provide strong quantitative evidence that the anions of ATMP tend to cluster, providing a route to proton transfer. However, one must be cautious in the extrapolation of such trends arising from isolated dimeric structures to bulk ensembles. Therein, numerous proximate ionic pairs modulate structural isomerism and together with environmental effects perturb the overall dimeric association. Even though we presented results based on the use of an implicit solvent treatment to mimic the bulk solvation of the isolated pairs, it remains an imprecise representation of the solvent effect, particularly due to its failure to account for directional solute–solvent interactions such as H-bonding. Towards remedying this shortcoming, we combined the precision of these ab initio determinations, with a set of bulk simulations that we will explore in the next sections.
3.2. Cluster MD Simulations: Four Ionic Pairs Cluster
To map the isolated calculations to the bulk character of the [ATMP]−[EMIM]+ material, we begin by presenting results from the MD simulation on an isolated cluster of four ionic pairs, that represents the first step in increasing the system size. The large part of the interaction between the [EMIM]+ and [ATMP]− ions was due to electrostatics with the minor addition of a stabilising H-bond between the slightly acidic imidazole ring hydrogens [49,50] and the oxygen atoms on the phosphonic acid groups. The short-range structure that results from these interactions is shown in Figure 4 where we report various O–H radial distribution functions (RDFs). The black line is the anion–cation O–H distance. The maximum of this RDF is around 2 Å and its overall shape and the lack of sharp structures indicates a weakly coordinating interaction.
Much sharper structures can be seen in the RDFs relative to the other H-bonds which are formed between anions (red line) and within a single anion (green line). The red line represents the O–H distance distribution between the (initially) deprotonated oxygen on a given anion and the protons (initially) on the other ones. The green line was computed in the same way, but within a single anion. The presence of a strong peak between 0.9 and 1.1 Å in both the anionic RDFs suggests that ATMP protons, can not only strongly interact with the deprotonated oxygen, but that, in several instances, they have migrated onto it (i.e., at covalent bonding distances). Hence, this evidences the formation of novel O–H bonds in the anions due to the migration of protons. This negatively charged oxygen atom is pivotal in initiating a cascade of proton jumps as it is the sole proton-deficient site.
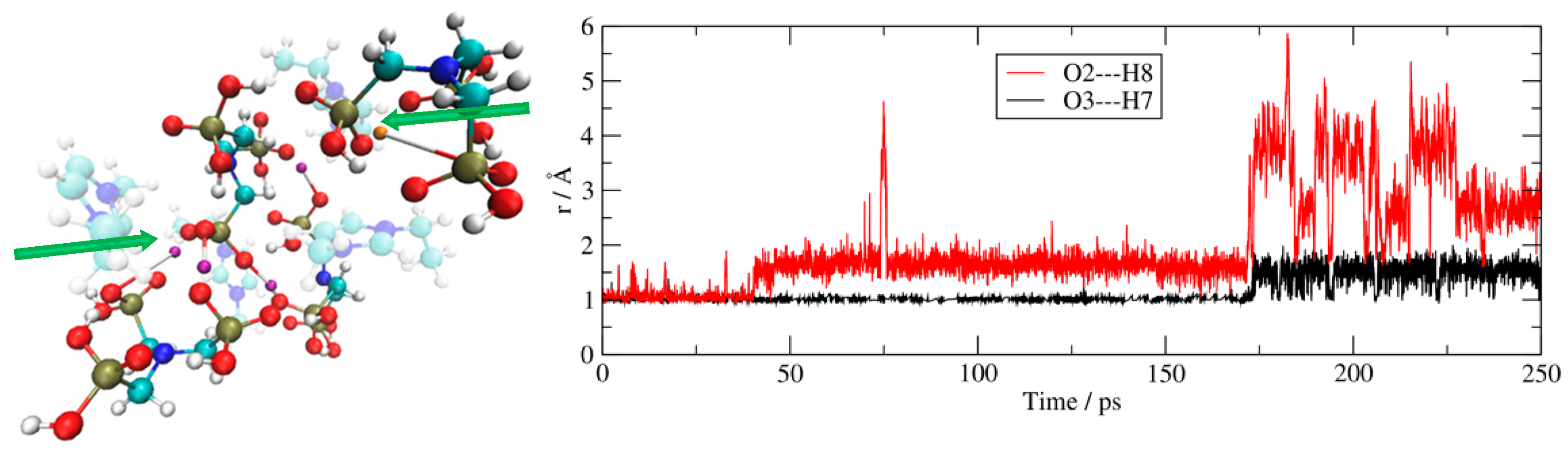
Further evidence is presented in Figure 5 (right panel) which shows two selected O–H bond distances as a function of simulation time. It is evident that both the tracked protons are abandoning the anion. One of the two distances (the red line) represents an initially O–H bound proton, dissociating to distances as great as 6 Å, whilst the other (the black line) represents a proton that migrates onto an acceptor oxygen, albeit remaining strongly bound to the donor via an H-bond. These proton transfers are essentially energy-resonant processes due to the correspondent protic functionality being the same, thus leading to a negligible energetic variation upon the proton movement.
The final structural snapshot of this cluster at the end of the MD trajectory is shown in the left panel of Figure 5 and reveals that various protons have migrated through a series of manifold inter-anionic and intra-anionic proton exchanges (purple and orange H atoms, respectively). Proton jumps appear throughout the simulation, reminiscent of a Grotthuss-type proton hopping mechanism.
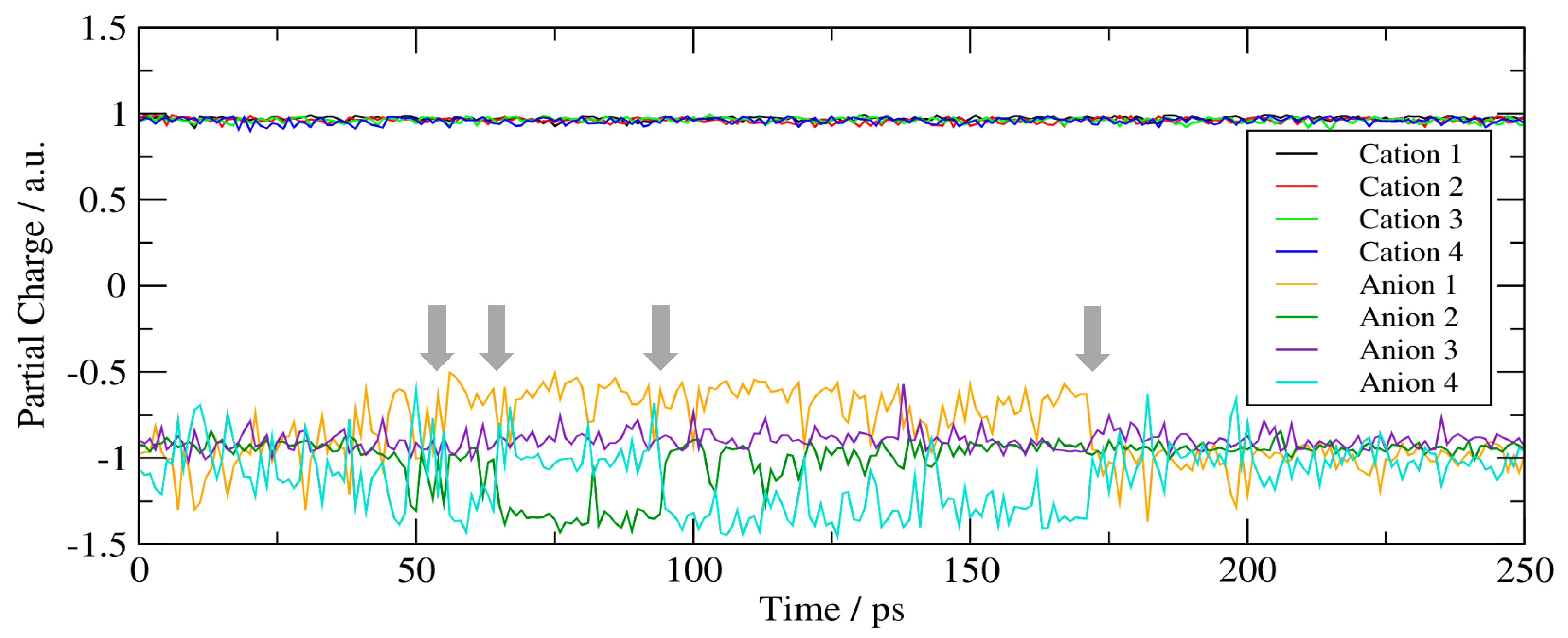
Towards a more quantitative characterisation of these proton transfer processes, we investigated how the movement of protons actually transports a charge through the system. This can be revealed through the analyses of the partial charges of each ion as a function of time throughout the entirety of their MD trajectories (Figure 6). The cations, which do not partake in the proton transfer process, manifest a fixed partial charge of +1 a.u., supporting our conclusion that they are merely ‘spectator’ ions. Three of the anions (orange, cyan and green lines in Figure 6) exhibit varied charges over the trajectories, due either to the partial loss or gain of a proton, whilst one of the anions (purple line, Figure 6) maintains a constant −1 a.u. charge. We note that the loss or acquisition of a proton does not necessarily lead to a dianionic or neutral species, respectively, but rather to a partial charge depletion or increase of ±0.5 a.u. This electrostatic character is due to the migration that takes place through H-bonding where the proton remains fractionally and dynamically shared between the acceptor and the donor sites over the course of the trajectory. Together, these trends support our initial assumption that charge transfer is rapid, reversible and promoted by the formation of like charge aggregates.
