Absolute Asymmetric Synthesis Involving Chiral Symmetry Breaking in Diels–Alder Reaction
by

Naohiro Uemura
1, 
Seiya Toyoda
1,
Waku Shimizu
1,
Yasushi Yoshida
1,2,
Takashi Mino
1,2 and
Masami Sakamoto
1,2,* 1
Department of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba University, Yayoi-Cho, Inage-Ku, Chiba 263-8522, Japan
2
Molecular Chirality Research Center, Chiba University, Yayoi-Cho, Inage-Ku, Chiba 263-8522, Japan
*
Author to whom correspondence should be addressed.
Symmetry 2020, 12(6), 910; https://doi.org/10.3390/sym12060910
Submission received: 27 April 2020
/
Revised: 14 May 2020
/
Accepted: 18 May 2020
/
Published: 1 June 2020
(This article belongs to the Special Issue Chemical Symmetry Breaking)
Abstract
:Efficient generation and amplification of chirality from prochiral substrates in the Diels–Alder reaction (DA reaction) followed by dynamic crystallization were achieved without using an external chiral source. Since the DA reaction of 2-methylfuran and various maleimides proceeds reversibly, an exo-adduct was obtained as the main product as the reaction proceeded. From single crystal X-ray structure analysis, it was found that five of ten exo-adducts gave conglomerates. When 2-methylfuran and various maleimides with a catalytic amount of TFA were reacted in a sealed tube, the exo-DA adducts were precipitated from the solution, while the reaction mixtures were continuously ground and stirred using glass beads. Deracemization occurred and chiral amplification was observed for four of the substrates. Each final enantiomeric purity was influenced by the crystal structure, and when enantiomers were included in the disorder, they reached an enantiomeric purity reflecting the ratio of the disorder. The final ee value of the 3,5-dimethylphenyl derivative after chiral amplification was 98% ee.
1. Introduction
The Diels–Alder (DA) reaction is one of the most important and fundamental organic synthesis reactions, achieving the concerted [4 + 2] cycloaddition of a diene and an alkene [1,2]. Because it can form two C-C single bonds in one step, it is used to create many cyclic compounds including polycyclic compounds (Figure 1) [3,4,5,6,7,8,9,10]. In addition, a number of asymmetric reactions have been reported, since these can theoretically construct four asymmetric centers at once. Excellent catalytic asymmetric synthesis [11,12,13,14,15,16,17,18,19,20] and diastereoselective reactions of chiral substrates have also been reported [21,22,23,24]. Each of these reactions is an asymmetric induction method using an enantiomerically active catalyst or substrate and constructs a diastereoselective environment in the transition state of the reaction.
In contrast to reactions using chiral sources as starting materials and catalysts, asymmetric synthesis using chirality that occurs naturally when organic compounds crystallize has been reported in recent years (Figure 2) [25]. In this method, a compound having a chiral center is generated from a prochiral substrate, and a dynamic preferential crystallization accompanied by the racemization of the generated chiral center is performed continuously without using any external asymmetric source. It is possible to obtain a crystal of the product having a high enantiomeric purity. The racemization process of the product includes a reaction that regenerates a prochiral starting material via a reverse reaction or a process via a direct racemization reaction of an asymmetric center.
These synthetic reactions are absolute asymmetric syntheses that provide enantiomerically active compounds from prochiral substrates without using an external chiral source; this is a phenomenon that is of wide interest to researchers in many academic fields [26,27,28,29,30]. Successful absolute asymmetric synthesis by fusion of the reaction that forms the chiral center from prochiral starting materials and the subsequent dynamic crystallization process have been reported in several reaction systems. For example, the Mannich-type reaction [31], aldol reaction [32], stereoisomerization of succinimide [25], synthesis of isoindolinone [33], aza-Michael addition [34,35,36], Strecker reaction [37,38], and a photochemical reaction [39,40] have all been achieved under achiral conditions. In these limited examples, crystals with high ee were obtained from prochiral materials, and this method is expected to be applied to many reaction systems.
We recently reported the asymmetric Diels–Alder reaction of prochiral starting materials leading to a conglomerate crystal of the adduct in enantiomerically active form [41]. When 2-methylfuran and N-phenylmaleimide were reacted with a small amount of solvent and a catalytic amount of trifluoroacetic acid in a sealed tube at 80 °C, the racemic exo-adduct quickly precipitated. Subsequently, the continuous suspension of the reaction mixture with glass beads promoted the chiral amplification to 90% ee by attrition-enhanced deracemization (Figure 3). In this phenomenon, a racemic product having a chiral center is first formed by a DA reaction from a prochiral substance, followed by preferential crystallization of conglomerate crystals. In the mother liquor, by returning to the prochiral substrate by the reverse reaction (retro-DA reaction), the racemic condition is always maintained, preventing the excessive formation of the enantiomer.
In this asymmetric amplification by dynamic crystallization, attrition-enhanced deracemization was quite effective in promoting Viedam ripening by continuous grinding of the crystals using glass beads, and finally, the enantiomer crystals converged to high enantiomeric purity [42]. This technique was followed in the deracemization reaction from a racemic mixture of NaClO3 and has recently been applied to deracemization of conglomerate crystals of organic compounds such as amino acids and pharmaceutical and agricultural chemical intermediates [43,44,45,46,47,48,49,50,51,52,53,54]. A key feature of this technique is that it can be applied to deracemization in a system with a relatively low racemization rate, as compared with the method of promoting crystallization using a solvent evaporation method or a temperature gradient [55,56,57,58].
The bottleneck of deracemization by the dynamic crystallization method is whether or not the target substrate crystallizes as a stable conglomerate. The occurrence of racemic mixtures crystallizing as a conglomerate is approximately 5–10% [59,60,61,62]. However, some substrates form conglomerates or chiral crystals at a very high rate due to the effects of molecular shapes and intermolecular interactions. We succeeded in the asymmetric DA reaction utilizing the fact that the DA adduct of 2-methylfuran and N-phenylmaleimide was a conglomerate [41]. However, to investigate the generality of this methodology, we synthesized and analyzed a variety of DA adducts with various substituents on the nitrogen atom (Figure 4).
Analysis of the crystal structures of the adducts revealed that they exhibited a very high probability of affording conglomerates. For these substrates, we achieved an asymmetric DA reaction without using an external chiral source and clarified the relationship between the crystal structure and the enantiomeric purity of the products.
2. Results and Discussion
In order to investigate the effect of the substituent on the nitrogen atom of the DA adduct on the crystal structure and the enantiomeric purity of the crystal in dynamic crystallization, the adducts exo-3b–j were synthesized by changing the substituent as shown in Table 1.
In the DA reaction between 2-methylfuran 1 and maleimides 2b–j, the formation of endo-isomers was confirmed at the beginning of the reaction as in the case of the reaction with 2a [41]. When the reaction time was extended, the products predominantly converged to exo-isomers, because the crystalline exo-adducts were excluded from the reaction system in solution. Therefore, the crystals of the exo-adducts were analyzed by single crystal X-ray structure analysis. When the racemate of each exo-isomer was recrystallized, single crystals suitable for crystal structure analysis were obtained for exo-3b–g. The crystal space groups of these seven types of crystals are shown in Table 1. All of them had 21 helices in the crystal lattices. The space group of 3a–3c and 3e was the orthorhombic P212121, and 3d and 3f–g were in the monoclinic space group. Surprisingly, five of the ten synthesized substrates, 3a-3e, formed conglomerates of a chiral crystal space group.
In all cases, interactions such as CH--π, C=O--HC and O--HC with relatively small energies were present, which controlled the molecular arrangement (Figures S1–S8). These molecules had nearly spherical shapes and were closely packed in the crystal. Even when alcohol or benzene-based solvents were used, solvent molecules were not incorporated into the crystals.
For 3h–j, single crystals suitable for X-ray crystallography could not be obtained, thus the detailed molecular arrangements were unknown. However, the second harmonic generation (SHG) of these crystals was inactive by irradiation with 1064 nm light from an Nd-YAG pulsed laser, indicating that they might be racemic crystals [63,64]. However, if the emission of the SHG is weak, the possibility of a conglomerate may have been overlooked.
For asymmetric synthesis by the proposed method, the product crystals must be conglomerates. For the four new substrates 3b–3e determined to be in a chiral crystal space group from the above crystal structure analysis, we investigated the absolute asymmetric DA reaction via asymmetric amplification by crystallization.
