Lipid Vesicles Loaded with an HIV-1 Fusion Inhibitor Peptide as a Potential Microbicide

 , , and
, , and
Abstract
: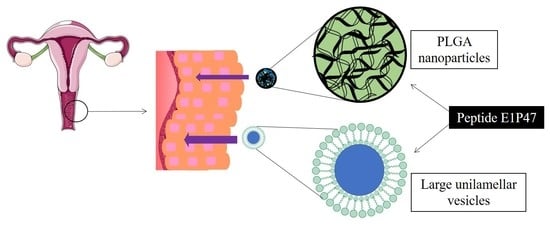
1. Introduction
2. Materials and Methods
2.1. Synthesis of E1P47 Peptide
2.2. Preparation of Large Unilamellar Vesicles
2.3. Preparation of Polymeric Nanoparticles
2.4. Characterization of Formulations
2.4.1. Particle Size, Zeta Potential and Polydispersity
2.4.2. Entrapment Efficiency (EE)
2.5. Short-Term Stability of LUVs and PLGA Nanoparticles
2.6. Toxicity Assessment
2.7. Permeability Assessment
2.8. Monolayer Integrity Assessment
2.9. In Vitro Release Study
2.10. Ex Vivo Permeation Study
2.11. Quantification of E1P47 by UPLC-MS/MS
3. Results and Discussion
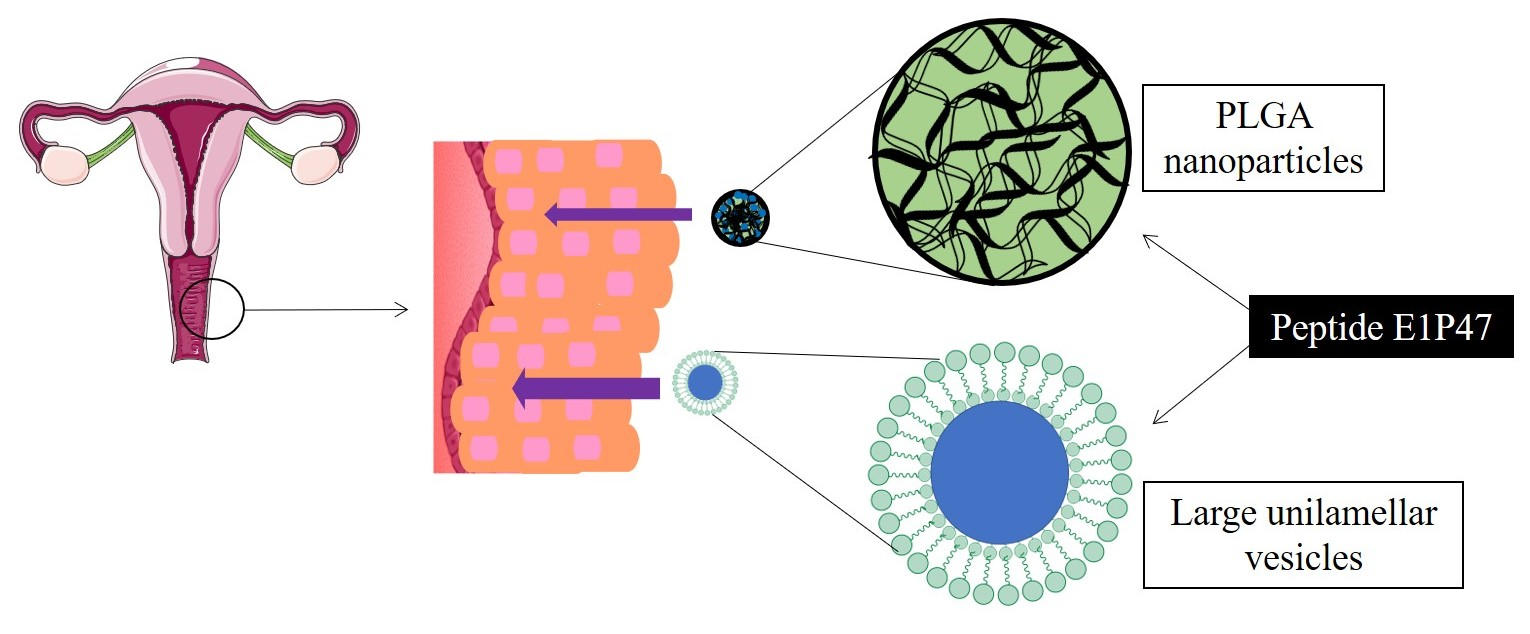
3.1. Characterization of the Nanocarriers
3.2. Short-Term Stability of the Nanocarriers
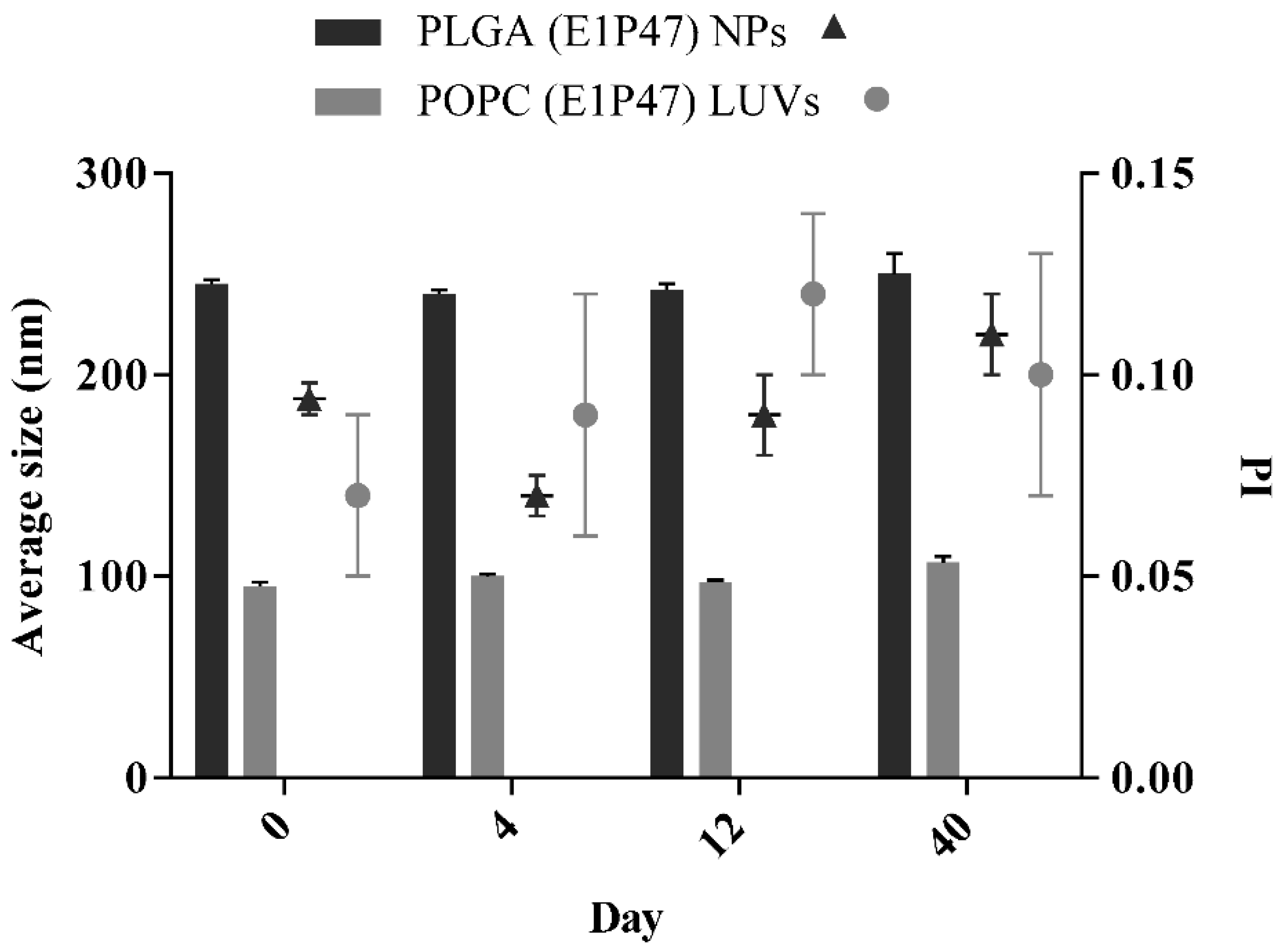
3.3. In Vitro Drug Release
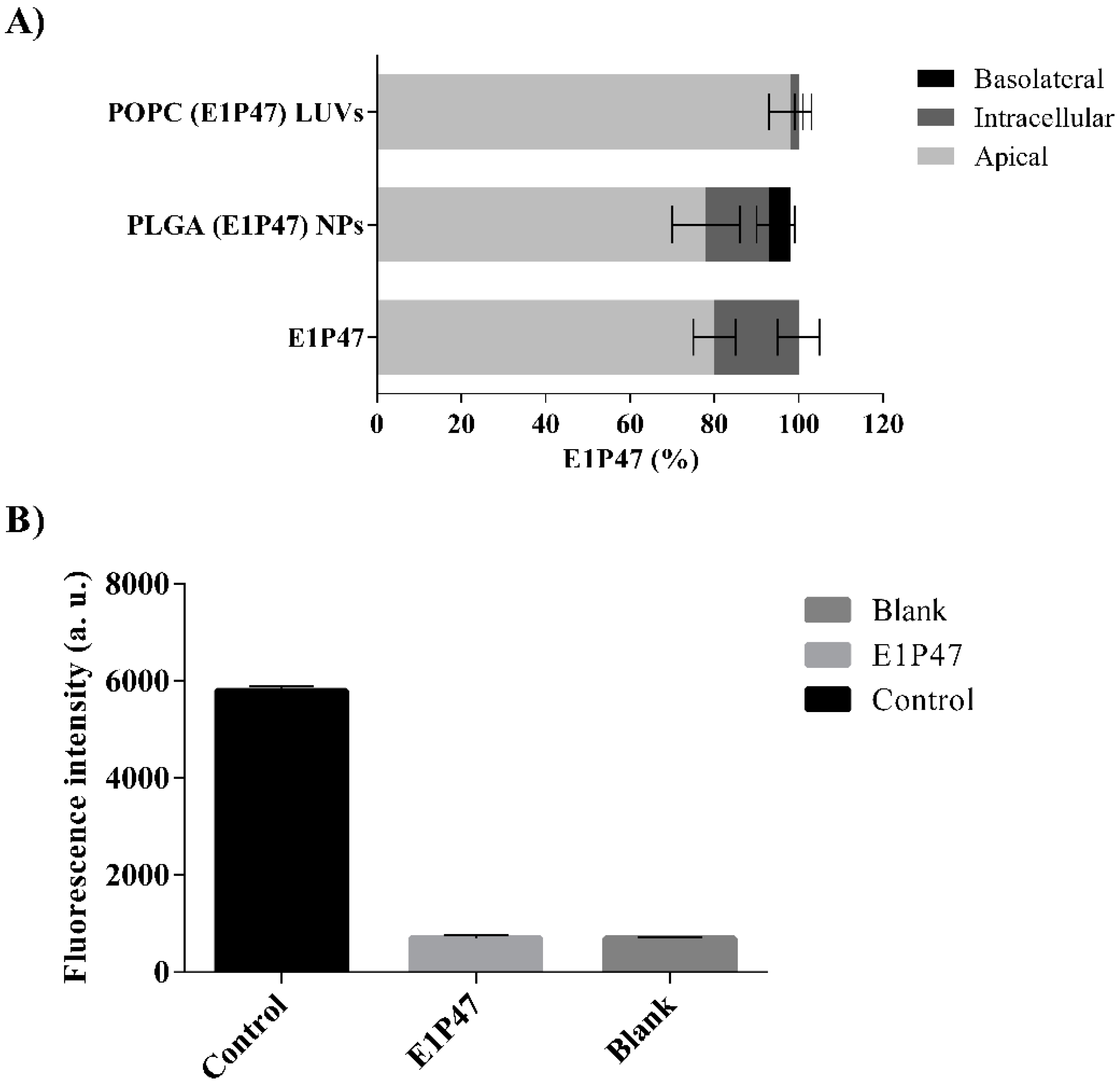
3.4. In Vitro Permeability Assay
3.5. Ex Vivo Permeation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lindl, K.A.; Marks, D.R.; Kolson, D.L.; Jordan-Sciutto, K.L. HIV-Associated Neurocognitive Disorder: Pathogenesis and Therapeutic Opportunities. J. Neuroimmune Pharmacol. 2010, 5, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Notario-Pérez, F.; Ruiz-Caro, R.; Veiga, M.-D. Historical development of vaginal microbicides to prevent sexual transmission of HIV in women: From past failures to future hopes. Drug Des. Dev. Ther. 2017, 11, 1767–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, A.; Reis, C.C.; Araújo, F.; Nunes, R.; Seabra, V.; Ferreira, D.; Das Neves, J.; Sarmento, B. Development and in vivo safety assessment of tenofovir-loaded nanoparticles-in-film as a novel vaginal microbicide delivery system. Acta Biomater. 2016, 44, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Cheu, R.; Birse, K.; Zevin, A.S.; Perner, M.; Noël-Romas, L.; Grobler, A.C.; Westmacott, G.; Xie, I.Y.; Butler, J.; et al. Vaginal bacteria modify HIV tenofovir microbicide efficacy in African women. Science 2017, 356, 938–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, T.; Zhang, W.; Parniak, M.A.; Graebing, P.W.; Moncla, B.; Gupta, P.; Empey, K.M.; Rohan, L.C. Preformulation and Vaginal Film Formulation Development of Microbicide Drug Candidate CSIC for HIV Prevention. J. Pharm. Innov. 2017, 12, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Patel, S.K.; Parniak, M.A.; Ballou, B.; Rohan, L.C. Nanocrystal Formulation Improves Vaginal Delivery of CSIC for HIV Prevention. AAPS PharmSciTech 2019, 20, 286. [Google Scholar] [CrossRef] [PubMed]
