Engineered Cationic Antimicrobial Peptides (eCAPs) to Combat Multidrug-Resistant Bacteria



1
Department of Environmental and Occupational Health, University of Pittsburgh Graduate School of Public Health, Pittsburgh, PA 15261, USA
2
Department of Microbiology and Molecular Genetics, University of Pittsburgh School of Medicine, Pittsburgh, PA 15219, USA
3
Department of Orthopaedic Surgery, University of Pittsburgh, Pittsburgh, PA 15213, USA
*
Author to whom correspondence should be addressed.
Pharmaceutics 2020, 12(6), 501; https://doi.org/10.3390/pharmaceutics12060501
Submission received: 13 April 2020
/
Revised: 25 May 2020
/
Accepted: 27 May 2020
/
Published: 30 May 2020
(This article belongs to the Special Issue Drug Delivery for Anti-Infective Agents)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The increasing rate of antibiotic resistance constitutes a global health crisis. Antimicrobial peptides (AMPs) have the property to selectively kill bacteria regardless of resistance to traditional antibiotics. However, several challenges (e.g., reduced activity in the presence of serum and lack of efficacy in vivo) to clinical development need to be overcome. In the last two decades, we have addressed many of those challenges by engineering cationic AMPs de novo for optimization under test conditions that typically inhibit the activities of natural AMPs, including systemic efficacy. We reviewed some of the most promising data of the last two decades in the context of the advancement of the field of helical AMPs toward clinical development.
1. Introduction
The development and the clinical use of the traditional classes of antibiotics, both prophylactically and therapeutically, have facilitated many of the advances in modern medicine. However, over the past three decades, the alarming rate of multidrug-resistance (MDR) has threatened to end the antibiotic era [1,2]. If this trend continues without increasingly effective countermeasures, it may become very difficult to perform many medical and surgical procedures that were challenging to perform prior to the commercialization of traditional antibiotics. In addition, secondary infections by MDR bacteria may increase mortality in conditions that are already difficult to manage such as COVID-19 and other diseases requiring hospitalization on a massive scale [3,4], which increase the risk for MDR-related infections [5,6,7]. Thus, there is an urgency to develop new antimicrobial strategies to address the pressing problems associated with MDR-related infectious diseases. Expanding the impact of antimicrobial resistance is the decrease in the pharmaceutical industry research pipeline for novel antimicrobial agents. In light of these challenges, several federal agencies and other organizations with specific programs to combat antibiotic resistance have inspired hope for novel antimicrobial discoveries. These organizations have formed partnerships with drug development companies to facilitate or accelerate pre-clinical and clinical testing requirements for novel antimicrobial agents. They include, but are not limited to, the Broad Spectrum Antimicrobials program of the Biomedical Advanced Research and Development Authority (BARDA), Combating Antibiotic Resistant Bacteria (CARB-X), European Gram Negative Antibacterial Engine (ENABLE), and the Repair Impact Fund. Noteworthy is the Joint Programming Initiative on Antimicrobial Resistance, which established the Virtual Research Institute (JPIAMR/VRI) with the mission to improve and coordinate research networks on antimicrobial resistance in seven countries in Europe and North America. Accordingly, the programs established by these organizations may create new funding opportunities for a robust exploration of antimicrobial peptides (AMPs), a promising source of new antimicrobial agents. It is in this context that we revisit some of the most promising data on engineered cationic antimicrobial peptides (eCAPs) to reframe efforts on the advances of helical AMPs toward clinical development.
2. Properties and Limitations of Natural Cationic Antimicrobial Peptides (AMPs)
To appreciate the logic in engineering synthetic AMPs, it is essential to understand the structure–function relationship of AMPs and the obstacles that have prevented their use in clinical treatment. Since their initial discoveries in the 1980s [8,9,10], AMPs have been shown to occur in almost all life forms as short peptides (10–50 amino acids long) with an amphipathic (e.g., cationic or anionic and hydrophobic domains) structure in the context of host defense [11,12,13,14]. Although classical AMPs are ribosomally synthesized (composed of the standard proteinogenic amino acids) [15,16], AMPs can be extended to include well-known peptides that display a cationic (or the less commonly known anionic) amphipathic structure (e.g., the cationic lipopeptide polymyxins or the anionic daptomycin) [17,18,19]. Hence, ubiquitous in nature, AMPs represent the first line of defense against a variety of microbial pathogens (e.g., bacteria, fungi, parasites, viruses), depending on amino acid composition of the amphipathic motif [20,21,22,23,24]. AMPs are structurally diverse (α-helix, β-sheets, loop structures, cyclic, etc.), indicating that the cationic amphipathic structure, rather than a particular secondary structure, is the most fundamental determinant of activity. In a classical sense, the most well-known structural classes are the α-helix and β-sheets through the pioneering work of Lehrer, Ganz, Boman, Zasloff, Hancock, and others [8,9,10,25,26,27]. In contrast, AMPs synthesized through extraribosomal pathways (e.g., the cyclic lipopeptides, polymyxins) [28,29], were known for several decades prior to the discovery of ribosomally synthesized AMPs [13,30,31,32]. While AMPs like the polymyxins and daptomycin are widely used clinically, those made exclusively of some of the 20 (excluding selenocysteine) conventional amino acids (including engineered derivatives of these AMPs) are yet to be clinically available. An important group comprises the primate theta-defensins (e.g., retrocyclins), not found in humans due to a premature stop codon [33,34]. In general, cyclic AMPs are significant for their enhanced stability compared to non-cyclic peptides, which partly explains a considerable interest in their clinical development [35,36,37,38]. Helical structures can be represented by magainins, cecropins, cathelicidins, and others [9,10,39,40,41]. β-sheet structures are exemplified by the α- and β-defensins (theta-defensins already mentioned), a highly ubiquitous class of AMPs with characteristic disulfide bridges that stabilize their secondary structures [26,42]. Importantly, there are many groups of AMPs that fall into one or more structural classes (including loop structures) and are well described elsewhere [43,44,45,46]. Cationic AMPs generally recognize their bacterial targets via electrostatic interactions with negatively charged bacterial membrane lipids [47,48,49,50,51,52,53,54,55,56]. This bacterial recognition is typically due to the cationic content in the context of the amphipathic structure and may result in the perturbation of the bacterial membrane in a concentration-dependent manner. Notably, the polymyxins are well known for their interactions with lipopolysaccharide (LPS), and that recognition may be reduced or abrogated with LPS modification (e.g., addition of ethanolamine or LPS deficiency) [57,58,59]. Daptomycin is another clinically used lipopeptide that kills gram-positive organisms (particularly S. aureus); it also permeabilizes bacterial membranes [60,61,62]. However, other antimicrobial mechanisms of AMPs have been demonstrated including bacterial cell penetration (e.g., proline-rich AMPs) and interference with vital intracellular processes [63,64,65,66]. Further, new antimicrobial mechanisms have been demonstrated in the last decade, notably the LPS-transport protein D (LptD) targeted by the cyclic peptide murepavadin [67,68,69,70].