Another requisite for chiral amplification by dynamic crystallization is rapid racemization under crystallization conditions. Adducts 3 have four chiral centers determined uniquely in the one-step concerted reaction and these cannot directly racemize. However, apparent racemization occurs due to the equilibrium reaction with a reverse DA reaction to regenerate achiral furan and maleimide [65] (Figure 5).
Utilizing this reversible DA reaction, a deracemization reaction by dynamic crystallization was developed. Many dynamic crystallization methods require rapid racemization under crystallization conditions. However, attrition-enhanced deracemization, which is a method based on crystal grinding, has been reported in many successful cases even in systems with a low racemization rate compared to dynamic preferential crystallization methods using a temperature gradient or solvent evaporation [55,56,57,58]. However, fast deracemization under the racemization conditions suppresses side reactions, and crystals with high enantiomeric purity can be obtained efficiently.
In the case of the asymmetric synthesis of exo-3a, we already found out that trifluoroacetic acid (TFA) was the best catalyst for both of the forward and reverse reactions. NMR spectroscopy was used to follow the reversible reaction at 60 °C in deuterated chloroform. Even when the substituents were changed, the catalytic activity of TFA was effective, and the reaction rate was improved in both the DA and retro-DA reversible reactions without side reactions. Table 2 shows the half-life of each substrate. For 1a, as reported in a previous paper, the addition reaction of maleimide reached 50% in about 1 h (Table 2), and an endo-adduct was formed at the initial stage of the reaction, which eventually converged to an exo-adduct over time [41]. In other cases, almost the same courses in the reactions were observed (Figures S9–S11). The change with time, in these cases, is a reaction in a homogeneous system, but the actual reaction is performed at a higher concentration, where the highly crystalline exo-adduct is crystallized and removed from the reaction system.
The reaction in the NMR tube was also examined for the reverse reaction. The degradation of exo-3 at low concentration (0.05 M) in deuterated chloroform at 60 °C was followed. Maleimide 2 and methylfuran 1 were quantitatively regenerated in all cases. In order to perform highly efficient deracemization, it was necessary to accelerate both the forward and reverse reactions. Specifically, it was necessary to search for a catalyst that greatly accelerated the reverse reaction.
When various maleimides 2a-d were reacted with 2-methylfuran in the presence of TFA, the DA reaction was accelerated about 4 times for most substrates. Regarding the reverse reaction, it was found that when exo-3a–d were reacted with TFA in the same concentration of 0.03 M, the reaction was accelerated by 1.5 to 2 times compared to the reaction without TFA (Table 2, Figures S9–S11). Table 2 shows the results at 60 °C, but the actual reaction can be run at 80 °C, whereupon the rate is expected to be several times faster, and a sufficient reversible reaction rate is ensured.
Once the formation of conglomerates and the progress of racemization were confirmed, the asymmetric synthesis involving the dynamic crystallization process was examined. In a sealed tube, N-arylmaleimide 2 (100 mg), 2-methylfuran 1 (15.0 equiv), TFA (0–1.0 equiv) as the catalyst, heptane (1.0 mL) as a solvent, and glass beads (2 mmΦ, 250 mg) were added to crush the crystals and the mixture was stirred at 80 °C for several days. The DA reaction proceeded immediately after the start of the reaction, and within a few minutes, crystals of the adducts precipitated and the reaction solution was suspended. After that, deracemization occurred by continuously stirring the obtained suspension. The change in ee value of exo-3 by deracemization was monitored by HPLC using a CHIRALPAK IA (Daicel Ind.) column.
In our previous paper, when 0.5 equiv of TFA was used, the enantiomeric purity started to increase after six days from the start of the reaction and reached 90% ee after 14 days (Figure 6) [41]. Thereafter, the suspension was filtered to isolate the crystals, and exo-3a was obtained with a yield of 80% and 90% ee. The plot of enantiomeric purity versus time showed a non-linear curve. This sigmoid-like increase in enantiomeric purity is typical for Viedma ripening, and the population balance model [66,67] and existing formulas were extended in view of the effects of Ostwald ripening and autocatalytic enantioselective crystal growth. Theoretical analysis by fitting [68,69] has also been performed. On the other hand, when no glass beads were used, deracemization did not occur.
Based on the results of the asymmetric reaction of 2a to exo-3a, asymmetric DA reactions for 2b-2e with 1 were also examined leading to conglomerates exo-3b–3e. As in the case of the reaction of 2a, 2-methylfuran 1, various N-arylmaleimides 2b–2e, 0.5 equiv of TFA as a catalyst for promoting racemization, heptane as a solvent, and glass beads for grinding the precipitated solids were stirred in a sealed tube at 80 °C for several days while tracking the change of the ee value of the solid exo-3 (Table 3 and Figure 6).
When 2b was used, crystals of DA adduct were precipitated immediately after the reaction, and the continuous stirring while suspending the solids led to an increase in the enantiomeric purity of exo-3b from the 7th day, reaching 40% ee after 15 days. However, no further asymmetric amplification occurred. This is attributable to the crystal structure of exo-3b. Disordered packing was indicated by single-crystal structure analysis, and the ratio of the enantiomers was 73:27, a result that exactly reflected the ee value of the converged enantiomorphic crystals.
In the case of 2c, the crystallinity of the produced exo-3c was also good, and a stable suspension by glass beads and the stir bar was obtained. The ee value of exo-3c increased after 3 days and reached 98% after 9 days, achieving the most efficient asymmetric amplification among all the substrates.
When 2d was used, immediately after the reaction, crystals were precipitated, a suspension was obtained, and asymmetric amplification started 4 days later. Eight days later, the ee reached 49%, after which further amplification did not occur. The reason for this limitation is that, similar to the case of exo-3b, due to the crystal packing of exo-3d, the enantiomer contained in the single crystal is disordered in a ratio of 74:24, which exactly reflected the maximum ee value of exo-3d.
The asymmetric synthesis of exo-3e using maleimide 2e was also examined, but exo-3e was obtained as a racemate without asymmetric amplification. The powdered X-ray crystal structure analysis of the obtained solid was different from the analysis pattern simulated from the conglomerate crystal P212121 (Figure 7). Conglomerate crystals of exo-3e gradually changed to a racemic crystal system during suspension with glass beads. The crystal does not contain a crystallization solvent and it showed a polymorphism with a phase transition.
It was unable to control the handedness of the crystals obtained after the DA reaction; solids of both types of handedness were obtained in approximately the same number of times for every ten experiments. However, we were able to control the handedness by starting the attrition-enhanced deracemization from a DA adduct with low ee (5% ee). The deracemization started immediately and the same handedness of the enantiomer as the slightly excess stereoisomer could be efficiently obtained.
3. Conclusions
Asymmetric DA reactions from prochiral starting materials involving dynamic preferential crystallization were achieved under achiral conditions. Since the DA reaction of 2-methylfuran and various maleimide derivatives proceeded reversibly, exo-adducts were obtained as major products as the reaction proceeded. Nine exo-adducts were newly synthesized by the DA reaction using maleimides with various substituents on the nitrogen atom. Single crystal X-ray structure analysis revealed that four derivatives gave conglomerates.
When 2-methylfuran and each maleimide in the presence of a catalytic amount of TFA were reacted in a sealed tube, DA adducts precipitated as crystals. The mixture was continuously ground and stirred using glass beads. Deracemization of exo-type DA adducts occurred and asymmetric amplification was observed for four substrates. Each final enantiomeric purity was greatly influenced by the crystal structure, and when enantiomers were included in the disorder, they reached an enantiomeric purity reflecting the ratio. In addition, it was found that the chirality of the 3,5-dimethylphenyl derivative was deracemized to 98% ee. We have developed an absolute asymmetric DA reaction that can obtain enantiomerically active DA adducts without using an external asymmetric source.
4. Experimental
General Information. NMR spectra were recorded in CDCl3 solutions on a Bruker DPX 300 and DPX 400 spectrometers for 1H- and 13C-NMR. Chemical shifts are reported in parts per million (ppm) relative to TMS as an internal standard. IR spectra were recorded on a JASCO FT/IR-230 spectrometer. HPLC analyses were performed on a JASCO HPLC system (JASCO PU-1580 pump, DG-1580-53, LG-2080-02, MD-2015, UV-2075 and CD-2095 detector). Single crystal X-ray structure analysis was conducted using a SMART APEX II (Bruker AXS) and APEX II ULTRA (Bruker AXS). Powder X-ray crystallographic analysis was performed using D8 ADVANCE (BRUKER AXS). Commercially available N-phenylmaleimide and 2-methylfuran were used without further purification. Other maleimides 2b-j were provided according to the reported procedure [70,71]. (Figures S18–S35)
Synthesis of Exo-3a–j.