- Herrera, C.; Shattock, R.J. Candidate Microbicides and Their Mechanisms of Action. In Molecular Aspects of Myeloid Stem Cell Development; Springer Science and Business Media LLC: Berlin, Germany, 2013; Volume 383, pp. 1–25. [Google Scholar]
- Harman, S.; Herrera, C.; Armanasco, N.; Nuttall, J.; Shattock, R.J. Preclinical Evaluation of the HIV-1 Fusion Inhibitor L’644 as a Potential Candidate Microbicide. Antimicrob. Agents Chemother. 2012, 56, 2347–2356. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Ben, Y.; Yuan, S.; Jiang, S.; Xu, J.; Zhang, X. Efficacy, Stability, and Biosafety of Sifuvirtide Gel as a Microbicide Candidate against HIV-1. PLoS ONE 2012, 7, e37381. [Google Scholar] [CrossRef]
- Wu, H.; Yao, C.; Su, B.; Lu, H.; Sun, Y.; Wang, M.; Wang, H.; Zheng, Y.; Zhu, B.; Yu, J.; et al. Efficacy and Safety of Long Acting HIV Fusion Inhibitor Albuvirtide in Antiretroviral-Experienced Adults with HIV-1: Interim 48 Week Results from the Randomized, Controlled, Phase 3 Trial, Non-Inferiority TALENT Study. SSRN Electron. J. 2018, 23–26. [Google Scholar] [CrossRef]
- Mesquita, L.; Galante, J.; Nunes, R.; Sarmento, B.; Das Neves, J. Pharmaceutical Vehicles for Vaginal and Rectal Administration of Anti-HIV Microbicide Nanosystems. Pharmaceutics 2019, 11, 145. [Google Scholar] [CrossRef] [Green Version]
- Gómara, M.J.; Sanchez-Merino, V.; Paús, A.; Merino-Mansilla, A.; Gatell, J.; Yuste, E.; Haro, I. Definition of an 18-mer Synthetic Peptide Derived from the GB virus C E1 Protein as a New HIV-1 Entry Inhibitor. Biochim. Biophys. Acta (BBA) Gen. Subj. 2016, 1860, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Ariza-Sáenz, M.; Espina, M.; Bolaños, N.; Calpena-Campmany, A.C.; Gómara, M.J.; Haro, I.; García, M.L. Penetration of polymeric nanoparticles loaded with an HIV-1 inhibitor peptide derived from GB virus C in a vaginal mucosa model. Eur. J. Pharm. Biopharm. 2017, 120, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Vaghasiya, K.; Gupta, P.; Gupta, U.D.; Verma, R.K. Reclaiming hijacked phagosomes: Hybrid nano-in-micro encapsulated MIAP peptide ensures host directed therapy by specifically augmenting phagosome-maturation and apoptosis in TB infected macrophage cells. Int. J. Pharm. 2018, 536, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Allémann, E. Polymeric nano- and microparticles for the oral delivery of peptides and peptidomimetics. Adv. Drug Deliv. Rev. 1998, 34, 171–189. [Google Scholar] [CrossRef]