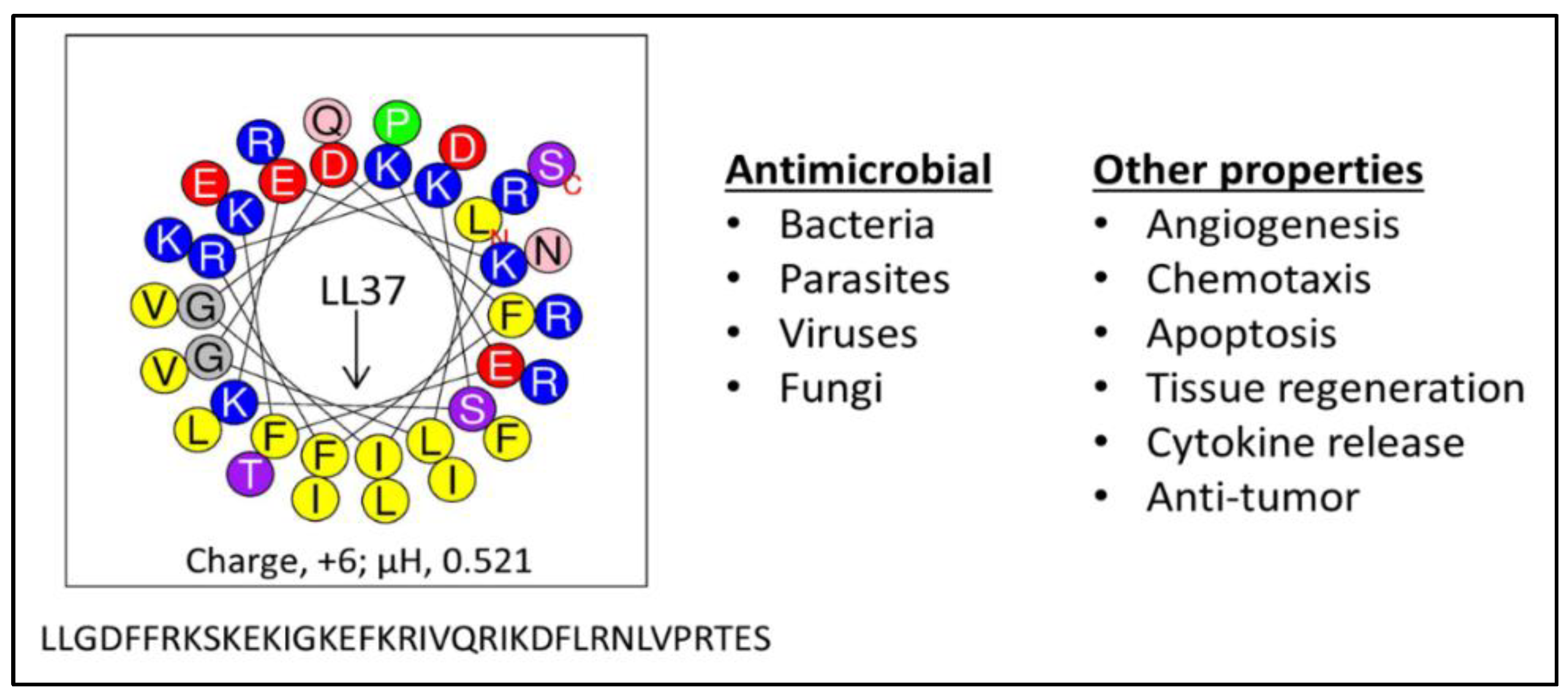
AMPs are considered a highly promising therapeutic resource in the fight against bacterial resistance to traditional antibiotics because they have a number of properties that confer the ability to overcome the common resistance mechanisms of MDR pathogens. AMPs do not require metabolic processes for antimicrobial activity [71,72], as they target a pre-existing bacterial cell structure: the bacterial membrane. Therefore, they are effective against both quiescent and actively growing bacteria. As previously shown, the human AMP LL37 (Figure 1) rapidly kills P. aeruginosa in phosphate-buffered saline where there is no carbon source to support bacterial growth or an active metabolic state [55]. The majority of the bacterial cells were killed within the first minute, and killing of all 106 cfu/mL was complete by 2.5 min. Thus, such rapid killing (less time for bacteria to divide and generate mutations) and disruption of a vital pre-existing structure (the bacterial membrane) make it difficult for mutations (requiring extensive bacterial division) to occur during AMP treatment and for ensuing selection of resistance compared to traditional antibiotics, which target specific metabolic pathways. Hence, in sharp contrast to conventional antibiotics, AMPs demonstrate a low propensity to select for bacterial resistance when bacterial cells grow in the presence of subinhibitory concentrations of AMPs [55,73,74,75,76,77]. A plausible explanation is that a fundamental reduction in electronegative density of the bacterial surface that would readily confer resistance to AMPs is likely to affect membrane integrity or fitness of the cells, thus rendering resistance to AMPs less common, although not impossible. Of note, resistance to several AMPs has been observed, including resistance against polymyxins [76,78,79], daptomycin [60,62,80,81], LL37 [73,82], and other AMPs [83,84,85].
AMPs have other interesting properties as well. The ability of select AMPs to display activity against multiple types of organisms (e.g., bacteria, viruses, fungi, and parasites) indicates the potential for application to a wide range of communicable or polymicrobial diseases [71,86,87,88,89,90,91,92,93,94]. In the context of the COVID-19 pandemic, the exploration of antiviral activity of AMPs is essential [91,95], as their antiviral mechanisms are not as well known as the electrostatic recognition of their target bacterial cell membranes. AMPs may also elicit an anti-infective host immune response and possess the ability to neutralize endotoxins, suggesting potential efficacy in septic shock [51,56,96,97,98,99,100]. Further, their anti-biofilm properties may confer efficacy against infections associated with wounds, medical implants, and chronic respiratory illnesses [82,91,101,102,103,104]. Antitumor properties also suggest a potential application against cancer [105]. Evidently, AMPs have the potential to display broad-spectrum efficacy in a wide range of applications, although some subsets of microorganisms may display various degrees of susceptibility to AMPs. An important trend in AMP development is the potential of AMPs to enhance the efficacy of traditional antibiotics when used in combination. Such synergistic properties may considerably contribute to the fight against antibiotic resistance by rendering the already-existing antibiotic arsenal more effective against MDR bacteria, as demonstrated by the polymyxins, murepavadin, and helical AMPs [106,107,108,109,110].