The corresponding maleimides 2 (1.0 g) and 2-methylfuran 1 (15 equiv) were added to 10 mL of hexane, and the mixture was stirred at 60 °C for 24 h. Thereafter, the solvent and extra amount of methylfuran were removed under reduced pressure, and the crude crystalline products were recrystallized from chloroform/hexane to isolate exo-3. The structures of known adducts 3a were determined by comparing their spectral data to literature values. The adducts, 3e-g, and 3j are commercially available; however, these materials were easily obtained by the above method.
(3aS*,4R*,7S*,7aR*)-4-Methyl-2-phenyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3a) [41]
Colorless prism; 96% yield; mp: 144–146 °C; 1H NMR (CDCl3) δ 1.80 (s, 3H), 2.88 (d, J = 6.6 Hz, 1H), 3.14 (d, J = 6.3 Hz, 1H), 5.32, (d, J = 1.5 Hz, 1H), 6.38 (d, J = 5.7 Hz, 1H), 6.57 (dd, J = 1.5 and 5.4 Hz, 1H), 7.27–7.30 (m, 2H), 7.37–7.51 (m,3H); 13C NMR (CDCl3) δ 15.7, 49.5, 50.6, 81.1, 88.6, 126.5, 128.7, 129.1, 131.7, 137.1, 140.7, 174.0, 175.3. (Figures S36 and S37) The enantiomeric purity of the solid was determined by HPLC using a CHIRALPAK IA (Daicel Ind.); column. tR(1) = 20 min for (+)-3a, tR(2) = 30.5 min for (–)-3a. Eluent: hexane/EtOH = 80:20 (v/v); flow rate: 0.7 mL/min.
(3aS*,4R*,7S*,7aR*)-2-(4-Chlorophenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3b)
Colorless crystal; m.p. 120 °C; 90% yield; 1H NMR (CDCl3) δ 1.78 (s, 3H), 2.85 (d, J = 6.5 Hz, 1H), 3.11 (d, J = 6.5 Hz, 1H), 5.29 (d, J = 1.8 Hz, 1H), 6.37 (d, J = 5.6 Hz, 1H), 6.55 (dd, J = 5.6, 1.6 Hz, 1H), 7.23–7.26 (m, 2H), 7.42–7.44 (m, 2H); 13C NMR (CDCl3) δ 15.7, 49.5, 50.6, 81.1, 88.6, 127.7, 129.2, 130.2, 134.4, 137.0, 140.7, 173.7, 175.0; IR (cm−1, KBr) 1701; HRMS (ESI-MS) m/z calcd for C15H12ClNO3 + H 290.0578, found 290.0577. (Figures S38 and S39) The enantiomeric purity of the solid was determined by HPLC using a CHIRALPAK IA (Daicel Ind.); column. tR(1) = 20 min for (+)-3b, tR(2) = 22 min for (–)-3b. Eluent: hexane/EtOH = 90: 10 (v/v); flow rate: 1.0 mL/min. (Figures S12 and S13)
(3aS*,4R*,7S*,7aR*)-2-(3,4-Dimethylphenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3c)
Colorless crystal; m.p. 140 °C; 91% yield; 1H NMR (CDCl3) δ 1.78 (s,3H), 2.27 (s, 3H), 2.27 (s, 3H), 2.84 (d, J = 6.5 Hz, 1H), 3.10 (d, J = 6.5 Hz, 1H), 5.30 (d, J = 1.6 Hz, 1H), 6.36 (d, J = 5.6 Hz, 1H), 6.54 (dd, J = 5.6, 1.6 Hz, 1H), 6.96-7.01 (m, 2H), 7.21–7.26 (m, 1H); 13C NMR (CDCl3) δ 15.7, 19.5, 19.8, 49.4, 50.6, 81.0, 88.5, 123.9, 127.5, 129.2, 130.2, 137.0, 137.6, 137.7, 140.7, 174.2, 175.6; IR (cm−1, KBr) 1708; HRMS (ESI-MS) m/z calcd for C17H17NO3 + H 284.1281, found 284.1276. (Figures S40 and S41) The enantiomeric purity of the solid was determined by HPLC using a CHIRALPAK IA (Daicel Ind.); column. tR(1) = 29.5 min for (+)-3c, tR(2) = 33 min for (–)-3c. Eluent: hexane/EtOH = 80: 20 (v/v); flow rate: 0.5 mL/min. (Figures S14 and S15)
(3aS*,4R*,7S*,7aR*)-2-(3,5-Dichlorophenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3d)
Colorless crystal; m.p. 136 °C; 99% yield; 1H NMR (CDCl3) δ 1.79 (s, 3H), 2.87 (d, J = 6.5 Hz, 1H), 3.13 (d, J = 6.5 Hz, 1H), 5.30 (d, J = 1.8 Hz, 1H), 6.38 (d, J = 5.6 Hz, 1H), 6.57 (dd, J = 5.6, 1.6 Hz, 1H), 7.26 (m, 2H), 7.39–7.40 (m, 1H); 13C NMR (CDCl3) δ 15.7, 49.5, 50.6, 81.2, 88.7, 125.1, 128.8, 133.3, 135.2, 137.1, 140.7, 173.2, 174.5; IR (cm−1, KBr) 1712. (Figures S42 and S43) The enantiomeric purity of the solid was determined by HPLC using a CHIRALPAK IA (Daicel Ind.); column. tR(1) = 13.5 min for (+)-3d, tR(2) = 16 min for (–)-3d. Eluent: hexane/EtOH = 90 : 10 (v/v); flow rate: 1.0 mL/min. (Figures S16 and S17)
(3aS*,4R*,7S*,7aR*)-4-Methyl-2-(4-tolyl)-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3e)
Colorless crystal; m.p. 132 °C; 90% yield; 1H NMR (CDCl3) δ 1.79 (s, 3H), 2.38 (s, 3H), 2.86 (d, J = 6.6 Hz, 1H), 3.12 (d, J = 6.6 Hz, 1H), 5.31 (d, J = 1.8 Hz, 1H), 6.37 (d, J = 5.5 Hz, 1H), 6.56 (dd, J = 5.6, 1.7 Hz, 1H), 7.13–7.28 (m, 4H); 13C NMR (CDCl3) δ 15.7, 21.2, 49.4, 50.6, 81.0, 88.5, 126.3, 129.1, 129.7, 137.0, 138.7, 140.7, 174.1, 175.4; IR (cm−1, KBr) 1707. (Figures S44 and S45)
(3aS*,4R*,7S*,7aR*)-2-(4-Fluorophenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3f)
Colorless crystal; m.p. 133 °C; 88% yield; 1H NMR (CDCl3) δ 1.78 (s, 3H), 2.86 (d, J = 6.5 Hz, 1H), 3.11 (d, J = 6.5 Hz, 1H), 5.29 (d, J = 1.8 Hz, 1H), 6.37 (d, J = 5.6 Hz, 1H), 6.55 (dd, J = 5.7, 1.7 Hz, 1H), 7.13–7.29 (m, 4H); 13C NMR (CDCl3) δ 15.7, 49.4, 50.6, 81.1, 88.6, 116.0, 116.2, 127.6, 128.3, 128.4, 137.0, 140.7, 160.9, 163.4, 173.9, 175.2. (Figures S46 and S47)
(3aS*,4R*,7S*,7aR*)-2-(4-Bromophenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3g)
Colorless crystal; m.p. 127 °C; 94% yield; 1H NMR (CDCl3) δ 1.78 (s, 3H), 2.86 (d, J = 6.5 Hz, 1H), 3.12 (d, J = 6.5 Hz, 1H), 5.29 (d, J = 1.8 Hz, 1H), 6.37 (d, J = 5.6 Hz, 1H), 6.56 (dd, J = 5.7, 1.7 Hz, 1H), 7.18–7.20 (m, 2H), 7.58–7.60 (m, 2H); 13C NMR (CDCl3) δ 15.7, 49.5, 50.6, 81.1, 88.6, 122.5, 128.0, 130.6, 132.2, 137.0, 140.7, 173.6, 174.9; IR (cm−1, KBr) 1705. (Figures S48 and S49)
(3aS*,4R*,7S*,7aR*)-4-Methyl-2-(3-tolyl)-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3h)
Colorless crystal; m.p. 128 °C; 93% yield; 1H NMR (CDCl3) δ 1.79 (s, 3H), 2.38 (s, 3H), 2.86 (d, J = 6.5 Hz, 1H), 3.12 (d, J = 6.5 Hz, 1H), 5.31 (d, J = 1.6 Hz, 1H), 6.37 (d, J = 5.6 Hz, 1H), 6.55 (dd, J = 5.6, 1.6 Hz, 1H), 7.05–7.37 (m, 4H); 13C NMR (CDCl3) δ 15.7, 21.3, 49.5, 50.6, 81.1, 88.6, 123.6, 127.1, 128.9, 129.6, 131.6, 137.0, 139.2, 140.7, 174.1, 175.4; IR (cm−1, KBr) 1707; HRMS (ESI-MS) m/z calcd for C16H15NO3 - H 268.0979, found 288.0992. (Figures S50 and S51)
(3aS*,4R*,7S*,7aR*)-2-(4-Ethylphenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3i)
Colorless crystal; m.p. 114 °C; 88% yield; 1H NMR (CDCl3) δ 1.24 (t, J = 7.7 Hz, 3H), 1.79 (s, 3H), 2.67 (q, J = 7.6 Hz, 2H), 2.85 (d, J = 6.5 Hz, 1H), 3.11 (d, J = 6.5 Hz, 1H), 5.30 (d, J = 1.8 Hz, 1H), 6.36 (d, J = 6.5 Hz, 1H), 6.55 (dd, J = 5.6, 1.6 Hz, 1H), 7.16–7.30 (m, 4H); 13C NMR (CDCl3) δ 15.3, 15.7, 28.6, 49.5, 50.6, 81.1, 88.6, 126.4, 128.6, 129.3, 137.0, 140.7, 144.9, 174.2, 175.5; IR (cm−1, KBr) 1702; HRMS (ESI-MS) m/z calcd for C17H17NO3 + H 284.1281, found 284.1277. (Figures S52 and S53)
(3aS*,4R*,7S*,7aR*)-2-(4-Methoxyphenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3j)