- A Haggag, Y.; Matchett, K.; Dakir, E.H.; Buchanan, P.; Osman, M.A.; Elgizawy, S.A.; El-Tanani, M.; Faheem, A.M.; McCarron, P.A.; El-Habib, D. Nano-encapsulation of a novel anti-Ran-GTPase peptide for blockade of regulator of chromosome condensation 1 (RCC1) function in MDA-MB-231 breast cancer cells. Int. J. Pharm. 2017, 521, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, M.; Amir Jalali, S.; Badiee, A.; Shariat, S.M.; Mercedeh Arabi, L.; Abbasi, A.; Saberi, Z.; Reza Jaafari, M. Stimulation of tumor-specific immunity by p5 HER-2/neu generated peptide encapsulated in nano-liposomes with high phase transition temperature phospholipids. Curr. Drug Deliv. 2017, 14, 492–502. [Google Scholar] [CrossRef]
- Ariza-Sáenz, M.; Espina, M.; Calpena, A.; Gómara, M.J.; Pérez-Pomeda, I.; Haro, I.; García, M.L. Design, Characterization, and Biopharmaceutical Behavior of Nanoparticles Loaded with an HIV-1 Fusion Inhibitor Peptide. Mol. Pharm. 2018, 15, 5005–5018. [Google Scholar] [CrossRef] [Green Version]
- Gómara, M.J.; Pérez-Pomeda, I.; Gatell, J.M.; Sanchez-Merino, V.; Yuste, E.; Haro, I. Lipid raft-like liposomes used for targeted delivery of a chimeric entry-inhibitor peptide with anti-HIV-1 activity. Nanomed. Nanotechnol. Boil. Med. 2017, 13, 601–609. [Google Scholar] [CrossRef]
- Gómara, M.J.; Perez, Y.; Martinez, J.P.; Barnadas-Rodriguez, R.; Schultz, A.; Von Briesen, H.; Peralvarez-Marin, A.; Meyerhans, A.; Haro, I. Peptide Assembly on the Membrane Determines the HIV-1 Inhibitory Activity of Dual-Targeting Fusion Inhibitor Peptides. Sci. Rep. 2019, 9, 3257. [Google Scholar] [CrossRef]
- Rossi, G.; Barnoud, J.; Monticelli, L. Polystyrene Nanoparticles Perturb Lipid Membranes. J. Phys. Chem. Lett. 2013, 5, 241–246. [Google Scholar] [CrossRef]
- Mijajlovic, M.; Wright, D.; Zivkovic, V.; Bi, J.; Biggs, M.J. Microfluidic hydrodynamic focusing based synthesis of POPC liposomes for model biological systems. Colloids Surf. B Biointerfaces 2013, 104, 276–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, J.A.; Pinazo, A.; Carilla, J.; Infante, M.R.; Alsina, M.A.; Haro, I.; Clapés, P. Interaction of Antimicrobial Arginine-Based Cationic Surfactants with Liposomes and Lipid Monolayers. Langmuir 2004, 20, 3379–3387. [Google Scholar] [CrossRef] [PubMed]
- E Ryman, B.; A Tyrrell, D. Liposomes—Methodology and applications. Front. Boil. 1979, 48, 549–574. [Google Scholar]
- Franzè, S.; Marengo, A.; Stella, B.; Minghetti, P.; Berlier, G.; Cilurzo, F. Hyaluronan-decorated liposomes as drug delivery systems for cutaneous administration. Int. J. Pharm. 2018, 535, 333–339. [Google Scholar] [CrossRef]