3. Overcoming the Limitations of AMPs
AMPs display several limitations that have delayed their successful development for clinical use.
3.1. Contextual Activity
Over the last several decades and since the early years of AMP discoveries, it has become increasingly clear that certain test conditions are challenging to most natural AMPs. Most classical AMPs typically demonstrate optimal bacterial killing activity in phosphate buffer, whereas their activity could be inhibited in the presence of varying concentrations of salt [55,73,75,77]. AMPs tend to show reduced activity under acidic conditions and in blood, plasma, or serum as they may bind to plasma proteins [55,75]. Therefore, in the context of a vast literature on antimicrobial properties in vitro, evidence for in vivo efficacy is still rare after almost four decades of discoveries. Furthermore, due to their peptidic property, AMPs are susceptible to protease digestion or even poor absorption (e.g., colistin in intestinal absorption) [111], which may limit their clinical applications only to parenteral routes of administration.
In addition to the contextual activity of many AMPs, there are other important concerns such as unclear pharmacokinetic (PK) properties, potential immunogenicity, and host toxicity. Protease digestion can be addressed with l-to-d enantiomerization and the use of unnatural amino acids or peptoids and other types of amino acid mimics [100,112,113,114,115,116,117]. Importantly, the short sequences of AMPs provide the advantage of conferring poor immunogenicity; however, it is important to rule it out for specific AMPs in clinical development. Another significant concern is the potential for host toxicity. Altogether, we and others have demonstrated that most of these limitations can be significantly overcome by structural design optimization, as shown by engineered peptides [28,38,55,75,104,118,119,120].
3.2. Advances in Helical AMP Engineering: eCAPs
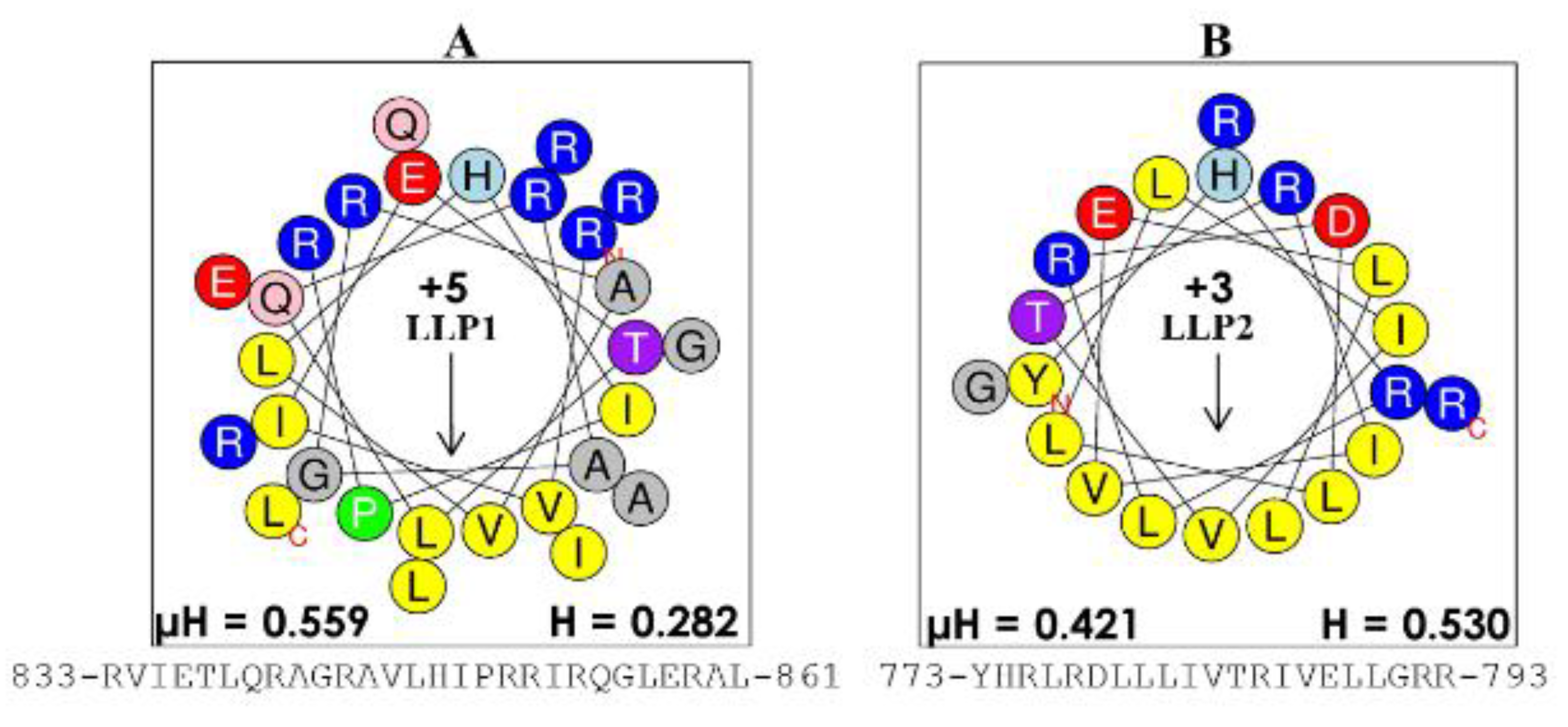
There are, of course, several pioneers who paved the way for our development of engineered helical peptides by their outstanding work (e.g., Boman, Zasloff, and others), as previously mentioned. These early and subsequent discoveries as well as our studies of the last fifteen years have led to the understanding that the cationic amphipathic structure of helical AMPs is a consensus motif necessary for substantial activity [10,55,75,121,122]. In the late 1990s, the Mietzner–Montelaro group began a number of structure–function studies of the human immunodeficiency virus 1 (HIV-1) envelope proteins, specifically the C-terminal tail of HIV-1 transmembrane protein gp41 that contains two domains, termed lentivirus lytic peptides (LLPs), whose sequences have characteristics that are similar to those of natural AMPs (Figure 2). Secondary structure predictions suggested that the LLP sequences would form cationic amphipathic helices, an important characteristic of a large class of natural AMPs. These LLP analogs killed both gram-negative and gram-positive bacteria in vitro at low μM concentrations, confirming predicted antibacterial properties [41,123]. Further studies of these LLP structures revealed a selective incorporation and conservation of arginine, not lysine, suggesting a critically unique role for arginine in the LLP. Mutational studies of LLPs showed that membrane perturbation and antibacterial properties could be enhanced by adding Arg residues to the hydrophilic side of the helix, Trp residues to the hydrophobic side, and extending the length of the amphipathic helix via computationally optimized sequences using Arg, Val, and Trp [41,123,124,125,126]. These initial studies produced the first generation of de novo-engineered peptides from our group, which we referred to as eCAPs; hence, these studies initiated some of the earliest paradigms for engineering synthetic AMPs to improve activity against specific pathogens. Based on the LLP and parallel structure–function studies by the research groups of McLaughlin [127] and Degrado [128,129], we concluded that the remarkable diversity of natural AMPs reflects the fact that the evolution of specific structures of AMPs is highly influenced by host–pathogens interactions in the context of a particular biological environment, referred to as contextual activity of AMPs. An illustration of this restriction to the environment is the suppression of AMP activity observed in the lungs of cystic fibrosis (CF) patients associated with changes (e.g., changes in salt concentration and pH) in surface secretions [130,131]. Thus, when one either changes the target pathogen or environment (e.g., normal mucosal surface, abnormal airway, blood), AMP activity is diminished, as shown by the differential activities of LL37 against P. aeruginosa and S. aureus (Figure 3) in the presence or absence of saline.