Colorless crystal; m.p. 120–121 °C; 94% yield; 1H NMR (CDCl3) δ 1.78 (s, 3H), 2.84 (d, J = 6.5 Hz, 1H), 3.10 (d, J = 6.5 Hz, 1H), 3.82 (s, 3H), 5.29 (d, J = 1.8 Hz, 1H), 6.36 (d, J = 5.7 Hz, 1H), 6.54 (dd, J = 5.6, 1.6 Hz, 1H), 6.94–7.26 (m, 4H); 13C NMR (CDCl3) δ 15.7, 49.4, 50.5, 55.4, 81.0, 88.5, 114.4, 124.4, 127.7, 137.0, 140.7, 159.5, 174.3, 175.5. (Figures S54 and S55)
Single crystal X-ray structure analysis of (3aS,4R,7S,7aR)-2-(4-chlorophenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3b)
Disordered crystal of the ratio of 73:27, exhibiting (-)-CD sign at 254 nm for major isomer. Colorless prism (0.20 × 0.20 × 0.01 mm3), orthorhombic space group P212121, a = 6.5485(4) Å, b = 12.3747(6) Å, c = 16.8764(9) Å, V = 1367.59(13) Å3, Z = 4, λ (CuKα) = 1.54178 Å, ρ = 1.407 g/cm3, μ (CuKα) = 2.539 cm, 3954 reflections measured (T = 173 K, 4.430° < θ < 68.264°), nb of independent data collected: 2213 nb of independent data used for refinement: 2127 in the final least-squares refinement cycles on F2, the model converged at R1 = 0.0505, wR2 = 0.1993 [I > 2σ(I)], R1 = 0.0522, wR2 = 0.1408 (all data), and GOF = 1.065, H-atom parameters constrained, absolute Flack parameter = 0.505(8). (CCDC 1985809). (Figure S1)
Single crystal X-ray structure analysis of (3aS,4R,7S,7aR)-2-(4-chlorophenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3b)
Enantiomerically pure crystal provided by optical resolution using HPLC, exhibiting (-)-CD sign at 254 nm. Colorless prism (0.20 × 0.20 × 0.10 mm3), orthorhombic space group P212121, a = 6.5975(2) Å, b = 12.3565(4) Å, c = 16.7223(6) Å, V = 1363.24(8) Å3, Z = 4, λ (CuKα) = 1.54178 Å, ρ = 1.412 g/cm3, μ (CuKα) = 2.547 cm, 12970 reflections measured (T = 173 K, 4.449° < θ < 68.213°), nb of independent data collected: 2427, nb of independent data used for refinement: 2397 in the final least-squares refinement cycles on F2, the model converged at R1 = 0.0369, wR2 = 0.0985 [I > 2σ(I)], R1 = 0.0371, wR2 = 0.0989 (all data), and GOF = 1.059, H-atom parameters constrained, absolute Flack parameter = 0.051(3). (CCDC 1985810). (Figure S2)
Single crystal X-ray structure analysis of (3aS,4R,7S,7aR)-2-(3,4-dimethylphenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3c)
Exhibiting (-)-CD sign at 254 nm. Colorless prism (0.20 × 0.10 × 0.10 mm3), orthorhombic space group P212121, a = 7.9707(3) Å, b = 12.9386(5) Å, c = 13.7305(5) Å, V = 1416.02(9) Å3, Z = 4, λ (CuKα) = 1.54178 Å, ρ = 1.329 g/cm3, μ (CuKα) = 0.741 cm, 21885 reflections measured (T = 173 K, 4.696° < θ < 68.241°), nb of independent data collected: 2593, nb of independent data used for refinement: 2576 in the final least-squares refinement cycles on F2, the model converged at R1 = 0.0334, wR2 = 0.0855 [I > 2σ(I)], R1 = 0.0335, wR2 = 0.0856 (all data), and GOF = 1.065, H-atom parameters constrained, absolute Flack parameter = 0.099(16). (CCDC 1985811). (Figure S3)
Single crystal X-ray structure analysis of (3aS,4R,7S,7aR)-2-(3,5-dichlorophenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3d)
Disordered crystal of the ratio of 76:24, exhibiting (-)-CD sign at 254 nm for major isomer. Colorless prism (0.30 × 0.20 × 0.20 mm3), monoclinic space group P21, a = 5.4032(5) Å, b = 13.9697(10) Å, c = 9.6673(8) Å, β = 105.179(4)°, V = 704.24(10) Å3, Z = 2, λ (CuKα) = 1.54178 Å, ρ = 1.529 g/cm3, μ (CuKα) = 4.237 cm, 2350 reflections measured (T = 173 K, 5.7024° < θ < 68.068°), nb of independent data collected: 1457, nb of independent data used for refinement: 1452 in the final least-squares refinement cycles on F2, the model converged at R1 = 0.0532, wR2 = 0.1391 [I > 2σ(I)], R1 = 0.0532, wR2 = 0.1392 (all data), and GOF = 1.125, H-atom parameters constrained, absolute Flack parameter = 0.12(3). (CCDC 1985812). (Figure S4)
Single crystal X-ray structure analysis of (3aR,4S,7R,7aS)-2-(3,5-dichlorophenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-(+)-3d)
Enantiomerically pure crystal provided by optical resolution using HPLC. Colorless prism (0.30 × 0.20 × 0.20 mm3), monoclinic space group P21, a = 5.3713(9) Å, b = 13.958(2) Å, c = 9.5751(14) Å, β = 103.609(5)°, V = 697.72(19) Å3, Z = 2, λ (CuKα) = 1.54178 Å, ρ = 1.529 g/cm3, μ (CuKα) = 4.277 cm, 10993 reflections measured (T = 173 K, 4.752° < θ < 72.198°), nb of independent data collected: 2558, nb of independent data used for refinement: 2519 in the final least-squares refinement cycles on F2, the model converged at R1 = 0.0429, wR2 = 0.1034 [I > 2σ(I)], R1 = 0.0431, wR2 = 0.1036 (all data), and GOF = 1.101, H-atom parameters constrained, absolute Flack parameter = 0.110(5). (CCDC 1985813). (Figure S5)
Single crystal X-ray structure analysis of analysis of (3aR,4S,7R,7aS)-4-methyl-2-(4-tolyl)-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3e)
Colorless prism (0.20 × 0.10 × 0.10 mm3), orthorhombic space group P212121, a = 9.3007(6) Å, b = 10.6635(6) Å, c = 13.7010(9) Å, V = 1358.84(15) Å3, Z = 4, λ (CuKα) = 1.54178 Å, ρ = 1.316 g/cm3, μ (CuKα) = 0.746 cm, 4911 reflections measured (T = 173 K, 6.314° < θ < 68.278°), nb of independent data collected: 2263, nb of independent data used for refinement: 2225 in the final least-squares refinement cycles on F2, the model converged at R1 = 0.0458, wR2 = 0.1278 [I > 2σ(I)], R1 = 0.0462, wR2 = 0.1284 (all data), and GOF = 1.048, H-atom parameters constrained, absolute Flack parameter = 0.39(7). (CCDC 1985814). (Figure S6)
Single crystal X-ray structure analysis of analysis of (3aS*,4R*,7S*,7aR*)-2-(4-fluorophenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3f)
Colorless prism (0.20 × 0.10 × 0.03 mm3), monoclinic space group P21/c, a = 9.9813(13) Å, b = 13.0236(15) Å, c = 9.7580(8) Å, β = 101.379(9)°, V = 1243.5(2) Å3, Z = 4, λ (CuKα) = 1.54178 Å, ρ = 1.460 g/cm3, μ (CuKα) = 0.945 cm, 2238 reflections measured (T = 173 K, 5.655° < θ < 68.328°), nb of independent data collected: 2238, nb of independent data used for refinement: 1687 in the final least-squares refinement cycles on F2, the model converged at R1 = 0.0687, wR2 = 0.1787 [I > 2σ(I)], R1 = 0.0915, wR2 = 0.1875 (all data), and GOF = 1.172, H-atom parameters constrained. (CCDC 1985815). (Figure S7)
Single crystal X-ray structure analysis of (3aS*,4R*,7S*,7aR*)-2-(4-bromophenyl)-4-methyl-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (exo-3g).