3.3. Cluster MD Simulations: Six Ionic Pairs Cluster
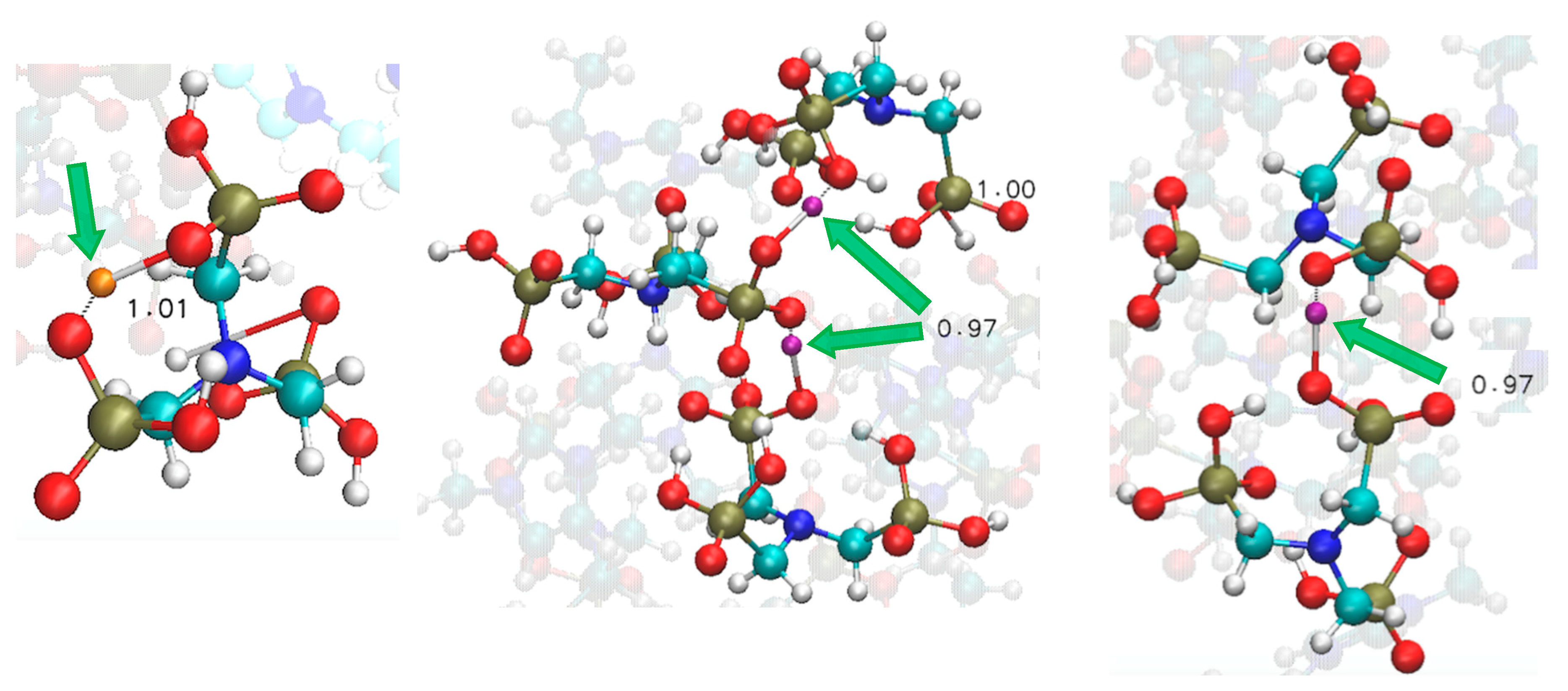
Larger aggregates comprised of six ionic pairs display novel structural patterns particularly in the way ATMP aggregates, with respect to the isolated dimers and four-pair clusters. Firstly, we ascertained that the anions tend to be in contact throughout the entirety of the simulation. Therein, phosphonic acid acts as a strong proton donor and for such a system rich in H-bond terminals it is evident that proton transfer necessitates anionic aggregation during the temporal evolution of the system. As we show in the snapshots reported in Figure 7, ATMP anions can form dimeric and even trimeric complexes. We presented selected geometric poses along the MD trajectory; wherein exemplary snapshots of both intra-molecular and intermolecular proton transfer occurred.
Those leading to the charge transfer process take place between differing anions (middle and right panels of Figure 7) with the anionic trimer manifesting a particular efficacy with post-transfer proton shifts amid adjacent anions actioning stabilising charge compensation. Hence, the role of ATMP anions in this system could be described as ‘equalisers’ as the protons move to maintain equally charged species. For a snapshot of the entire cluster see the SI, Figure S2.
In summary, with the evolution of system size, anionic ATMP trimers emerge as structurally and dynamically functional contributors to mesoscopic charge transfer and bulk phenomena.
3.4. Bulk Phase MD Simulations
Towards characterising ever larger ensembles, another MD simulation was performed, effectively scaling the system size up to nine ionic pairs to characterise the structural features of ATMP extending further into the nano- and mesoscopic regimes. As in the smaller clusters, intermolecular proton migration between the ATMP anions remains the dominant chemical process, as exemplified by the selected results presented in Figures S3 and S4. Therein, the pronounced O–H bond length variation signals the ongoing bond formation and breaking throughout the trajectories.