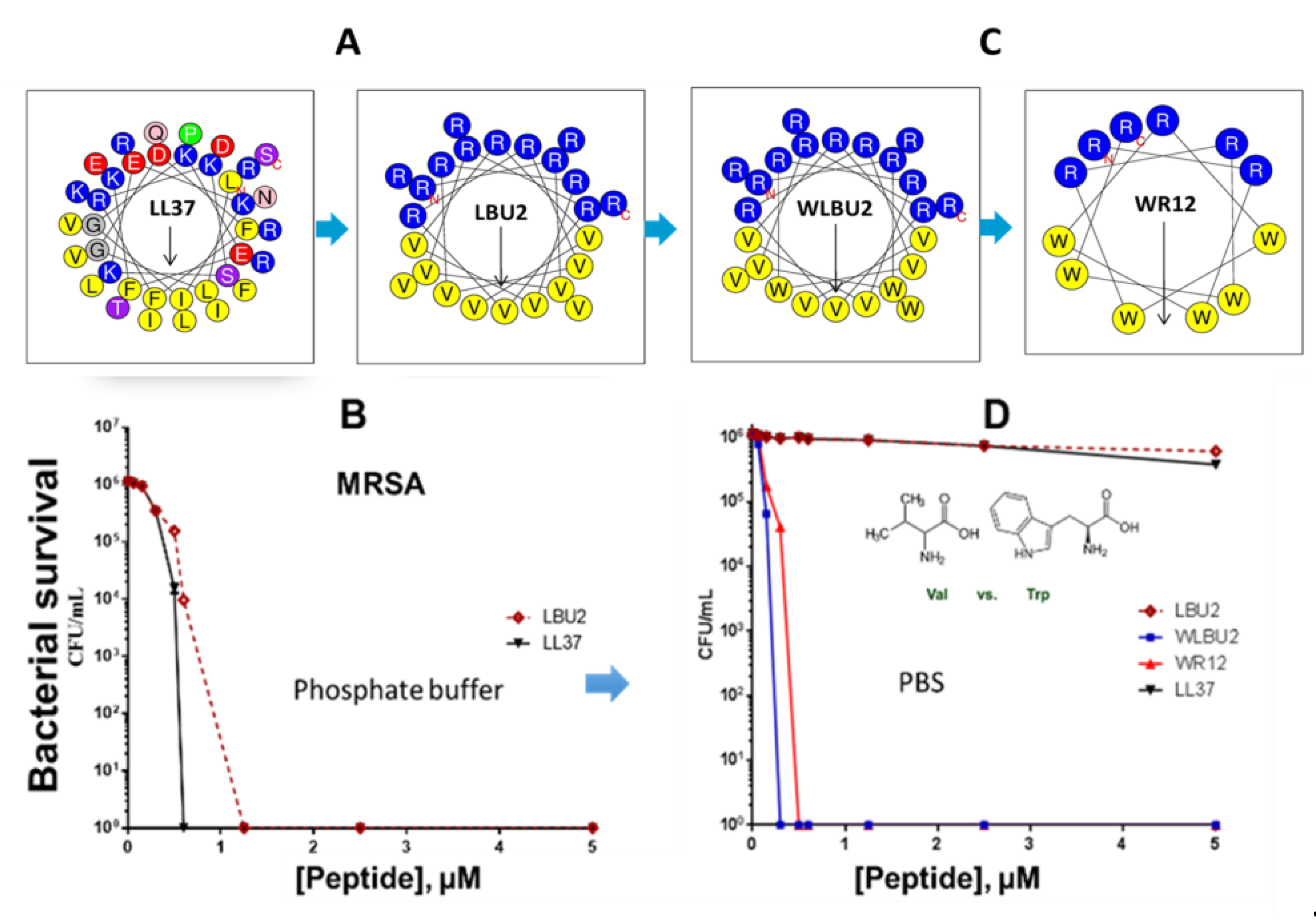
An important concept is the generally accepted view that antibacterial activity is not due to a particular consensus sequence but rather to the secondary motif of a cationic amphipathic structure. The database of thousands of AMPs strongly supports the notion of consensus amphipathic secondary structure vs. consensus sequence [43,132,133]. However, prior to the discoveries of all these peptides, it was not that obvious. Thus, we initially addressed this hypothesis by reducing the diversity in amino acid composition observed in natural AMPs (e.g., 14 different amino acids in the human AMP LL37, Figure 1) to just two (LBU2, R and V; WR12, R and W) or three (WLBU2, R, W, and V) different amino acids (Figure 3). A diverse amino acid composition, while determining multiple functions displayed by LL37 (Figure 1) [134,135,136,137,138,139,140] is certainly not necessary to establish a cationic amphipathic motif. In fact, it may interfere with the idealization of a cationic amphipathic structure. Thus, LL37 structure includes 11 positively charged residues, but it has a net charge of +6 due to five electronegative residues. In addition, the hydrophilic and hydrophobic domains are not completely segregated compared to an idealized helical structure revealed by helical wheel analysis. In contrast, LBU2, a peptide made only of Arg and Val, requires a length of only 24 residues (compared to 37 in LL37) to display an antibacterial activity profile similar to that of LL37, including the reduced activity against S. aureus in phosphate-buffered saline (PBS) compared to activity in phosphate buffer alone [75]. Importantly, our group and others have shown that such a susceptibility to salt could be overcome by sequence optimization within the framework of cationic amphipathicity. Hence, in sharp contrast to LL37 and LBU2, Trp-rich eCAPs WLBU2 (made of Arg, Trp, and Val) and WR12 (made of Arg and Trp) retain activity in saline, divalent cations, human blood, and even acidic pH [55,71,75,77]. Of note, the eCAPs kill bacteria by rapidly disrupting the membrane as shown by biochemical assays (access of the impermeant ortho-Nitrophenyl-β-galactoside (ONPG) to the intracellular enzyme β-galactosidase in P. aeruginosa) [71] and transmission electron microscopy as well as propidium iodide (PI) incorporation of peptide-treated bacteria (unpublished data).