Colorless prism (0.20 × 0.20 × 0.20 mm3), monoclinic space group P21/n, a = 20.838(6) Å, b = 6.485(2) Å, c = 20.853(6) Å, β = 104.364(3)°, V = 2729.9(15) Å3, Z = 8, λ (MoKα) = 0.71073 Å, ρ = 1.626 g/cm3, μ (MoKα) = 3.018 cm, 5224 reflections measured (T = 173 K, 1.593° < θ < 27.570°), nb of independent data collected: 5224, nb of independent data used for refinement: 3679 in the final least-squares refinement cycles on F2, the model converged at R1 = 0.0511, wR2 = 0.1118 [I > 2σ(I)], R1 = 0.0928, wR2 = 0.1310 (all data), and GOF = 1.025, H-atom parameters constrained. (CCDC 1985818). (Figure S8)
Reaction conditions for asymmetric DA reaction via dynamic crystallization
In a sealed tube (L = 200 mm, Φ = 25 mm), N-arylmaleimide 2 (100 mg), 2-methylfuran 1 (15 eq.), TFA (0–1.0 eq), and heptane (1.0 mL) were stirred with or without glass beads (250 mg) using a stir bar at 80 °C. The crystalline adduct 3 appeared after a few minutes, and the solution was kept in suspension by stirring at 600 rpm for several days at 80 °C. The change of ee value of crystalline 3 was monitored by HPLC using CHIRALPAK IA (Daicel Ind.) column; eluent: n-hexane/EtOH. Finally, crystalline exo-3 was isolated by filtration. The same procedure was performed for all substrates.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-8994/12/6/910/s1, Figure S1: Single crystal X-Ray crystallographic analysis of exo-3b (disordered), Figure S2: Single crystal X-Ray crystallographic analysis of exo-3b (enantiopure), Figure S3: Single crystal X-Ray crystallographic analysis of exo-3c (enantiopure), Figure S4: Single crystal X-Ray crystallographic analysis of exo-3d (disordered), Figure S5: Single crystal X-Ray crystallographic analysis of exo-3d (enantiopure), Figure S6: Single crystal X-Ray crystallographic analysis of exo-3e (enantiopure), Figure S7: Single crystal X-Ray crystallographic analysis of exo-3f, Figure S8: Single crystal X-Ray crystallographic analysis of exo-3g, Figure S9: Time course for DA reaction of 1 (0.5 M) and 2b–d (0.05 M) at 60 °C in CDCl3 monitored by 1H NMR, Figure S10: Time course for reverse-DA reaction of exo-3b–d (0.05 M) at 60 °C in CDCl3 monitored by 1H NMR, Figure S11: Time course for reverse-DA reaction of exo-3b–d (0.03 M) in the presence of TFA (0.03 eq) at 60 °C in CDCl3 monitored by 1H NMR, Figure S12: HPLC analysis of racemic exo-3b, Figure S13: HPLC analysis of 40% ee of exo-(-)-3b, Figure S14. HPLC analysis of racemic of exo-3c, Figure S15. HPLC analysis of 98% ee of exo-(-)-3c, Figure S16. HPLC analysis of racemic of exo-3d, Figure S17. HPLC analysis of 49% ee of exo-(+)-3d, Figure S18: 1H NMR spectrum of maleimide 2b, Figure S19: 13C NMR spectrum of maleimide 2b, Figure S20: 1H NMR spectrum of maleimide 2c, Figure S21: 13C NMR spectrum of maleimide 2c, Figure S22: 1H NMR spectrum of maleimide 2d, Figure S23: 13C NMR spectrum of maleimide 2d, Figure S24: 1H NMR spectrum of maleimide 2e, Figure S25: 13C NMR spectrum of maleimide 2e, Figure S26: 1H NMR spectrum of maleimide 2f, Figure S27: 13C NMR spectrum of maleimide 2f, Figure S28: 1H NMR spectrum of maleimide 2g, Figure S29: 13C NMR spectrum of maleimide 2g, Figure S30: 1H NMR spectrum of maleimide 2h, Figure S31: 13C NMR spectrum of maleimide 2h, Figure S32: 1H NMR spectrum of maleimide 2i, Figure S33: 13C NMR spectrum of maleimide 2i, Figure S34: 1H NMR spectrum of maleimide 2j, Figure S35: 13C NMR spectrum of maleimide 2j, Figure S36: 1H NMR spectrum of exo-3a, Figure S37: 13C NMR spectrum of exo-3a, Figure S38: 1H NMR spectrum of exo-3b, Figure S39: 13C NMR spectrum of exo-3b, Figure S40: 1H NMR spectrum of exo-3c, Figure S41: 13C NMR spectrum of exo-3c, Figure S42: 1H NMR spectrum of endo-3d, Figure S43: 13C NMR spectrum of endo-3d, Figure S44: 1H NMR spectrum of exo-3e, Figure S45: 13C NMR spectrum of exo-3e, Figure S46: 1H NMR spectrum of endo-3f, Figure S47: 13C NMR spectrum of endo-3f, Figure S48: 1H NMR spectrum of exo-3g, Figure S49: 13C NMR spectrum of exo-3g, Figure S50: 1H NMR spectrum of endo-3h, Figure S51: 13C NMR spectrum of endo-3h, Figure S52: 1H NMR spectrum of exo-3i, Figure S53: 13C NMR spectrum of exo-3i, Figure S54: 1H NMR spectrum of endo-3j, Figure S55: 13C NMR spectrum of endo-3j.