In Figure 8 (and in Figure S5 in the SI) we report the total net charge on each of the nine anions over the course of the trajectory. In contrast to cations, which maintain a +1 charge all along the simulation, the anions exhibit rapid fluctuation in partial charge due to the inter-anionic proton transfer equilibria at work. Time-dependent analyses of each anion over this ‘bulk’ nine-pair simulation provides quantitative tracking of the proton diffusions, with the persistent charge fluctuations observed being characteristic of a reversible microscopic process. These ongoing proton transfer processes do not neutralize the participant systems although they do momentarily modulate the net charge of an anion by ±0.5 a.u. Each anion does preserve an overall negative charge as the system tends to remain completely ionised, thus retaining the typical properties of an ionic liquid. Overall, the ATMP moiety remains ionised with proton motion driven to counterbalance any dramatic changes in anionic charge and is dominated by the following two transient species: a nearly neutral one with a −0.5 a.u. charge, and a nearly dianionic one with a −1.5 a.u. charge. Hence, despite being dominated by proton transfers, ATMP tends to remain ionised, yet does not populate dianionic or fully neutral states.
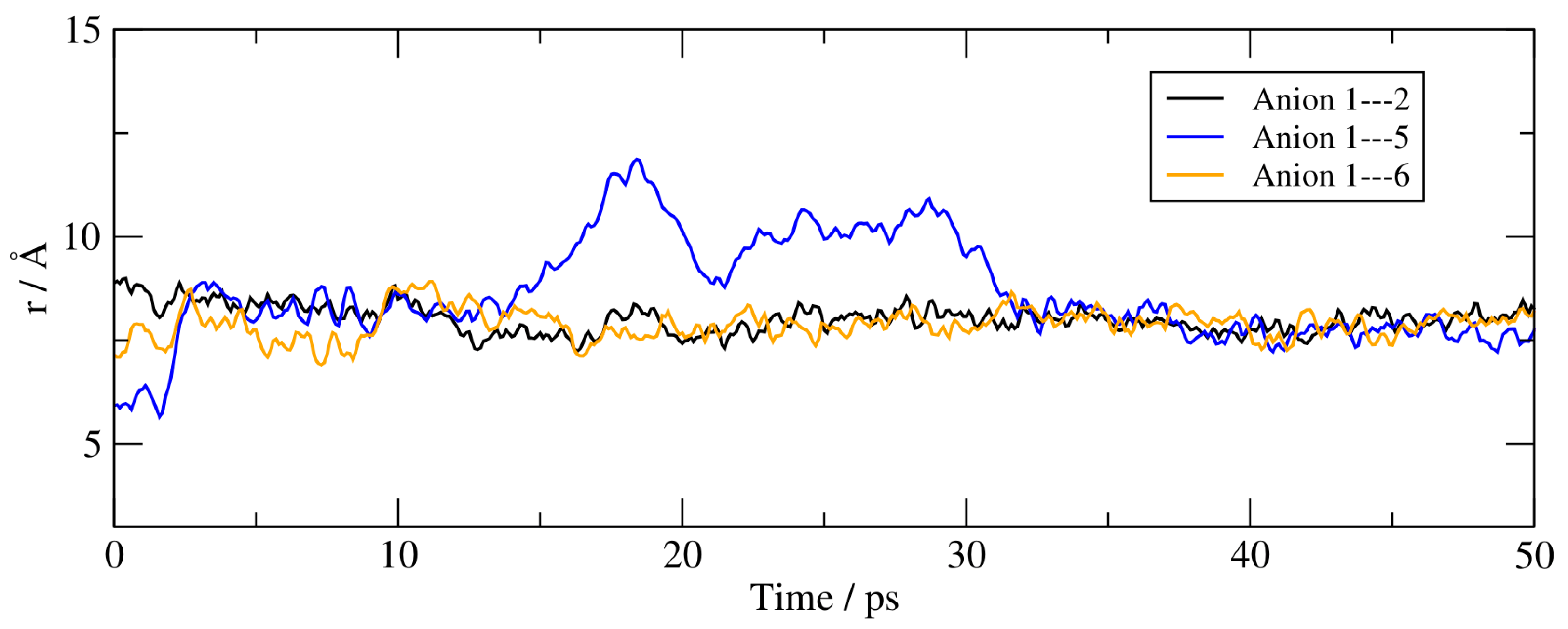
Finally, the spatial aggregation of the anions in the bulk was characterised by the variation in the distances between the centres of mass of the anions over the course of the trajectory. These are reported in Figure 9 where we have limited the information to the distance between anion 1 and its immediate first neighbours. Anion 1 is part of a stable anionic tetramer and is surrounded by three other anions with average distances below 10 Å.
3.5. Doubly Deprotonated ATMP Anion Cluster
All the simulation features detailed above also relate to the doubly deprotonated ATMP anions. The system we have simulated, initially, contains three [ATMP]2− and six [EMIM]+. Each [ATMP]2− has four protons, of which only one is mobile (pKa~2) whilst the other three are much more tightly bound to the phosphate groups (pKa~7) and thus extremely unlikely to undergo any transfer. Despite the less abundant protons, the ATMP dianion still manifests a high degree of self-interaction and short-range structuring as shown in Figure 10 on the left. The structural pattern of the distance distributions in this cluster closely resembles the same structural indicators we have highlighted in Figure 4 for the singly deprotonated system. Short-range anion–anion interactions upheld by strong H-bonds overcome the cation–anion aggregation driven by weaker H-bonds and by Coulombic attraction. These strong intra-anion associations are the crucial ingredient facilitating proton transfer, a process demonstrated herein also occurring between the doubly deprotonated variant of the ATMP anions. The final geometry of the cluster at the end of the trajectory is reported in Figure 10 (right hand side) revealing the various proton transfer processes that have taken place.
4. Conclusions
In this work, we presented a computational study utilising ab initio calculations and semi empirical molecular dynamics to explore a possible anionic partner for conducting materials such as protic ionic liquids. Two conditions must be fulfilled by a conductive PIL: (i) mobile protons must exist and (ii) the molecular ions must remain ionised. As a corollary to these conditions we need a ‘spectator’ cation that does not partake in the proton transfer processes which are therefore expected to take place exclusively between anions, hence avoiding mutual neutralisation. We chose the ATMP anion which exhibits the formation of stable anionic complexes through intricate H-bonding patterns and spontaneous proton transfer events. The presence of three phosphonic acid side chains (one is deprotonated) serves to drive proton motion among anions with like charge repulsion weakened via cooperative hydrogen bonding. Through the use of the efficient DFTB method we were able to simulate this system on sufficiently long time spans and to comprehend the anionic arrangements, dynamics and the feasibility of proton migration.
We showed that, in our models, proton transfer phenomena are present through intra- and intermolecular processes, but the local charge distribution of anionic species is seen to preserve the high-ionisation degree which is expected to maintain the ionic liquid properties. The mechanistic behaviour of this prototype ATMP-based material can be summarised by the following features:
- The EMIM cation, lacking basicity, it is merely a spectator which does not participate in the proton transfer processes. In addition, it binds only weakly with the ATMP anions thereby promoting the segregation of anionic and cationic moieties;
- The three phosphonic acid side chains of the anions provide six potential terminals for the formation of strong hydrogen bonds leading to the generation of stable anionic dimers as well as larger oligomers;
- The anionic dimers of ATMP anions have association energies which are competitive with their oppositely charged counterparts which is a rare condition in typical IL systems, one that allows anions to cluster and to promote proton transfer;
- Phosphonic acid groups are effective proton donors which can drive coupled proton motion to counterbalance changes in charge distribution. Phosphonic terminals play the role of ‘equalisers’ with intermolecular proton transfer a prevalent (and dominant) event that tends to maintain ionised species;
- Due to the lack of basicity in this system, proton transfer events can advance freely without risk of quenching;
- Fast dry proton conductivity may be achievable utilising ATMP because proton migration is a spontaneous reversible process over extended time scales.