- López, E.S.; Ettcheto, M.; Egea, M.A.; Espina, M.; Calpena-Campmany, A.C.; Folch, J.; Camins, A.; García, M.L. New potential strategies for Alzheimer’s disease prevention: Pegylated biodegradable dexibuprofen nanospheres administration to APPswe/PS1dE9. Nanomed. Nanotechnol. Boil. Med. 2017, 13, 1171–1182. [Google Scholar] [CrossRef]
- López, E.S.; Egea, M.; Cano, A.; Espina, M.; Calpena, A.; Ettcheto, M.; Camins, A.; Souto, E.; Silva, A.; García, M.L. PEGylated PLGA nanospheres optimized by design of experiments for ocular administration of dexibuprofen—In vitro, ex vivo and in vivo characterization. Colloids Surf. B Biointerfaces 2016, 145, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Caddeo, C.; Pucci, L.; Gabriele, M.; Carbone, C.; Fernàndez-Busquets, X.; Valenti, N.; Pons, R.; Vassallo, A.; Fadda, A.M.; Manconi, M. Stability, biocompatibility and antioxidant activity of PEG-modified liposomes containing resveratrol. Int. J. Pharm. 2018, 538, 40–47. [Google Scholar] [CrossRef]
- López, E.S.; Egea, M.A.; Davis, B.; Guo, L.; Espina, M.; Silva, A.M.; Calpena-Campmany, A.C.; Souto, E.; Ravindran, N.; Ettcheto, M.; et al. Memantine-Loaded PEGylated Biodegradable Nanoparticles for the Treatment of Glaucoma. Small 2017, 14, 1701808. [Google Scholar] [CrossRef]
- Neves, A.; Queiroz, J.F.; Reis, S. Brain-targeted delivery of resveratrol using solid lipid nanoparticles functionalized with apolipoprotein E. J. Nanobiotechnology 2016, 14, 27. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Li, X.; Li, T.; Wang, X.; Yang, Y.; Xiao, L.; Shen, H. Combination effects of paclitaxel with signaling inhibitors in endometrial cancer cells. Asian Pac. J. Cancer Prev. 2011, 12, 2951–2957. [Google Scholar]
- Grammen, C.; Augustijns, P.; Brouwers, J. In vitro profiling of the vaginal permeation potential of anti-HIV microbicides and the influence of formulation excipients. Antivir. Res. 2012, 96, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Grammen, C.; Ariën, K.K.; Venkatraj, M.; Joossens, J.; Van Der Veken, P.; Heeres, J.; Lewi, P.; Haenen, S.; Augustyns, K.; Vanham, G.; et al. Development and in vitro evaluation of a vaginal microbicide gel formulation for UAMC01398, a novel diaryltriazine NNRTI against HIV-1. Antivir. Res. 2014, 101, 113–121. [Google Scholar] [CrossRef] [PubMed]
- López, E.S.; Ettcheto, M.; Egea, M.A.; Espina, M.; Cano, A.; Calpena-Campmany, A.C.; Camins, A.; Carmona, N.; Silva, A.; Souto, E.; et al. Memantine loaded PLGA PEGylated nanoparticles for Alzheimer’s disease: In vitro and in vivo characterization. J. Nanobiotechnology 2018, 16, 32. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Narai, A.; Shimizu, M. Purification and cDNA cloning of a protein derived from Flammulina velutipes that increases the permeability of the intestinal Caco-2 cell monolayer. JBIC J. Boil. Inorg. Chem. 1999, 262, 850–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, L.; Calpena-Campmany, A.C.; Silva-Abreu, M.; Espinoza, L.C.; Rincón, M.; Bozal, N.; Domènech, Ò.; Rodríguez-Lagunas, M.J.; Clares-Naveros, B. Thermoreversible Gel-Loaded Amphotericin B for the Treatment of Dermal and Vaginal Candidiasis. Pharmaceutics 2019, 11, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.W.; Dhanawat, M.; Rathbone, M.J. Vaginal drug delivery: Strategies and concerns in polymeric nanoparticle development. Expert Opin. Drug Deliv. 2014, 11, 1419–1434. [Google Scholar] [CrossRef]
- Caramella, C.; Rossi, S.; Ferrari, F.; Bonferoni, M.C.; Sandri, G. Mucoadhesive and thermogelling systems for vaginal drug delivery. Adv. Drug Deliv. Rev. 2015, 92, 39–52. [Google Scholar] [CrossRef]
- Das Neves, J.; Nunes, R.; Machado, A.; Sarmento, B. Polymer-based nanocarriers for vaginal drug delivery. Adv. Drug Deliv. Rev. 2015, 92, 53–70. [Google Scholar] [CrossRef] [Green Version]
- Abriata, J.; Turatti, R.C.; Luiz, M.T.; Raspantini, G.L.; Tofani, L.B.; Amaral, R.L.F.D.; Swiech, K.; Marcato, P.D.; Marchetti, J.M. Development, characterization and biological in vitro assays of paclitaxel-loaded PCL polymeric nanoparticles. Mater. Sci. Eng. C 2019, 96, 347–355. [Google Scholar] [CrossRef]
- Yang, M.; Lai, S.K.; Yu, T.; Wang, Y.-Y.; Happe, C.; Zhong, W.; Zhang, M.; Anonuevo, A.; Fridley, C.; Hung, A.; et al. Nanoparticle penetration of human cervicovaginal mucus: The effect of polyvinyl alcohol. J. Control. Release 2014, 192, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Pizarro, R.; Parrotta, G.; Vera, R.; Sánchez-López, E.; Galindo, R.; Kjeldsen, F.; Badia, J.; Baldoma, L.; Espina, M.; García, M.L. Ocular penetration of fluorometholone-loaded PEG-PLGA nanoparticles functionalized with cell-penetrating peptides. Nanomedicine 2019, 14, 3089–3104. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Yamada, T.; Kimura, M.; Oka, T.; Ohshima, H.; Kondo, T. Temperature- and ionic strength-induced conformational changes in the lipid head group region of liposomes as suggested by zeta potential data. Biophys. Chem. 1991, 41, 175–183. [Google Scholar] [CrossRef]