Author Contributions
Conceptualization, M.S.; methodology, N.U., S.T., W.S., M.S.; validation, Y.Y. and T.M.; formal analysis, N.U., S.T., W.S.; investigation, N.U., S.T., W.S.; data curation, Y.Y., T.M., M.S.; writing—original draft preparation, N.U., S.T., M.S.; writing—review and editing, M.S.; supervision, M.S.; project administration, M.S.; funding acquisition, M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Grants-in-Aid for Scientific Research (No. 19H02708) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of the Japanese Government. Uemura acknowledges financial support from the Frontier Science Program of the Graduate School of Science and Engineering, Chiba University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Diels, O.; Alder, K. Synthesen in der Hydroaromatischen Reihe. Justus Liebigs Ann. Chem. 1928, 460, 98–122. [Google Scholar] [CrossRef]
- Diels, O.; Alder, K. Synthesen in der Hydroaromatischen Reihe, IV. Mitteilung: Über die Anlagerung von Maleinsäure-anhydrid an Arylierte Diene, Triene und Fulvene. Ber. Dtsch. Chem. Ges. 1929, 62, 2081–2087. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels-Alder Reaction in Total Synthesis. Angew. Chem. Int. Ed. 2002, 41, 1668–1698. [Google Scholar] [CrossRef]
- Takao, K.-I.; Munakata, R.; Tadano, K. Recent advances in natural product synthesis by using intramolecular Diels-Alder reactions. Chem. Rev. 2005, 105, 4779–4807. [Google Scholar] [CrossRef]
- Min, L.; Liu, X.; Li, C.-C. Total Synthesis of Natural Products with Bridged Bicyclo[m.n.1] Ring Systems via Type II [5 + 2] Cycloaddition. Acc. Chem. Res. 2020, 53, 703–718. [Google Scholar] [CrossRef]
- Breunig, M.; Yuan, P.; Gaich, T. An Unexpected Transannular [4 + 2] Cycloaddition during the Total Synthesis of (+)-Norcembrene. Angew. Chem. Int. Ed. 2020, 59, 5521–5525. [Google Scholar] [CrossRef]
- Chavan, S.P.; Kadam, A.L.; Gonnade, R.G. Enantioselective Formal Total Synthesis of (-)-Quinagolide. Org. Lett. 2019, 21, 9089–9093. [Google Scholar] [CrossRef]
- Burns, A.S.; Rychnovsky, S.D. Total Synthesis and Structure Revision of (-)-Illisimonin A, a Neuroprotective Sesquiterpenoid from the Fruits of Illicium simonsii. J. Am. Chem. Soc. 2019, 141, 13295–13300. [Google Scholar] [CrossRef]
- Zhou, S.; Xia, K.; Leng, X.; Li, A. Asymmetric Total Synthesis of Arcutinidine, Arcutinine, and Arcutine. J. Am. Chem. Soc. 2019, 141, 13718–13723. [Google Scholar] [CrossRef]
- Maurya, V.; Appayee, C. Enantioselective Total Synthesis of Potent 9β-11-Hydroxyhexahydrocannabinol. J. Org. Chem. 2020, 85, 1291–1297. [Google Scholar] [CrossRef]
- Corey, E.J.; Shibata, T.; Lee, T.W. Asymmetric Diels-Alder Reactions Catalyzed by a Triflic Acid Activated Chiral Oxazaborolidine. J. Am. Chem. Soc. 2002, 124, 3808–3809. [Google Scholar] [CrossRef] [PubMed]
- Choy, W.; Reed, L.A.; Masamune, S. Asymmetric Diels-Alder Reaction: Design of Chiral Dienophiles. J. Org. Chem. 1983, 48, 1137–1139. [Google Scholar] [CrossRef]
- Oppolzer, W. Asymmetric Diels-Alder and Ene Reactions in Organic Synthesis. New Synthetic Methods (48). Angew. Chem. Int. Ed. 1984, 23, 876–889. [Google Scholar] [CrossRef]
- Kagan, H.B.; Riant, O. Catalytic Asymmetric Diels-Alder Reactions. Chem. Rev. 1992, 92, 1007–1019. [Google Scholar] [CrossRef]
- Mehta, G.; Uma, R. Stereoelectronic Control in Diels−Alder Reaction of Dissymmetric 1,3-Dienes. Acc. Chem. Res. 2000, 33, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.M.; Jen, W.S.; MacMillan, D.W.C. Enantioselective Organocatalytic Intramolecular Diels−Alder Reactions. The Asymmetric Synthesis of Solanapyrone D. J. Am. Chem. Soc. 2005, 127, 11616–11617. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lin, L.; Liu, X.; Hu, X.; Lu, Y.; Zhang, X.; Feng, X. Catalytic Asymmetric Diels-Alder Reaction/[3,3] Sigmatropic Rearrangement Cascade of 1-Thiocyanatobutadienes. Angew. Chem. Int. Ed. 2018, 57, 9113–9116. [Google Scholar] [CrossRef]
- Li, M.; Carreras, V.; Jalba, A.; Ollevier, T. Asymmetric Diels-Alder Reaction of α,β-Unsaturated Oxazolidin-2-one Derivatives Catalyzed by a Chiral Fe(III)-Bipyridine Diol Complex. Org. Lett. 2018, 20, 995–998. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Lin, L.; Fu, K.; Zheng, H.; Liu, X.; Feng, X. Synthesis of Optically Pure Spiro[cyclohexane-oxindoline] Derivatives via Catalytic Asymmetric Diels-Alder Reaction of Brassard-Type Diene with Methyleneindolines. J. Org. Chem. 2015, 80, 8836–8842. [Google Scholar] [CrossRef]
- Chauhan, M.S.; Kumar, P.; Singh, S. Synthesis of MacMillan catalyst modified with ionic liquid as a recoverable catalyst for asymmetric Diels-Alder reaction. RSC Adv. 2015, 5, 52636–52641. [Google Scholar] [CrossRef]
- Walborsky, H.; Barash, L.; Davis, T. Communications- Partial Asymmetric Syntheses: The Diels-Alder Reaction. J. Am. Chem. Soc. 1988, 110, 1238–1256. [Google Scholar]
- Evans, D.A.; Chapman, K.T.; Bisaha, J. Asymmetric Diels-Alder Cycloaddition Reactions with Chiral α,β-Unsaturated N-Acyloxazolidinones. J. Am. Chem. Soc. 1988, 110, 1238–1256. [Google Scholar] [CrossRef]
- Robiette, R.; Cheboub-Benchaba, K.; Peeters, D.; Marchand-Brynaert, J. Design of a New and Highly Effective Chiral Auxiliary for Diels-Alder Reaction of 1-Aminodiene. J. Org. Chem. 2003, 68, 9809–9812. [Google Scholar] [CrossRef] [PubMed]
- Lakner, F.J.; Negrete, G.R. A new and convenient chiral auxiliary for asymmetric Diels-Alder cycloadditions in environmentally benign solvents. Synlett 2002, 4, 643–645. [Google Scholar] [CrossRef]
- Hachiya, S.; Kasashima, Y.; Yagishita, F.; Mino, T.; Masu, H.; Sakamoto, M. Asymmetric Transformation by Dynamic Crystallization of Achiral Succinimides. Chem. Commun. 2013, 49, 4776–4778. [Google Scholar] [CrossRef]
- Addadi, L.; Lahav, M. Origin of Optical Activity in Nature; Walker, D.C., Ed.; Elsevier: New York, NY, USA, 1979. [Google Scholar]
- Mason, S.F. Origins of Biomolecular Handedness. Nature 1984, 311, 19–23. [Google Scholar] [CrossRef]
- Bonner, W.A. The Origin and Amplification of Biomolecular Chirality. Orig. Life Evol. Biosph. 1991, 21, 59. [Google Scholar] [CrossRef]
- Avalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.L.; Palacios, J.C.; Barron, L.D. Absolute Asymmetric Synthesis under Physical Fields: Facts and Fictions. Chem. Rev. 1998, 98, 2391–2404. [Google Scholar] [CrossRef]
- Feringa, B.L.; van Delden, R. Absolute Asymmetric Synthesis: The Origin, Control, and Amplification of Chirality. Angew. Chem. Int. Ed. 1999, 38, 3418–3438. [Google Scholar] [CrossRef]
- Tsogoeva, S.B.; Wei, S.; Freund, M.; Mauksch, M. Deracemization with reversible Mannich type reaction. Angew. Chem. Int. Ed. 2009, 48, 590–594. [Google Scholar] [CrossRef]
- Flock, A.M.; Reucher, C.M.M.; Bolm, C. Enantioenrichment by Iterative Retro-Aldol/Aldol ReactionCatalyzed by an Achiral or Racemic Base. Chem. Eur. J. 2010, 16, 3918–3921. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, F.; Ishikawa, H.; Onuki, T.; Hachiya, S.; Mino, T.; Sakamoto, M. Total spontaneous resolution by deracemization of isoindolinones. Angew. Chem. Int. Ed. 2012, 51, 13023–13025. [Google Scholar] [CrossRef] [PubMed]
- Steendam, R.R.E.; Verkade, J.M.M.; Van Benthem, T.J.B.; Meekes, H.; Van Enckevort, W.J.P.; Raap, J.; Rutjes, F.P.J.T.; Vlieg, E. Emergence of Single-molecular Chirality from Achiral Reactants. Nat. Commun. 2014, 5, 5543. [Google Scholar]
- Kaji, Y.; Uemura, N.; Kasashima, Y.; Ishikawa, H.; Yoshida, Y.; Mino, T.; Sakamoto, M. Asymmetric Synthesis of an Amino Acid Derivative from Achiral Aroyl Acrylamide by Reversible Michael Addition and Preferential Crystallization. Chem. Eur. J. 2016, 22, 16429–16432. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Sano, K.; Matsumoto, A.; Yoshida, Y.; Mino, T.; Sakamoto, M. Absolute Asymmetric Synthesis of an Aspartic Acid Derivative from Prochiral Maleic Acid and Pyridine under Achiral Conditions. Chem. Asian J. 2019, 14, 4150–4153. [Google Scholar] [CrossRef]