The ATMP system also presents an additional degree of freedom since it can also be prepared in its doubly deprotonated state in materials in a 1:2 anion to cation ratio. In this state, as evidenced by calculation, the system still shows the presence of the necessary nanoscopic phenomena to sustain an efficient bulk conductivity.
This behaviour characteristic of ATMP reveals why it represents a component of task-specific material for dry proton conductivity and why it may be used as a molecular partner in novel ionic liquids tailored for electrochemical applications.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-8994/12/6/920/s1, Section S1: Validation of DFTB approach vs. DFT (B3LYP), Tables S1 and S2: Association energies in kcal/mol of the various dimeric conformers, Figure S1: DFT vs. DFTB association energies. Section S2: Additional data and figures relevant for discussion: Figure S2: DFTB MD snapshot of six ionic couples cluster, Figure S3: Bulk simulation, double proton transfer. Figure S4: Bulk simulation, triple proton transfer. Figure S5: Bulk simulation, partial charge vs. simulation time. Figures S6 and S7: Bulk simulation, inter-anionic centre of mass distances.
Author Contributions
Conceptualisation, E.B. and H.A.; methodology, E.B.; data analysis, H.A.; writing—original draft preparation, E.B.; writing, review and editing, G.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
G.C. thanks the Departments of Chemistry at Sapienza University, McMaster University and the University of Hong Kong for respectively supporting his visiting, adjunct and honorary professorships.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hayes, R.; Warr, G.W.; Atkin, R. Structure and nanostructure in ionic liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef] [Green Version]
- MacFarlane, D.R.; Seddon, K.R. Ionic Liquids—Progress on the Fundamental Issues. Aust. J. Chem. 2007, 60, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Hough, W.L.; Smiglak, M.; Rodrìguez, H.; Swatloski, R.P.; Spear, S.K.; Daly, D.Y.; Pernak, J.; Grisel, J.E.; Carliss, R.D.; Soutullo, M.D.; et al. The third evolution of ionic liquids: Active pharmaceutical ingredients. New J. Chem. 2007, 31, 1429–1436. [Google Scholar] [CrossRef]
- Vekariya., R.L. A review of ionic liquids: Applications towards catalytic organic transformations. J. Mol. Liq. 2017, 227, 44–60. [Google Scholar] [CrossRef]
- Fukaya, Y.; Iizuka, Y.; Sekikawa, K.; Ohno, H. Bio ionic liquids: Room temperature ionic liquids composed wholly of biomaterials. Green Chem. 2007, 9, 1155–1157. [Google Scholar] [CrossRef]
- Arunkumar, R.; Drummond, C.J.; Greaves, T.L. FTIR Spectroscopic Study of the Secondary Structure of Globular Proteins in Aqueous Protic Ionic Liquids. Front. Chem. 2019, 7, 74. [Google Scholar] [CrossRef]
- Mucsi, Z.; Chass, G.A.; Abranyi-Balogh, P.; Jojart, B.; Fang, D.C.; Ramirez-Cuesta, A.J.; Viskolcz, B.; Csizmadia, I.G. Penicillin’s catalytic mechanism revealed by inelastic neutrons and quantum chemical theory. Phys. Chem. Chem. Phys. 2013, 15, 20447–20455. [Google Scholar] [CrossRef]
- Campetella, M.; Gontrani, L.; Leonelli, F.; Bencivenni, L.; Caminiti, R. Two different models to predict ionic-liquid diffraction patterns: Fixed-charge versus polarizable potentials. Chem. Phys. Chem. 2015, 16, 197–203. [Google Scholar] [CrossRef]
- Bodo, E.; Mangialardo, S.; Capitani, F.; Gontrani, L.; Leonelli, F.; Postorino, P. Interaction of a long alkyl chain protic ionic liquid and water. J. Chem. Phys. 2014, 140, 204503. [Google Scholar] [CrossRef] [Green Version]
- Fumino, K.; Fossog, V.; Stange, P.; Paschek, D.; Hempelmann, R.; Ludwig, R. Controlling the subtle energy balance in protic ionic liquids: Dispersion forces compete with hydrogen bonds. Angew. Chem. Int. Ed. 2015, 54, 2792–2795. [Google Scholar] [CrossRef]
- Campetella, M.; Le Donne, A.; Daniele, M.; Gontrani, L.; Lupi, S.; Bodo, E.; Leonelli, F. Hydrogen bonding features in cholinium-based protic ionic liquids from molecular dynamics simulations. J. Phys. Chem. B 2018, 122, 2635–2645. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.; Wilson, T.; Murphy, P.J.; Refson, K.; Hannon, A.C.; Imberti, S.; Callear, S.K.; Chass, G.A.; Parker, S.F. How the Surface Structure Determines the Properties of CuH. Inorg. Chem. 2015, 54, 2213–2220. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.T.; Tian, K.V.; Dobo-Nagy, C.; Chass, G.A.; Greaves, G.N.; Yue, Y.Z. Phase separation in an ionomer glass: Insight from calorimetry and phase transitions. J. Non Cryst. Sol. 2015, 415, 24–29. [Google Scholar] [CrossRef]