- Chibowski, E.; Szczes, A. Zeta potential and surface charge of DPPC and DOPC liposomes in the presence of PLC enzyme. Adsorption 2016, 22, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Pérez, Y.; Gómara, M.J.; Yuste, E.; Gómez-Gutierrez, P.; Perez, J.J.; Haro, I. Structural Study of a New HIV-1 Entry Inhibitor and Interaction with the HIV-1 Fusion Peptide in Dodecylphosphocholine Micelles. Chem. A Eur. J. 2017, 23, 11703–11713. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.; Zhang, L.; Quan, Y.; Wei, K. Resveratrol-loaded PLGA nanoparticles: Enhanced stability, solubility and bioactivity of resveratrol for non-alcoholic fatty liver disease therapy. R. Soc. Open Sci. 2018, 5, 181457. [Google Scholar] [CrossRef] [Green Version]
- Chittasupho, C.; Posritong, P.; Ariyawong, P. Stability, Cytotoxicity, and Retinal Pigment Epithelial Cell Binding of Hyaluronic Acid-Coated PLGA Nanoparticles Encapsulating Lutein. AAPS PharmSciTech 2018, 20, 4. [Google Scholar] [CrossRef]
- Wu, I.Y.; Bala, S.; Škalko-Basnet, N.; Di Cagno, M.P. Interpreting non-linear drug diffusion data: Utilizing Korsmeyer-Peppas model to study drug release from liposomes. Eur. J. Pharm. Sci. 2019, 138, 105026. [Google Scholar] [CrossRef]
- Yacasi, G.R.R.; Lopéz, M.L.G.; García, M.E.; Coca, A.P.; Campmany, A.C.C. Influence of freeze-drying and γ-irradiation in preclinical studies of flurbiprofen polymeric nanoparticles for ocular delivery using d-(+)-trehalose and polyethylene glycol. Int. J. Nanomed. 2016, 11, 4093–4106. [Google Scholar] [CrossRef] [Green Version]
- Delbaldo, C.; Pierga, J.Y.; Dieras, V.; Faivre, S.; Laurence, V.; Vedovato, J.C.; Bonnay, M.; Mueser, M.; Nolting, A.; Kovar, A.; et al. Pharmacokinetic profile of cetuximab (ErbituxTM) alone and in combination with irinotecan in patients with advanced EGFR-positive adenocarcinoma. Eur. J. Cancer 2005, 41, 1739–1745. [Google Scholar] [CrossRef]
- Albarellos, G.A.; Montoya, L.; Landoni, M.F. Pharmacokinetics of marbofloxacin after single intravenous and repeat oral administration to cats. Vet. J. 2005, 170, 222–229. [Google Scholar] [CrossRef]
- García, M.L.; Pérez, Y.; Gómara, M.J.; Vasconcelos, A.; Vega, E.; Haro, I. Conjugation of cell-penetrating peptides with poly(lactic-co-glycolic acid)-polyethylene glycol nanoparticles improves ocular drug delivery. Int. J. Nanomed. 2015, 10, 609–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, P.M.; Batista, P.; Madureira, A.R.; Sarmento, B.; Pintado, M. Combination of PLGA nanoparticles with mucoadhesive guar-gum films for buccal delivery of antihypertensive peptide. Int. J. Pharm. 2018, 547, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Signorell, R.D.; Luciani, P.; Brambilla, D.; Leroux, J.-C. Pharmacokinetics of lipid-drug conjugates loaded into liposomes. Eur. J. Pharm. Biopharm. 2018, 128, 188–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassano, R.; Ferrarelli, T.; Mauro, M.V.; Cavalcanti, P.; Picci, N.; Trombino, S. Preparation, characterization and in vitro activities evaluation of solid lipid nanoparticles based on PEG-40 stearate for antifungal drugs vaginal delivery. Drug Deliv. 