- Kawasaki, T.; Takamatsu, N.; Aiba, S.; Tokunaga, Y. Spontaneous Formation and Amplification of an Enantioenriched α-Amino Nitrile: A Chiral Precursor for Strecker Amino Acid Synthesis. Chem. Commun. 2015, 51, 14377–14380. [Google Scholar] [CrossRef]
- Takamatsu, N.; Aiba, S.; Yamada, T.; Tokunaga, Y.; Kawasaki, T. Highly Stereoselective Strecker Synthesis Induced by a Slight Modification of Benzhydrylamine from Achiral to Chiral. Chem. Eur. J. 2018, 24, 1304–1310. [Google Scholar] [CrossRef]
- Sakamoto, M.; Shiratsuki, K.; Uemura, N.; Ishikawa, H.; Yoshida, Y.; Kasashima, Y.; Mino, T. Asymmetric Synthesis by Using Natural Sunlight under Absolute Achiral Conditions. Chem. Eur. J. 2017, 23, 1717–1721. [Google Scholar] [CrossRef]
- Ishikawa, H.; Uemura, N.; Yagishita, Y.; Baba, N.; Yoshida, Y.; Mino, T.; Kasashima, Y.; Sakamoto, M. Asymmetric Synthesis Involving Reversible Photodimerization of a Prochiral Flavonoid Followed by Crystallization. Eur. J. Org. Chem. 2017, 46, 6878–6881. [Google Scholar] [CrossRef]
- Uemura, N.; Toyoda, S.; Ishikawa, H.; Yoshida, Y.; Mino, T.; Kasashima, Y.; Sakamoto, M. Asymmetric Diels−Alder Reaction Involving Dynamic Enantioselective Crystallization. J. Org. Chem. 2018, 83, 9300–9304. [Google Scholar] [CrossRef]
- Viedma, C. Chiral Symmetry Breaking during Crystallization: Complete Chiral Purity Induced by Nonlinear Autocatalysis and Recycling. Phys. Rev. Lett. 2005, 94, 065504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coquerel, G. Crystallization of Molecular Systems from Solution: Phase Diagrams, Supersaturation and Other Basic Concepts. Chem. Soc. Rev. 2014, 43, 2286–2300. [Google Scholar] [CrossRef] [PubMed]
- Viedma, C.; Ortiz, J.E.; de Torres, T.; Izumi, T.; Blackmond, D.G.; Viedma, C.; Ortiz, J.E.; de Torres, T.; Izumi, T.; Blackmond, D.G. Evolution of Solid Phase Homochirality for a Proteinogenic Amino Acid. J. Am. Chem. Soc. 2008, 130, 15274–15275. [Google Scholar] [CrossRef] [PubMed]
- Noorduin, W.L.; Bode, A.A.C.; van der Meiden, M.; Meekes, H.; van Etteger, A.F.; van Enckevort, W.J.P.; Christianen, P.C.M.; Kaptein, B.; Kellogg, R.M.; Rasing, T.; et al. Complete Chiral Symmetry Breaking of an Amino Acid Derivative Directed by Circularly Polarized Light. Nature Chem. 2009, 1, 729–732. [Google Scholar] [CrossRef] [Green Version]
- Gherase, D.; Conroy, D.; Matar, O.K.; Blackmond, D.G. Experimental and Theoretical Study of the Emergence of Single Chirality in Attrition-Enhanced Deracemization. Cryst. Growth Des. 2014, 14, 928–937. [Google Scholar] [CrossRef]
- Sogutoglu, L.-C.; Steendam, R.R.E.; Meekes, H.; Vlieg, E.; Rutjes, F.P.J.T. Viedma Ripening: A Reliable Crystallisation Method to Reach Single Chirality. Chem. Soc. Rev. 2015, 44, 6723–6732. [Google Scholar] [CrossRef] [Green Version]
- Steendam, R.R.E.; Kulka, M.W.; Meekes, H.; van Enckevort, W.J.P.; Raap, J.; Vlieg, E.; Rutjes, F.P.J.T. One-Pot Synthesis, Crystallization and Deracemization of Isoindolinones from Achiral Reactants. Eur. J. Org. Chem. 2015, 7249–7252. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.P.T.; Cheung, P.S.M.; Werber, L.; Gagnon, J.; Sivakumar, R.; Lennox, C.; Sossin, A.; Mastai, Y.; Cuccia, L.A. Directing the Viedma ripening of ethylenediammonium sulfate using “Tailor-made” chiral additives. Chem. Commun. 2016, 52, 12626–12629. [Google Scholar] [CrossRef]
- Sivakumar, R.; Askari, M.S.; Woo, S.; Madwar, C.; Ottenwaelder, X.; Bohle, D.S.; Cuccia, L.A. Homochiral crystal generation via sequential dehydration and Viedma ripening. Cryst. Eng. Comm. 2016, 18, 4277–4280. [Google Scholar] [CrossRef]
- Breveglieri, F.; Maggioni, G.M.; Mazzotti, M. Deracemization of NMPA via Temperature Cycles. Cryst. Growth Des. 2018, 18, 1873–1881. [Google Scholar] [CrossRef] [Green Version]
- Engwerda, A.H.J.; Maassen, R.; Tinnemans, P.; Meekes, H.; Rutjes, F.P.J.T.; Vlieg, E. Attrition-Enhanced Deracemization of the Antimalaria Drug Mefloquine. Angew. Chem. Int. Ed. 2019, 58, 1670–1673. [Google Scholar] [CrossRef] [PubMed]
- Houk, K.N.; Blackmond, D.G. Isotopically Directed Symmetry Breaking and Enantioenrichment in Attrition-Enhanced Deracemization. J. Am. Chem. Soc. 2020, 142, 3873–3879. [Google Scholar]
- Ishikawa, H.; Ban, K.; Uemura, N.; Yoshida, Y.; Mino, T.; Kasashima, Y.; Sakamoto, M. Attrition-Enhanced Deracemization of Axially Chiral Nicotinamides. Eur. J. Org. Chem. 2020, 8, 1001–1005. [Google Scholar] [CrossRef]
- Havinga, E. Spontaneous Formation of Optically Active Substances. Biochim. Biophys. Acta 1954, 13, 171–174. [Google Scholar] [CrossRef]
- Frank, F.C. On Spontaneous Asymmetric Synthesis. Biochim. Biophys. Acta 1953, 11, 459–463. [Google Scholar] [CrossRef]
- Yoshioka, R. Racemization, Optical Resolution and Crystallization-induced Asymmetric Transformation of Amino Acids and Pharmaceutical Intermediates. Top. Curr. Chem. 2007, 269, 83–132. [Google Scholar]
- Sakamoto, M.; Mino, T. Asymmetric Reaction Using Molecular Chirality Controlled by Spontaneous Crystallization. Eur. J. Org. Chem. 2017, 6878–6881. [Google Scholar] [CrossRef]
- Jacques, J.; Collet, A.; Wilen, S.H. Enantiomers, Racemates and Resolution; Krieger: Malabar, FL, USA, 1994. [Google Scholar]
- Coquerel, G. Chiral Discrimination in the Solid State: Applications to Resolution and Deracemization; Springer: Tokyo, Japan, 2015; pp. 393–420. [Google Scholar]
- Kellogg, R.M. How to Use Pasteur’s Tweezers; Springer: Tokyo, Japan, 2015; pp. 421–443. [Google Scholar]
- Sakamoto, M.; Mino, T. Total Resolution of Racemates by Dynamic Preferential Crystallization; Springer: Tokyo, Japan, 2015; pp. 445–462. [Google Scholar]
- Petralli-Mallow, T.; Wong, T.M.; Byers, J.D.; Yee, H.I.; Hicks, J.M. Circular Dichroism Spectroscopy at Interfaces: A Surface Second Harmonic Generation Study. J. Phys. Chem. 1993, 97, 1383–1388. [Google Scholar] [CrossRef]
- Fischer, P.; Hache, F. Nonlinear Optical Spectroscopy of Chiral Molecules. Chirality 2005, 17, 421–437. [Google Scholar] [CrossRef]
- Kotha, S.; Banerjee, S. Recent developments in the retro-Diels–Alder reaction. RSC Adv. 2013, 3, 7642–7666. [Google Scholar] [CrossRef]
- Saito, Y.; Hyuga, H. Chiral Crystal Growth under Grinding. J. Phys. Soc. Jpn. 2008, 77, 113001/1. [Google Scholar] [CrossRef]
- Martin, I.; Marco, M.A. A Population Balance Model for Chiral Resolution via Viedma Ripening. Cryst. Growth Des. 2011, 11, 4611. [Google Scholar]
- Peter, J.S. Kinetics and Thermodynamics of Efficient Chiral Symmetry Breaking in Nearly Racemic Mixtures of Conglomerate Crystals. Cryst. Growth Des. 2011, 11, 1957–1965. [Google Scholar]
- Xiouras, C.; Van Cleemput, E.; Kumpen, A.; Ter Horst, J.H.; Van Gerven, T.; Stefanidis, G.D. Towards Deracemization in the Absence of Grinding through Crystal Transformation, Ripening, and Racemization. Cryst. Growth Des. 2017, 17, 882–890. [Google Scholar] [CrossRef] [Green Version]
- Matuszak, N.; Muccioli, G.G.; Labar, G.; Lambert, D.M. Synthesis and in Vitro Evaluation of N-Substituted Maleimide Derivatives as Selective Monoglyceride Lipase Inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1510–1515. [Google Scholar] [CrossRef]
- Eloh, K.; Demurtas, M.; Mura, M.G.; Deplano, A.; Onnis, V.; Sasanelli, N.; Maxia, A.; Caboni, P. Potent Nematicidal Activity of Maleimide Derivatives on Meloidogyne incognita. J. Agric. Food Chem. 2016, 64, 4876–4881. [Google Scholar] [CrossRef]
Figure 1.