- Martins, V.L.; Torresi, R.M. Ionic liquids in electrochemical energy storage. Curr. Opin. Electrochem. 2019, 9, 26–32. [Google Scholar] [CrossRef]
- Nakamoto, H.; Watanabe, M. Brønsted Acid-Base ionic liquids for fuel cell electrolytes. Chem. Commun. 2007, 24, 2539–2541. [Google Scholar] [CrossRef]
- Matsuoka, H.; Nakamoto, H.; Susan, M.A.B.H.; Watanabe, M. Brønsted acid base and poly base complexes as electrolytes for fuel cells under non-humidifying conditions. Electrochim. Acta 2005, 50, 4015–4021. [Google Scholar] [CrossRef]
- Stettner., T.; Walter. F., C.; Balducci., A. Imidazolium-Based Protic Ionic Liquids as Electrolytes for Lithium-Ion Batteries. Batter. Supercaps 2019, 2, 55–59. [Google Scholar] [CrossRef]
- Benedetto, A.; Bodo, E.; Gontrani, L.; Ballone, P.; Caminiti, R. Amino acid anions in organic ionic compounds. An ab-initio study of selected ion pairs. J. Phys. Chem. B 2014, 118, 2471. [Google Scholar] [CrossRef]
- Eftekhari, A. Supercapacitors utilising ionic liquids. Energy Storage Mater. 2017, 9, 47–69. [Google Scholar] [CrossRef]
- Shmukler, L.E.; Gruzdev, M.S.; Kudryakova, N.O.; Fadeeva, Y.A.; Kolker, A.M.; Safonova, L.P. Triethylammonium-based protic ionic liquids with sulfonic acids: Phase behavior and electrochemistry. J. Mol. Liq. 2018, 266, 139–146. [Google Scholar] [CrossRef]
- Doi, H.; Song, X.; Minofar, B.; Kanzaki, R.; Takamuku, T.; Umebayashi, Y. A new proton conductive liquid with no ions: Pseudo-protic ionic liquids. Chem. Eur. J. 2013, 19, 11522–11526. [Google Scholar] [CrossRef] [PubMed]
- Stoimenovski, J.; Izgorodina, E.I.; MacFarlane, D.R. Ionicity and proton transfer in protic ionic liquids. Phys. Chem. Chem. Phys. 2010, 12, 10341–10347. [Google Scholar] [CrossRef] [PubMed]
- Campetella, M.; Montagna, M.; Gontrani, L.; Scarpellini, E.; Bodo, E. Unexpected proton mobility in the bulk phase of cholinium-based ionic liquids: New insights from theoretical calculations. Phys. Chem. Chem. Phys. 2017, 19, 11869–11880. [Google Scholar] [CrossRef] [PubMed]
- Le Donne, A.; Bodo, E. Isomerization patterns and proton transfer in ionic liquids constituents as probed by ab-initio computation. J. Mol. Liq. 2018, 249, 1075–1082. [Google Scholar] [CrossRef]
- Le Donne, A.; Adenusi, H.; Porcelli, F.; Bodo, E. Structural features of cholinium based protic ionic liquids through molecular dynamics. J. Phys. Chem. B 2019, 123, 5568–5576. [Google Scholar] [CrossRef]
- Le Donne, A.; Adenusi, H.; Porcelli, F.; Bodo, E. Hydrogen Bonding as a Clustering Agent in Protic Ionic Liquids: Like-Charge vs Opposite-Charge Dimer Formation. ACS Omega 2018, 3, 10589–10600. [Google Scholar] [CrossRef] [Green Version]
- Adenusi, H.; Le Donne, A.; Porcelli, F.; Bodo, E. Ab Initio Molecular Dynamics Study of Phospho-Amino Acid-Based Ionic Liquids: Formation of Zwitterionic Anions in the Presence of Acidic Side Chains. J. Chem. Phys. B 2020, 124, 1955–1964. [Google Scholar] [CrossRef]
- Vilciauskas, L.; Tuckerman, M.E.; Bester, G.; Paddison, S.J.; Kreuer, K.D. The mechanism of proton conduction in phosphoric acid. Nat. Chem. 2012, 4, 461–466. [Google Scholar] [CrossRef]
- Abdurrokhman, I.; Elamin, K.; Danyliv, O.; Hasani, M.; Swenson, J.; Martinell, A. Protic Ionic Liquids Based on the Alkyl-Imidazolium Cation: Effect of the Alkyl Chain Length on Structure and Dynamics. J. Phys. Chem. B 2019, 123, 4044–4054. [Google Scholar] [CrossRef]
- Watanabe, H.; Hiroyuki, D.; Saito, S.; Matsugami, M.; Fujii, K.; Kanzaki, R.; Kameda, Y.; Umebayashi, Y. Hydrogen bond in imidazolium based protic and aprotic ionic liquids. J. Mol. Liq. 2016, 217, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Umecky, T.; Arai, N.; Nazet, A.; Takamuku, T.; Harris, K.R.; Kameda, Y.; Buchner, R.; Umebayashi, Y. Possible Proton Conduction Mechanism in Pseudo-Protic Ionic Liquids: A Concept of Specific Proton Conduction. J. Phys. Chem. B 2019, 123, 6244–6252. [Google Scholar] [CrossRef] [PubMed]