3.3. Overcoming Resistance by Recalcitrant Bacteria
A critical consideration in overall antimicrobial effectiveness is the potential for bacteria to evolve resistance to the agent(s) of interest, regardless of prior exposure to the antibiotic [18]. The molecular target of a traditional antibiotic requires a vital cellular process. Thus, for such an antibiotic to be effective, the bacterial cell must be in an active metabolic state that is advancing the life cycle of the cell. Ironically, an active metabolic state may permit the formation of new resistance-conferring mutations in the cell (during DNA replication). By contrast, a membrane-active antibiotic (e.g, WLBU2) targets a pre-existing cellular structure (e.g., LPS) and its activity is not necessarily affected by the metabolic state of the cell (no carbon source needed to be active). Similarly, the rapid membrane perturbation mechanisms of many helical AMPs are based on interactions with membrane lipids that may be essential to bacterial survival [71,77]. Resistance to AMPs typically occur by lipid modification, which reduces the net negative charge on the bacterial cell surface, while other resistance mechanisms are relatively less common (e.g., peptide digestion, inactivation, efflux) [141,142,143,144]. This membrane-active mechanism is the reason colistin is effective against MDR gram-negative bacteria (GNB) as a drug of last resort. Notably, this property is highly enhanced in rationally designed helical peptides [119,120,145,146]. The eCAPs WLBU2 and WR12 demonstrate effective antimicrobial activity against diverse clinical isolates of the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae (K. pneumoniae), Acinetobacter baumannii, Pseudomonas aeruginosa (P. aeruginosa), and Enterobacter species) that are resistant to LL37 and colistin and display lower propensity to select for resistant bacteria in vitro [2,73,74,147,148]. When bacteria are allowed to grow in the absence of selective pressure, no test strains resistant to Trp-rich eCAPs have been found (unpublished data). Once bacteria are allowed to grow by serial daily passages in the presence of sub-inhibitory concentrations of antibiotics or eCAPs, resistance to WLBU2 or WR12 was delayed by 28 days (resistance achieved within 28 days) in comparison to colistin (13 days) and traditional antibiotics (two days). These data are consistent with several reports on propensity of engineered AMPs to select for resistant bacteria compared to traditional antibiotics [73,120]. In addition, resistance to LL37 and colistin by ESKAPE pathogens typically does not result in resistance to the eCAPs. These results indicate the ability of these eCAPs to overcome the most common resistance mechanisms against current antibiotics and natural AMPs. However, while very uncommon, cross-resistance among these three types of membrane-active antimicrobial agents has been observed in our studies. In addition, although the differential mechanisms of AMPs and other antibiotics targeting specific metabolic pathways are obvious, the ability to remain active against colistin- and LL37-resistant strains does not necessary indicate a fundamentally different mechanism. Similarly, two drugs displaying different affinities for the same target does not necessarily mean their molecular targets are different. Differences in potency are not necessarily due to differences in mechanisms of action. In the case of optimized eCAPs, it could be that differences in potency are due to differential sequence optimization, using Trp in the context of the cationic amphipathic structure, resulting in enhanced effects on the bacterial membrane and cell death.
3.4. More Advanced Antimicrobial Properties
One of the biggest challenges to AMP development is their lack of in vivo efficacy, and this is consistent with their contextual activity in vitro. In that regard, eCAPs are advanced compared to most other AMPs. Similar to the development of pexiganan, most new AMPs in development continue to typically target topical applications. In contrast, more than a decade ago, we showed that systemic delivery was plausible. Aside from the in vivo efficacy in a vaginal Chlamydia trachomatis monkey model [149], WLBU2 demonstrates systemic efficacy in a murine P. aeruginosa septicemia model. This was the first evidence for systemic efficacy of a helical AMP [55,118]. To date, the idea of AMP development for systemic applications is still novel when compared to other advanced cationic helical AMPs in clinical trials for topical use (e.g., pexiganan, an analogue of magainin 2, which is a natural AMP isolated from frog skin) [150,151,152,153]. WLBU2 is able to protect mice against P. aeruginosa septicemia with a single dose of 3–4 mg/kg injected intravenously (i.v.) 1 h after or prior to P. aeruginosa exposure. Since WLBU2 displays a maximum tolerated dose (MTD) of 15–30 mg/kg in mice (slow i.v. injection) with a minimum therapeutic dose (mTd) of 3–4 mg/kg, the therapeutic index (TI = MTD/mTd) is ≤10). In that context, colistin, an antibiotic drug of last resort against infections due to MDR gram-negative bacteria (GNB), has a lower TI in similar animal models [154].
An important property of AMPs is the ability to prevent, disrupt, or eliminate bacterial biofilm growth. Surgical and medical device infections are typically associated with bacterial biofilms [155]. They are difficult to treat with traditional antibiotics due to the inherent tolerance of the biofilm to antibiotics. Traditional antibiotics require a high dose of antibiotics over an extended time period to eliminate biofilm [156,157]. As a result, antibiotics alone are often unable to treat an infection associated with a medical device, and removal of the implant is required [158,159,160]. WLBU2 displays activity against biofilm in both biotic and abiotic assays and against enveloped viruses either independently or in bacteria–virus co-culture models [82,91,101,102,104]. The anti-biofilm properties of eCAPs are further explored using S. aureus grown on a titanium rod and in murine models of trauma-associated infection. In comparison to cefazolin, WLBU2 was able to eradicate titanium-bound S. aureus antimicrobial resistant biofilm in vitro within 30 min [102]. This was the first evidence of a single antimicrobial agent capable of eliminating biofilm in a short time period. WLBU2 also displayed efficacy in an orthopedic implant biofilm animal model, indicating advanced antibiofilm properties relevant to saving implants and improving outcomes in these difficult-to-treat infections.