Valuable Diels–Alder reaction constructing numerous cyclic compounds.

Figure 2.
Absolute asymmetric synthesis involving dynamic crystallization from prochiral starting materials under achiral conditions.
Figure 2.
Absolute asymmetric synthesis involving dynamic crystallization from prochiral starting materials under achiral conditions.

Figure 3.
Diels–Alder (DA) reaction of 2-methylfuran and N-phenylmaleimide.

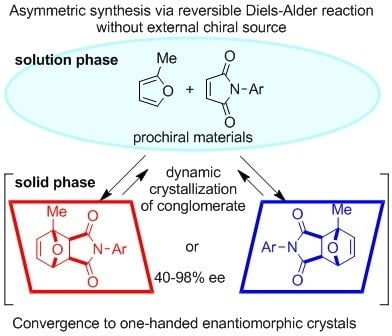
Figure 4.
Asymmetric Diels–Alder reaction of 2-methylfuran and various N-arylmaleimides converged to one-handed enantiomers.
Figure 4.
Asymmetric Diels–Alder reaction of 2-methylfuran and various N-arylmaleimides converged to one-handed enantiomers.

Figure 5.
Asymmetric synthesis of Diels–Alder reaction products involving racemization via reversible reaction and preferential crystallization.
Figure 5.
Asymmetric synthesis of Diels–Alder reaction products involving racemization via reversible reaction and preferential crystallization.

Figure 6.
Asymmetric DA reaction followed by attrition-enhanced deracemization applied to prochiral 2a–d (100 mg), 2-methylfuran 1 (5.0 equiv), TFA (0.5 equiv), heptane (1.00 mL), and glass beads (250 mg) at 80 °C in a sealed tube.
Figure 6.
Asymmetric DA reaction followed by attrition-enhanced deracemization applied to prochiral 2a–d (100 mg), 2-methylfuran 1 (5.0 equiv), TFA (0.5 equiv), heptane (1.00 mL), and glass beads (250 mg) at 80 °C in a sealed tube.

Figure 7.
PXRD pattern of exo-3e: (a) powder of exo-3e after grinding, (b) calculated pattern from a single crystal of exo-3e of the P212121 space group.
Figure 7.
PXRD pattern of exo-3e: (a) powder of exo-3e after grinding, (b) calculated pattern from a single crystal of exo-3e of the P212121 space group.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Space groups of exo-3.
| exo-3 | R | Space Group |
|---|---|---|
| 3a | C6H5 | P212121[a] |
| 3b | 4-ClC6H4 | P212121, (73:27) [b] |
| 3c | 3,4-Me2C6H3 | P212121 |
| 3d | 3,5-Cl2C6H3 | P21, (76:24) [b] |
| 3e | 4-MeC6H4 | P212121, polymorphism [c] |
| 3f | 4-FC6H4 | P21/c |
| 3g | 4-BrC6H4 | P21/n |
| 3h | 3-MeC6H4 | ND [d] |
| 3i | 4-EtC6H4 | ND [d] |
| 3j | 4-MeOC6H4 | ND [d] |
[a] Reference No. 41. [b] Disordered crystal consisting of the ratio of both enantiomers indicated in parentheses. [c] Determined by PXRD. [d] Not determined; however, SHG property was inactive by 1064 nm line from an Nd-YAG pulsed laser.
Table 2.
The half-life of DA and retro-DA reactions with or without trifluoroacetic acid (TFA) [a].
| Substrate | τ1/2 of 2 (h) | τ1/2 of exo-3 (h) | ||
|---|---|---|---|---|
| DA w/o TFA [b] | DA with TFA [c] | retro-DA w/o TFA [d] | retro-DA with TFA [e] | |
| a[f] | 0.92 | 0.25 | 4.5 | 3.0 |
| b | 0.30 | <0.1 | 4.0 | 2.0 |
| c | 0.42 | <0.1 | 6.5 | 3.2 |
| d | 0.30 | <0.1 | 2.0 | 1.2 |
[a] All reactions were monitored by 1H NMR spectroscopy. [b] Conditions: maleimide (0.05 M) and 2-methylfuran (0.5 M) in CDCl3 at 60 °C. [c] Conditions: maleimide (0.05 M), 2-methylfuran (0.5 M), and TFA (0.05 M) in CDCl3 at 60 °C. [d] Conditions: exo-3 (0.05 M) in CDCl3 at 60 °C. [e] Conditions: exo-3 (0.03 M) and TFA (0.03 M) in CDCl3 at 60 °C. [f] Reference No. 41.
Table 3.
Asymmetric DA reaction of 3a-d [a].
| 3 | Time (day) [b] | Yield (%) [c] | Ee (%) [d] |
|---|---|---|---|
| 3a[e] | 6–14 | 80 | 90 |
| 3b | 7–15 | 70 | 40 |
| 3c | 3–9 | 81 | 98 |
| 3d | 4–8 | 78 | 49 |
[a] Conditions: prochiral 2 (100 mg), 2-methylfuran 1 (5.0 equiv), TFA (0.5 equiv), heptane (1.00 mL), and glass beads (250 mg) were stirred at 80 °C in a sealed tube. [b] Time required for asymmetric expression and amplification. [c] Yields of crystalline 3 after filtration. [d] Enantiomeric excess of crystals of 3. [e] Reference No. 41.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Uemura, N.; Toyoda, S.; Shimizu, W.; Yoshida, Y.; Mino, T.; Sakamoto, M. Absolute Asymmetric Synthesis Involving Chiral Symmetry Breaking in Diels–Alder Reaction. Symmetry 2020, 12, 910. https://doi.org/10.3390/sym12060910
AMA Style
Uemura N, Toyoda S, Shimizu W, Yoshida Y, Mino T, Sakamoto M. Absolute Asymmetric Synthesis Involving Chiral Symmetry Breaking in Diels–Alder Reaction. Symmetry. 2020; 12(6):910. https://doi.org/10.3390/sym12060910
Chicago/Turabian StyleUemura, Naohiro, Seiya Toyoda, Waku Shimizu, Yasushi Yoshida, Takashi Mino, and Masami Sakamoto. 2020. "Absolute Asymmetric Synthesis Involving Chiral Symmetry Breaking in Diels–Alder Reaction" Symmetry 12, no. 6: 910. https://doi.org/10.3390/sym12060910
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.