2014, 23, 1037–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Eyk, A.D.; Van Der Bijl, P. Porcine vaginal mucosa as an in vitro permeability model for human vaginal mucosa. Int. J. Pharm. 2005, 305, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Leyva-Gómez, G.; Piñón-Segundo, E.; Mendoza-Munoz, N.; Zaragoza, M.D.L.L.Z.; Mendoza-Elvira, S.; Quintanar-Guerrero, D. Approaches in Polymeric Nanoparticles for Vaginal Drug Delivery: A Review of the State of the Art. Int. J. Mol. Sci. 2018, 19, 1549. [Google Scholar] [CrossRef] [Green Version]
- Blakney, A.K.; Jiang, Y.; Whittington, D.; Woodrow, K.A. Simultaneous measurement of etravirine, maraviroc and raltegravir in pigtail macaque plasma, vaginal secretions and vaginal tissue using a LC-MS/MS assay. J. Chromatogr. B 2016, 1025, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhang, J.; Shan, W.; Huang, Y. Developments of mucus penetrating nanoparticles. Asian J. Pharm. Sci. 2015, 10, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Lai, S.K.; Wang, Y.-Y.; Hanes, J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv. Drug Deliv. Rev. 2009, 61, 158–171. [Google Scholar] [CrossRef] [Green Version]
- Ensign, L.M.; Tang, B.C.; Wang, Y.-Y.; Tse, T.A.; Hoen, T.; Cone, R.; Hanes, J. Mucus-Penetrating Nanoparticles for Vaginal Drug Delivery Protect Against Herpes Simplex Virus. Sci. Transl. Med. 2012, 4, 138ra79. [Google Scholar] [CrossRef] [Green Version]
- Han, H.-K.; Jung, I.-W.; Lee, B.-J. Effective mucoadhesive liposomal delivery system for risedronate: Preparation and in vitro/in vivo characterization. Int. J. Nanomed. 2014, 9, 2299–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, C.; Clarke, J.; Gregoriadis, G. Effect of the cholesterol content of small unilamellar liposomes on their stability in vivo and in vitro. Biochem. J. 1980, 186, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sułkowski, W.; Pentak, D.; Nowak, K.; Sułkowska, A. The influence of temperature, cholesterol content and pH on liposome stability. J. Mol. Struct. 2005, 744, 737–747. [Google Scholar] [CrossRef]
- Socaciu, C.; Jessel, R.; A Diehl, H. Competitive carotenoid and cholesterol incorporation into liposomes: Effects on membrane phase transition, fluidity, polarity and anisotropy. Chem. Phys. Lipids 2000, 106, 79–88. [Google Scholar] [CrossRef]