4. Perspective
Despite the advancement of eCAPs and other engineered AMPs toward clinical development [161], the work required to establish AMPs as novel antibiotics is still enormous. Even if WLBU2 becomes clinically available, some important concerns remain to be addressed. One is the increased risk of toxicity upon sequence optimization with the intent to increase antimicrobial potency. If the goal is to develop antibiotics that are sufficiently safe to use clinically, then the efficacy will be limited. Once a drug is being used for a relatively long period, bacteria will evolve to strains that are at least less susceptible. In the face of just a moderate decrease in susceptibility at best, the clinical use of such a drug will become severely limited, as the therapeutic window is not sufficiently wide to increase dosage in order to overcome such variations in susceptibility. The most effective way to overcome potential host toxicity of AMPs is to establish structure–function guidelines based on differential roles of the cationic and hydrophobic domains in the amphipathic structure of AMPs. Thus, it is important to determine how each domain uniquely contributes to microbial as opposed to host toxicity. Such structure–function studies may inform the prediction of amino acid composition, proportion of cationic and hydrophobic residues, length, and the arrangement of such residues that would more selectively target bacterial cells as opposed to host cells. Although some progress has been made in that regard, the studies have been incremental and not sufficiently systematic to provide guidelines that would substantially predict low risk of toxicity or other essential functions.
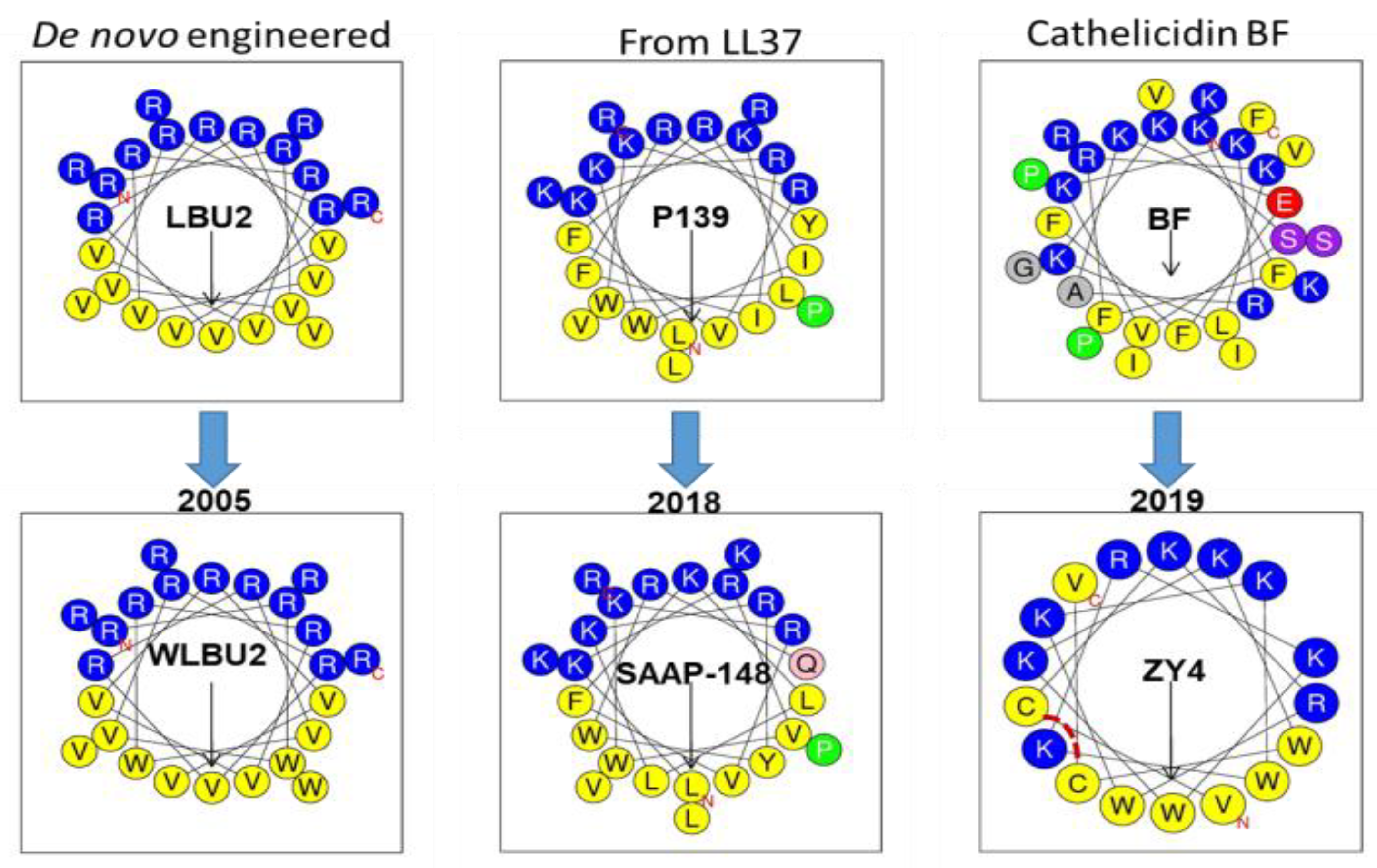
While the design of an AMP always begins with a rational basis, such design is typically implemented with a great deal of trial and error. To illustrate, let us compare WLBU2 with the most recent and promising AMPs. The structure of WLBU2 is based upon three main premises: (i) the optimization of the cationic amphipathic structure will enhance antibacterial potency; (ii) there must be an optimal length for highest antimicrobial potency; (iii) in the context of the helical amphipathic structure, Trp substitutions will enhance potency under physiological conditions [75]. Importantly, we did not have any information that would have predicted the sites for Trp substitution in relation to the potential toxicity to mammalian cells, except for the placement of the Trp residues in the hydrophobic domain of the secondary structure. Thus, while the design of WLBU2 began with a rational basis, an important part of the design resulted from an educated guess. The tendency for trial and error can also be demonstrated in AMP design that uses a template from a natural AMP for modification. As illustrated in Figure 4, two very recent AMPs were obtained from two natural amino acid sequences. Similarly to the use of Trp and few amino acids in WLBU2, SAAP-148 (LKRVWKRVFKLLKRYWRQLKKPVR), although derived from the cathelicidin LL37 (composed of 14 different amino acids), has an optimized cationic amphipathic structure with 24 residues in length [120]. This peptide also displays broad activity against MDR bacteria and moderate lytic effects on red blood cells at concentrations surpassing 10 µM. Similarly, the ZY4 peptide (VCKRWKKWKRKWKKWCV) was derived from a cathelicidin template with a very diverse amino acid composition, which was reduced to a composition of five amino acids (R, K, C, V, and W) and 17 residues in length. Interestingly, the cysteine residues provide a bridge that does not substantially disrupt the helical structure (an important lesson), as shown by the helical wheel projection (Figure 4) and by the three-dimensional (3D) structure [119]. In this case, the disulfide bridge may have a similar effect to that of stapling the peptide [162,163], which may contribute to its activity. Thus, both SAAP-148 and ZY4 show some structural similarities to WLBU2 and could not have been obtained without some level of trial and error 13–14 years after the first study on WLBU2 was published. These three examples (Figure 4) illustrate conceptually the incremental progress in the design of helical AMPs. Therefore, more transformative structure–function studies are necessary despite bold efforts in clinical development (LL37 for melanoma) [161]. Of note, the use of D-enantiomerization and unnatural amino acids, including peptoids, and cyclization of non-helical AMPs are additional strategies that could be effective in enhancing the pharmacological utility of AMPs [100,114,115,116].