- Arumugama, V.; Sriramb, P.; Yen, T.J.; Redhia, G.G.; Gengana, R.M. Nano-material as an excellent catalyst for reducing a series of nitroanilines and dyes: Triphosphonated ionic liquid-CuFe2O4-modified boron nitride. Appl. Catal. B Environ. 2018, 222, 99–114. [Google Scholar] [CrossRef]
- Jalili, J.; Geppi, M.; Tricoli, V. Organic protic ionics based on Nitrilo(trimethylenephosphonic acid) as water-free, proton-conducting materials. J. Solid State Electrochem. 2015, 19, 1643–1650. [Google Scholar] [CrossRef]
- Shah, B.; Chudasama, U. Synthesis and Characterization of a Novel Hybrid Material Titanium Amino Tris(methylenephosphonic acid) and Its Application as a Cation Exchanger. Ind. Eng. Chem. Res. 2014, 53, 17454–17467. [Google Scholar] [CrossRef]
- Knorr, A.; Stange, P.; Fumino, K.; Weinhold, F.; Ludwig, F. Spectroscopic evidence for clusters of like-charged ions in ionic liquids stabilized by cooperative hydrogen bonding. ChemPhysChem 2016, 17, 458–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemann, T.; Stange, P.; Strate, A.; Ludwig, R. Like-likes-Like: Cooperative hydrogen bonding overcomes Coulomb repulsion in cationic clusters with net charges up to Q=+6e. ChemPhysChem 2018, 19, 1691–1695. [Google Scholar] [CrossRef] [Green Version]
- Knorr, A.; Fumino, K.; Bonsa, A.M.; Ludwig, R. Spectroscopic evidence of ‘jumping and pecking’ of cholinium and H-bond enhanced cation−cation interaction in ionic liquids. Phys. Chem. Chem. Phys. 2015, 17, 30978–30982. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar]
- Wojnarowska, Z.; Paluch, M. Recent progress on dielectric properties of protic ionic liquids. J. Phys. Condens. Matter. 2015, 27, 073202. [Google Scholar] [CrossRef]
- Huang, M.M.; Jiang, Y.; Sasisanker, P.; Driver, G.W.; Weingartner, H. Static Relative Dielectric Permittivities of Ionic Liquids at 25 °C. J. Chem. Eng. Data 2011, 56, 1494–1499. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian-basis sets for molecular calculations. 1. 2nd row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Addicoat, M.A.; Stefanovic, R.; Webber, G.B.; Atkin, R.; Page, A.J. Assessment of the Density Functional Tight Binding Method for Protic Ionic Liquids. J. Chem. Theory Comput. 2014, 10, 4633–4643. [Google Scholar] [CrossRef] [Green Version]
- Zentel, T.; Kühn, O. Properties of hydrogen bonds in the protic ionic liquid ethylammonium nitrate DFT versus DFTB molecular dynamics. Theor. Chem. Acc. 2017, 136, 87. [Google Scholar]
- Zentel, T.; Kühn, O. Hydrogen bonding in the protic ionic liquid triethylammonium nitrate explored by density functional tight binding simulations. J. Chem. Phys. 2016, 145, 234504. [Google Scholar] [CrossRef] [PubMed]
- Joswig, J.O.; Hazebroucq, S.; Seifert, G. Properties of the phosphonic-acid molecule and the proton transfer in the phosphonic-acid dimer. J. Mol. Struct. 2007, 816, 119–123. [Google Scholar] [CrossRef]
- Gaus, M.; Cui, Q.; Elstner, M. DFTB3: Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB). J. Chem. Theory Comput. 2011, 7, 931–948. [Google Scholar] [CrossRef] [Green Version]
- Xi, N.; Huang, Q.; Liu, L. Comprehensive Heterocyclic Chemistry Chemistry. Mol. Sci. Chem. Eng. 2008, 4, 143–364. [Google Scholar]
- Baek, C.S.; Lee, Y.J.; Lee, S.J.; Lee, S.G.; Kim, H.C.; Jeong, S.W. C2-Functionalized 1,3-dialkylimidazolium ionic liquids for efficient cellulose dissolution. J. Mol. Liq. 2017, 234, 111–116. [Google Scholar] [CrossRef]
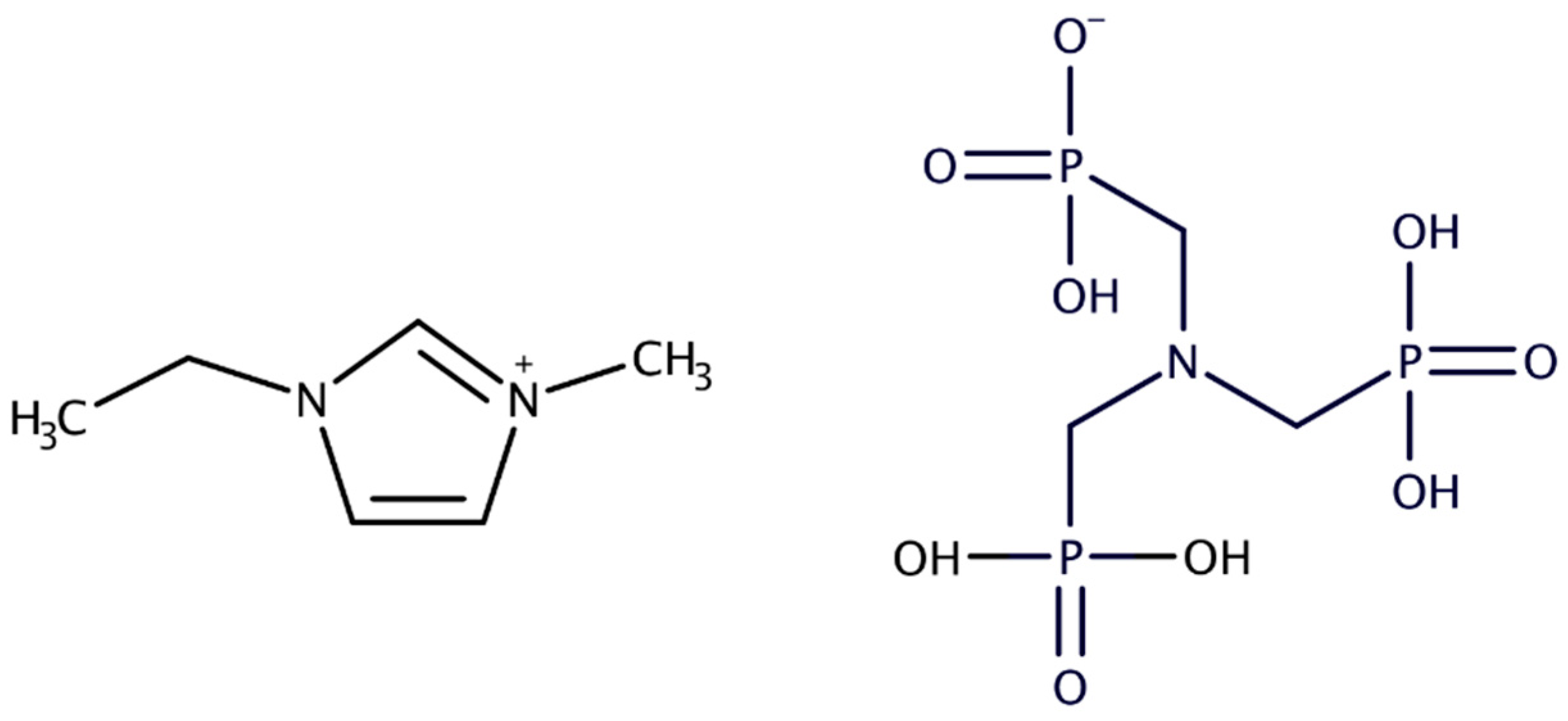
Figure 1.
Molecular structures of 1-ethyl-3-methyl-imidazole (EMIM) (cation) (left) and Aminotris(methylenephosphonic) acid (ATMP) anion (right).
Figure 1.
Molecular structures of 1-ethyl-3-methyl-imidazole (EMIM) (cation) (left) and Aminotris(methylenephosphonic) acid (ATMP) anion (right).

Figure 2.
Optimised geometries of the oppositely charged dimers of EMIM and ATMP. [EMIM]+[ATMP]− on the left and [EMIM]+[ATMP]2− on the right. Cation–anion H-bond distances (Å) are shown.
Figure 2.
Optimised geometries of the oppositely charged dimers of EMIM and ATMP. [EMIM]+[ATMP]− on the left and [EMIM]+[ATMP]2− on the right. Cation–anion H-bond distances (Å) are shown.

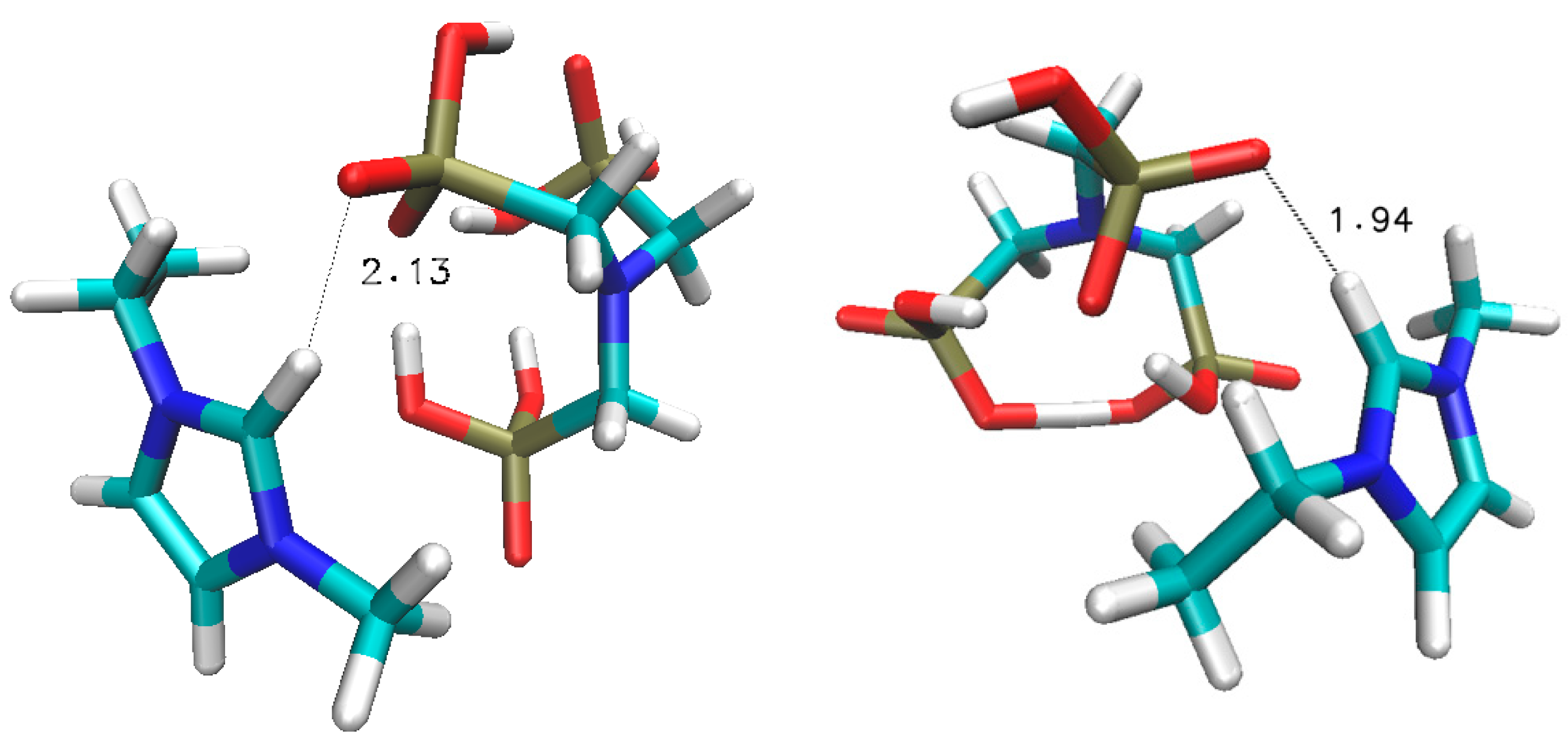
Figure 3.