- Cunha-Reis, C.; Machado, A.; Barreiros, L.; Araújo, F.; Nunes, R.; Seabra, V.; Ferreira, D.; Segundo, M.A.; Sarmento, B.; Das Neves, J. Nanoparticles-in-film for the combined vaginal delivery of anti-HIV microbicide drugs. J. Control. Release 2016, 243, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Almaraz, M.T.; Gref, R.; Agostoni, V.; Kreuz, C.; Clayette, P.; Serre, C.; Couvreur, P.; Horcajada, P. Towards improved HIV-microbicide activity through the co-encapsulation of NRTI drugs in biocompatible metal organic framework nanocarriers. J. Mater. Chem. B 2017, 5, 8563–8569. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Zav ± SD (nm) | PI ± SD | ZP ± SD (mV) | EE (%) |
|---|---|---|---|---|
| Free E1P47 | - | - | 10.6 ± 0.6 | - |
| PLGA NPs | 245 ± 2 | 0.09 ± 0.00 | −13.0 ± 1.1 | - |
| PLGA (E1P47) NPs | 240 ± 2 | 0.07 ± 0.05 | −0.5 ± 0.3 | 69% |
| POPC LUVs | 98 ± 1 | 0.05 ± 0.02 | −13.0 ± 1.0 | |
| POPC (E1P47) LUVs | 95 ± 2 | 0.07 ± 0.02 | −3.5 ± 0.1 | 93% |
| Formulation | Ka (h−1) | K10 (h−1) | Ltime (h) | Qmax (µg) | Tmax (h) |
|---|---|---|---|---|---|
| Free E1P47 | (1.9 ± 0.6) × 10−1 | (6.0 ± 4.9) × 10−2 | 1.4 ± 0.1 | 1.8 ± 0.4 | 11.1 ± 2.1 |
| PLGA (E1P47) NPs | (6.0 ± 0.8) × 10−1 | (2.8 ± 0.4) × 10−2 | 1.8 ± 1.5 | 8.0 ± 5.4 | 7.0 ± 3.4 |
| POPC (E1P47) LUVs | (1.1 ± 0.5) ×·10−1 | (1.4 ± 0.6) × 10−2 | 3.0 ± 2.5 | 1.5 ± 0.1 | 23.0 ± 1.7 |
| Formulation | Peptide Retained in the Membrane (µg) | Peptide Retained (%) |
|---|---|---|
| Free E1P47 | 0.1 ± 0.5 | 0.1 |
| PLGA (E1P47) NPs | 2.2 ± 1.5 | 1.5 |
| POPC (E1P47) LUVs | 1.5 ± 0.5 | 1.0 |
| Formulation | Cp (µg/mL) | Wp (g) | Peptide Retained (Mean ± SD; µg/g × cm−2) | Concentration of Peptide Retained (µM) |
|---|---|---|---|---|
| Free E1P47 | 0.5 ± 0.4 | 0.4 ± 0.1 | 10.0 ± 7.0 | 3.0 ± 2.0 |
| PLGA (E1P47) NPs | 0.2 ± 0.1 | 0.4 ± 0.1 | 3.6 ± 0.1 | 1.0 ± 0.0 |
| POPC (E1P47) LUVs | 2.5 ± 0.2 | 0.4 ± 0.1 | 40.0 ± 8.0 | 11.0 ± 2.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-López, E.; Paús, A.; Pérez-Pomeda, I.; Calpena, A.; Haro, I.; Gómara, M.J. Lipid Vesicles Loaded with an HIV-1 Fusion Inhibitor Peptide as a Potential Microbicide. Pharmaceutics 2020, 12, 502. https://doi.org/10.3390/pharmaceutics12060502
Sánchez-López E, Paús A, Pérez-Pomeda I, Calpena A, Haro I, Gómara MJ. Lipid Vesicles Loaded with an HIV-1 Fusion Inhibitor Peptide as a Potential Microbicide. Pharmaceutics. 2020; 12(6):502. https://doi.org/10.3390/pharmaceutics12060502
Chicago/Turabian StyleSánchez-López, Elena, Anna Paús, Ignacio Pérez-Pomeda, Ana Calpena, Isabel Haro, and María José Gómara. 2020. "Lipid Vesicles Loaded with an HIV-1 Fusion Inhibitor Peptide as a Potential Microbicide" Pharmaceutics 12, no. 6: 502. https://doi.org/10.3390/pharmaceutics12060502