5. Conclusions
While the field of AMPs is not progressing as fast as initially expected, the promising therapeutic impact on MDR-related infections is still relevant and the need for AMP development even more urgent. The eCAP and other AMP engineering technologies have not advanced the field fast enough amid lack of federal funding due to skepticism stemming from many limitations of AMPs, including past failed attempts at clinical development in previous decades. However, efforts to revive the field are still on the way, as several AMPs are in clinical trials [161], including WLBU2 (PLG0206), now in phase I trials for treatment of prosthetic joint infection associated with gram-positive bacteria. Similar to other AMPs in clinical trials [164], WLBU2 could become one of the first success stories carried out to completion. If so, the perception that classical AMPs are failed antimicrobial agents will change, which will open up more funding opportunities for consistent efforts to turn the prospect of AMPs as effective antimicrobials into a reality.
6. Patents
R.C.M. holds a patent on WLBU2 (PLG0206), which is licensed by Peptilogics.
Funding
This review was supported by NIH awards GM125917 (to B.D.) and AI133351 (to Y.P.D.).
Conflicts of Interest
R.C.M. holds stock in Peptilogics and serves on an advisory board for Peptilogics. Although a financial conflict of interest was identified on the basis of R.C.M.’s relationship with Peptilogics, the opinions included in this publication are not related to the interests of Peptilogics. All other authors declare that they have no competing interests. The funders had no role in the conception or the writing of the manuscript.
References
- Barra, A.L.C.; Dantas, L.O.C.; Morao, L.G.; Gutierrez, R.F.; Polikarpov, I.; Wrenger, C.; Nascimento, A.S. Essential metabolic routes as a way to eskape from antibiotic resistance. Front. Public Health 2020, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No eskape! An update from the infectious diseases society of america. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.; Xiao, Y.; Kang, L.; Ma, W.; Shi, L.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.; Yang, D.; et al. Genomic diversity of sars-cov-2 in coronavirus disease 2019 patients. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Shin, W.I.; Pang, Y.X.; Meng, Y.; Lai, J.; You, C.; Zhao, H.; Lester, E.; Wu, T.; Pang, C.H. The first 75 days of novel coronavirus (sars-cov-2) outbreak: Recent advances, prevention, and treatment. Int. J. Environ. Res. Public Health 2020, 17, 2323. [Google Scholar] [CrossRef] [Green Version]
- Conlon-Bingham, G.M.; Aldeyab, M.; Scott, M.; Kearney, M.P.; Farren, D.; Gilmore, F.; McElnay, J. Effects of antibiotic cycling policy on incidence of healthcare-associated mrsa and clostridioides difficile infection in secondary healthcare settings. Emerg. Infect. Dis. 2019, 25, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Ballus, J.; Lopez-Delgado, J.C.; Sabater-Riera, J.; Perez-Fernandez, X.L.; Betbese, A.J.; Roncal, J.A. Surgical site infection in critically ill patients with secondary and tertiary peritonitis: Epidemiology, microbiology and influence in outcomes. BMC Infect. Dis. 2015, 15, 304. [Google Scholar] [CrossRef] [Green Version]
- Hota, S.; Hirji, Z.; Stockton, K.; Lemieux, C.; Dedier, H.; Wolfaardt, G.; Gardam, M.A. Outbreak of multidrug-resistant pseudomonas aeruginosa colonization and infection secondary to imperfect intensive care unit room design. Infect. Control Hosp. Epidemiol. 2009, 30, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Patterson-Delafield, J.; Martinez, R.J.; Lehrer, R.I. Microbicidal cationic proteins in rabbit alveolar macrophages: A potential host defense mechanism. Infect. Immun. 1980, 30, 180–192. [Google Scholar] [CrossRef] [Green Version]
- Marion, D.; Zasloff, M.; Bax, A. A two-dimensional nmr study of the antimicrobial peptide magainin 2. FEBS Lett. 1988, 227, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Merrifield, R.B.; Vizioli, L.D.; Boman, H.G. Synthesis of the antibacterial peptide cecropin a (1-33). Biochemistry 1982, 21, 5020–5031. [Google Scholar] [CrossRef]
- Barksdale, S.M.; Hrifko, E.J.; van Hoek, M.L. Cathelicidin antimicrobial peptide from alligator mississippiensis has antibacterial activity against multi-drug resistant acinetobacter baumanii and klebsiella pneumoniae. Dev. Comp. Immunol. 2017, 70, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Stephens, C.; Kazan, K.; Goulter, K.C.; Maclean, D.J.; Manners, J.M. The mode of action of the plant antimicrobial peptide miamp1 differs from that of its structural homologue, the yeast killer toxin wmkt. FEMS Microbiol. Lett. 2005, 243, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Benko-Iseppon, A.M.; Galdino, S.L.; Calsa, T., Jr.; Kido, E.A.; Tossi, A.; Belarmino, L.C.; Crovella, S. Overview on plant antimicrobial peptides. Curr. Protein Pept. Sci. 2010, 11, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Das, S. Mammalian antimicrobial peptides: Promising therapeutic targets against infection and chronic inflammation. Curr. Top. Med. Chem. 2016, 16, 99–129. [Google Scholar] [CrossRef]
- Singh, N.; Abraham, J. Ribosomally synthesized peptides from natural sources. J. Antibiot. 2014, 67, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Park, O.J.; Hong, S.H. Incorporation of non-standard amino acids into proteins: Challenges, recent achievements, and emerging applications. Appl. Microbiol. Biotechnol. 2019, 103, 2947–2958. [Google Scholar] [CrossRef]