Optimised geometries of the three anionic dimers studied ([ATMP]−[ATMP]−, [ATMP]2−[ATMP]−, [ATMP]2−[ATMP]2−). The selected inter-atomic H-bond acceptor–donor distances (Å) are shown.
Figure 3.
Optimised geometries of the three anionic dimers studied ([ATMP]−[ATMP]−, [ATMP]2−[ATMP]−, [ATMP]2−[ATMP]2−). The selected inter-atomic H-bond acceptor–donor distances (Å) are shown.

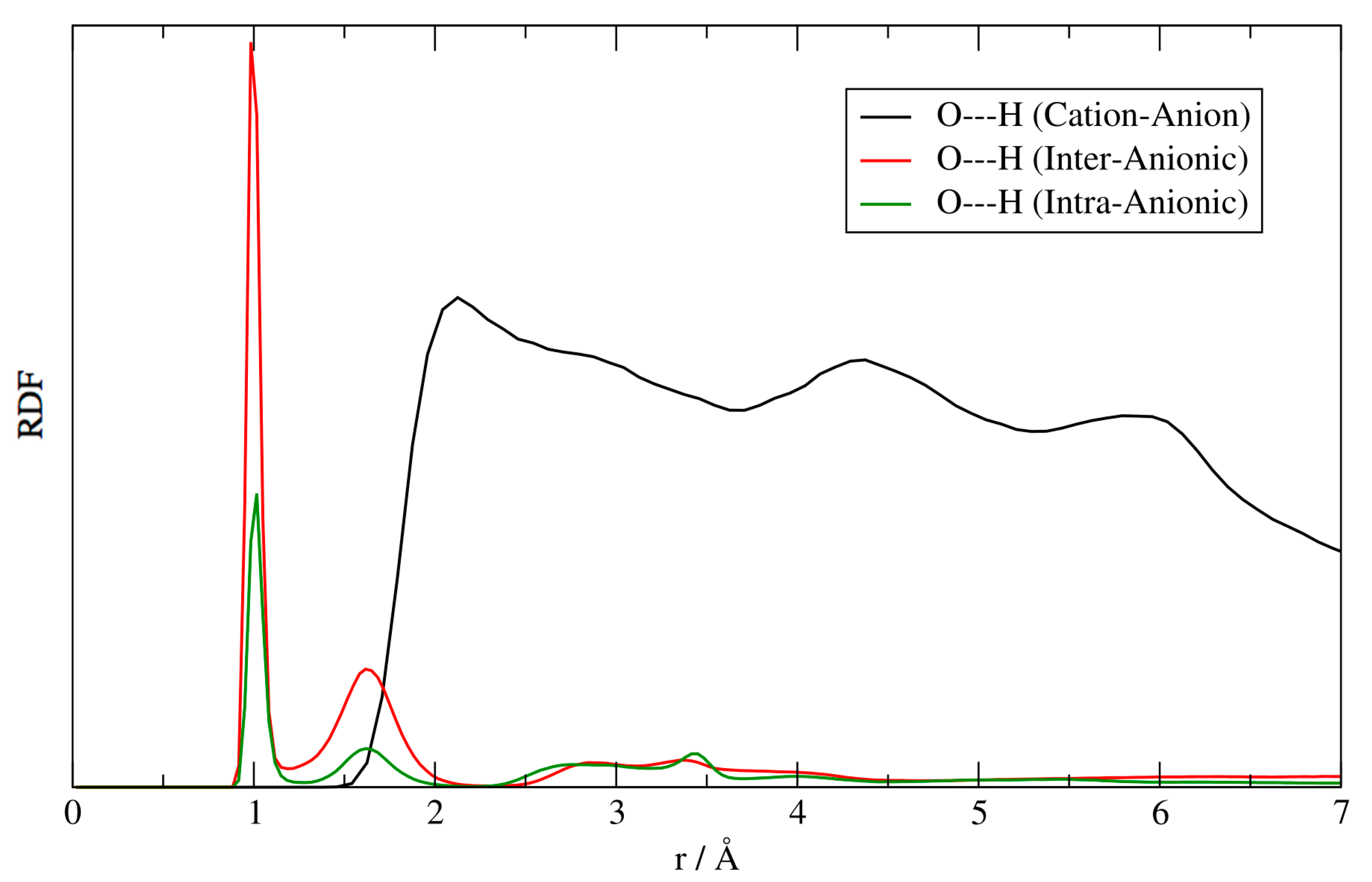
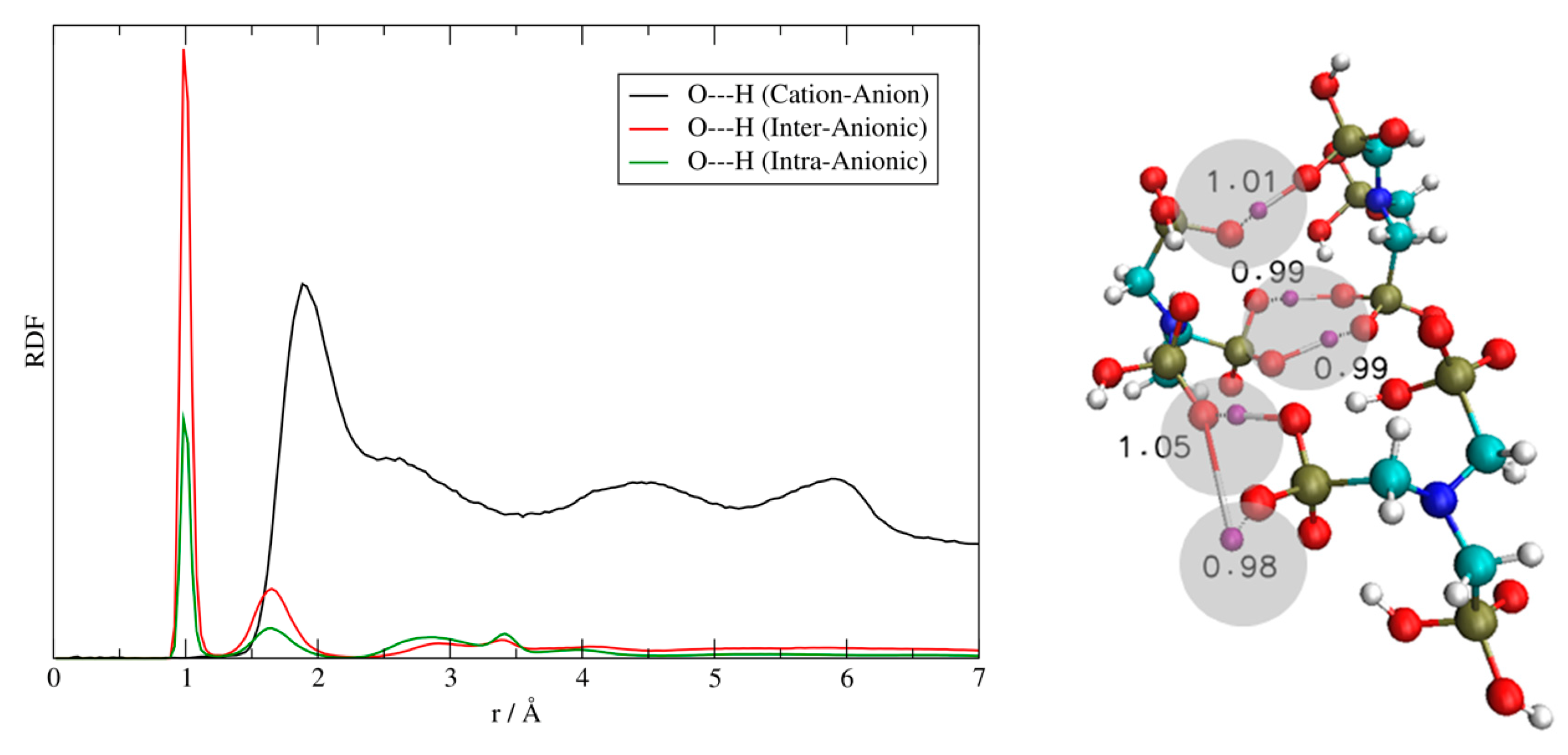
Figure 4.
Singly deprotonated ATMP, 4 ionic couples cluster. Black line: the anion–cation, O–H radial distribution function (RDF) between the oxygen atoms of [ATMP]− and the hydrogens on the imidazole ring of [EMIM]+. Red line: the intermolecular O–H RDF between the different [ATMP]− anions. Green line: the intra-molecular O–H RDF within the same [ATMP]− anion.
Figure 4.
Singly deprotonated ATMP, 4 ionic couples cluster. Black line: the anion–cation, O–H radial distribution function (RDF) between the oxygen atoms of [ATMP]− and the hydrogens on the imidazole ring of [EMIM]+. Red line: the intermolecular O–H RDF between the different [ATMP]− anions. Green line: the intra-molecular O–H RDF within the same [ATMP]− anion.

Figure 5.
Singly deprotonated ATMP, 4 ionic couples cluster. Left: structural snapshot of a 4 ionic pairs cluster at around 100 ps; the migrating protons are shown in orange (intramolecular) and in purple (intermolecular) highlighted by the green arrows, and the cations have been faded for clarity. Right: two selected O–H bond distances as a function of time.
Figure 5.
Singly deprotonated ATMP, 4 ionic couples cluster. Left: structural snapshot of a 4 ionic pairs cluster at around 100 ps; the migrating protons are shown in orange (intramolecular) and in purple (intermolecular) highlighted by the green arrows, and the cations have been faded for clarity. Right: two selected O–H bond distances as a function of time.

Figure 6.
Singly deprotonated ATMP, 4 ionic couples cluster. Partial charge vs. time. The trends highlight the localisation of the cation and anion charge at approximately 1 a.u. and −1 a.u., respectively. Times of specific cases of charge migration are indicated by grey arrows.
Figure 6.
Singly deprotonated ATMP, 4 ionic couples cluster. Partial charge vs. time. The trends highlight the localisation of the cation and anion charge at approximately 1 a.u. and −1 a.u., respectively. Times of specific cases of charge migration are indicated by grey arrows.

Figure 7.
Singly deprotonated ATMP, 6 ionic couples cluster. Snapshots along the MD trajectory. Only the relevant anions are highlighted with the green arrows showcasing the migrating protons. Left panel: the intramolecular proton transfer. Middle panel: an ATMP anionic trimer where two intermolecular proton transfers occurred simultaneously. Right panel: an ATMP anionic dimer with a single intermolecular proton transfer. Selected H–O distances (Ǻ) of the newly formed bonds are provided.
Figure 7.
Singly deprotonated ATMP, 6 ionic couples cluster. Snapshots along the MD trajectory. Only the relevant anions are highlighted with the green arrows showcasing the migrating protons. Left panel: the intramolecular proton transfer. Middle panel: an ATMP anionic trimer where two intermolecular proton transfers occurred simultaneously. Right panel: an ATMP anionic dimer with a single intermolecular proton transfer. Selected H–O distances (Ǻ) of the newly formed bonds are provided.

Figure 8.
Singly deprotonated ATMP, bulk simulation. Partial charge vs. time of 9 ATMP anions. Grey arrows visually signal the instances of proton transfer.
Figure 8.
Singly deprotonated ATMP, bulk simulation. Partial charge vs. time of 9 ATMP anions. Grey arrows visually signal the instances of proton transfer.

Figure 9.
Singly deprotonated ATMP, bulk simulation. Centre-of-mass distance between anion 1 and other selected anions vs. time. The panel shows the existence of an anionic tetramer. The full matrix of relative distances is presented in Figures S6 and S7 in the supporting information.
Figure 9.
Singly deprotonated ATMP, bulk simulation. Centre-of-mass distance between anion 1 and other selected anions vs. time. The panel shows the existence of an anionic tetramer. The full matrix of relative distances is presented in Figures S6 and S7 in the supporting information.

Figure 10.
Doubly deprotonated ATMP cluster. Left: black line: cation–anion, O–H RDF between the oxygen atoms of [ATMP]2− and the hydrogens on the imidazole ring of [EMIM]+; red line: intermolecular O–H RDF between the different [ATMP]2− anions; green line: intra-molecular O–H RDF within the same [ATMP]2− anion. Right: the final snapshot of the cluster simulation (~300 ps) with several occurrence of proton transfer highlighted by grey circles. In the latter, cations are omitted for clarity.
Figure 10.
Doubly deprotonated ATMP cluster. Left: black line: cation–anion, O–H RDF between the oxygen atoms of [ATMP]2− and the hydrogens on the imidazole ring of [EMIM]+; red line: intermolecular O–H RDF between the different [ATMP]2− anions; green line: intra-molecular O–H RDF within the same [ATMP]2− anion. Right: the final snapshot of the cluster simulation (~300 ps) with several occurrence of proton transfer highlighted by grey circles. In the latter, cations are omitted for clarity.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Enthalpies of association (ΔH, kcal/mol) of the differing dimers characterised, with respect to their separated components in both the gas-phase (vacuum) and the acetonitrile solvent (ε = 35.7).
Table 1.
Enthalpies of association (ΔH, kcal/mol) of the differing dimers characterised, with respect to their separated components in both the gas-phase (vacuum) and the acetonitrile solvent (ε = 35.7).
| Dimer | Gas-Phase | Acetonitrile Solvent (PCM ε = 35.7) |
|---|---|---|
| [EMIM]+[ATMP]− | −92.7 | −23.5 |
| [EMIM]+[ATMP]2− | −177.8 | −29.9 |
| [ATMP]−[ATMP]− | 25.6 | −37.6 |
| [ATMP]2−[ATMP]2− | 79.6 | −34.6 |
| [ATMP]−[ATMP]2− | 26.8 | −36.6 |
| [ATMP][ATMP]2− | 69.6 | −35.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Adenusi, H.; Chass, G.; Bodo, E. Theoretical Insights into the Structure of the Aminotris(Methylenephosphonic Acid) (ATMP) Anion: A Possible Partner for Conducting Ionic Media. Symmetry 2020, 12, 920. https://doi.org/10.3390/sym12060920
AMA Style
Adenusi H, Chass G, Bodo E. Theoretical Insights into the Structure of the Aminotris(Methylenephosphonic Acid) (ATMP) Anion: A Possible Partner for Conducting Ionic Media. Symmetry. 2020; 12(6):920. https://doi.org/10.3390/sym12060920
Chicago/Turabian StyleAdenusi, Henry, Gregory Chass, and Enrico Bodo. 2020. "Theoretical Insights into the Structure of the Aminotris(Methylenephosphonic Acid) (ATMP) Anion: A Possible Partner for Conducting Ionic Media" Symmetry 12, no. 6: 920. https://doi.org/10.3390/sym12060920
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.