- McPhee, J.B.; Lewenza, S.; Hancock, R.E. Cationic antimicrobial peptides activate a two-component regulatory system, pmra-pmrb, that regulates resistance to polymyxin b and cationic antimicrobial peptides in pseudomonas aeruginosa. Mol. Microbiol. 2003, 50, 205–217. [Google Scholar] [CrossRef]
- Yin, J.; Meng, Q.; Cheng, D.; Fu, J.; Luo, Q.; Liu, Y.; Yu, Z. Mechanisms of bactericidal action and resistance of polymyxins for gram-positive bacteria. Appl. Microbiol. Biotechnol. 2020, 104, 3771–3780. [Google Scholar] [CrossRef]
- Davis, C.A.; Janssen, E.M. Environmental fate processes of antimicrobial peptides daptomycin, bacitracins, and polymyxins. Environ. Int. 2020, 134, 105271. [Google Scholar] [CrossRef]
- Boulanger, N.; Munks, R.J.; Hamilton, J.V.; Vovelle, F.; Brun, R.; Lehane, M.J.; Bulet, P. Epithelial innate immunity. A novel antimicrobial peptide with antiparasitic activity in the blood-sucking insect stomoxys calcitrans. J. Biol. Chem. 2002, 277, 49921–49926. [Google Scholar] [CrossRef] [Green Version]
- Hancock, R.E. Cationic antimicrobial peptides: Towards clinical applications. Expert Opin. Investig. Drugs 2000, 9, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Diamond, G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 2000, 8, 402–410. [Google Scholar] [CrossRef]
- Kamysz, E.; Sikorska, E.; Karafova, A.; Dawgul, M. Synthesis, biological activity and conformational analysis of head-to-tail cyclic analogues of ll37 and histatin 5. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2012, 18, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.; Choi, S.Y.; Acharya, S.; Chun, Y.J.; Gurley, C.; Park, Y.; Armstrong, C.A.; Song, P.I.; Kim, B.J. Antimicrobial and anti-inflammatory effects of cecropin a(1-8)-magainin2(1-12) hybrid peptide analog p5 against malassezia furfur infection in human keratinocytes. J. Investig. Dermatol. 2011, 131, 1677–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, G.; Zasloff, M.; Eck, H.; Brasseur, M.; Maloy, W.L.; Bevins, C.L. Tracheal antimicrobial peptide, a cysteine-rich peptide from mammalian tracheal mucosa: Peptide isolation and cloning of a cdna. Proc. Natl. Acad. Sci. USA 1991, 88, 3952–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehrer, R.I.; Ganz, T. Antimicrobial peptides in mammalian and insect host defence. Curr. Opin. Immunol. 1999, 11, 23–27. [Google Scholar] [CrossRef]
- Arenas, I.; Ibarra, M.A.; Santana, F.L.; Villegas, E.; Hancock, R.E.W.; Corzo, G. In vitro and in vivo antibiotic capacity of two host defence peptides. Antimicrob. Agents Chemother. 2020. [Google Scholar] [CrossRef]
- Rabanal, F.; Cajal, Y. Recent advances and perspectives in the design and development of polymyxins. Nat. Prod. Rep. 2017, 34, 886–908. [Google Scholar] [CrossRef]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure--activity relationships of polymyxin antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef] [Green Version]
- Tajbakhsh, M.; Karimi, A.; Fallah, F.; Akhavan, M.M. Overview of ribosomal and non-ribosomal antimicrobial peptides produced by gram positive bacteria. Cell. Mol. Biol. 2017, 63, 20–32. [Google Scholar] [CrossRef]
- Sierra, J.M.; Fuste, E.; Rabanal, F.; Vinuesa, T.; Vinas, M. An overview of antimicrobial peptides and the latest advances in their development. Expert Opin. Biol. Ther. 2017, 17, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Meyer, K.J.; Iinishi, A.; Favre-Godal, Q.; Green, R.; Manuse, S.; Caboni, M.; Mori, M.; Niles, S.; Ghiglieri, M.; et al. Author correction: A new antibiotic selectively kills gram-negative pathogens. Nature 2020, 580, E3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryashova, E.; Seveau, S.; Lu, W.; Kudryashov, D.S. Retrocyclins neutralize bacterial toxins by potentiating their unfolding. Biochem. J. 2015, 467, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welkos, S.; Cote, C.K.; Hahn, U.; Shastak, O.; Jedermann, J.; Bozue, J.; Jung, G.; Ruchala, P.; Pratikhya, P.; Tang, T.; et al. Humanized theta-defensins (retrocyclins) enhance macrophage performance and protect mice from experimental anthrax infections. Antimicrob. Agents Chemother. 2011, 55, 4238–4250. [Google Scholar] [CrossRef] [Green Version]
- Yeoh, Y.Q.; Yu, J.; Polyak, S.W.; Horsley, J.R.; Abell, A.D. Photopharmacological control of cyclic antimicrobial peptides. Chembiochem 2018, 19, 2591–2597. [Google Scholar] [CrossRef]
- Bagheri, M.; Amininasab, M.; Dathe, M. Arginine/tryptophan-rich cyclic alpha/beta-antimicrobial peptides: The roles of hydrogen bonding and hydrophobic/hydrophilic solvent-accessible surface areas upon activity and membrane selectivity. Chemistry 2018, 24, 14242–14253. [Google Scholar] [CrossRef]
- He, R.; Di Bonaventura, I.; Visini, R.; Gan, B.H.; Fu, Y.; Probst, D.; Luscher, A.; Kohler, T.; van Delden, C.; Stocker, A.; et al. Design, crystal structure and atomic force microscopy study of thioether ligated d,l-cyclic antimicrobial peptides against multidrug resistant pseudomonas aeruginosa. Chem. Sci. 2017, 8, 7464–7475. [Google Scholar] [CrossRef] [Green Version]
- Cirac, A.D.; Torne, M.; Badosa, E.; Montesinos, E.; Salvador, P.; Feliu, L.; Planas, M. Rational design of cyclic antimicrobial peptides based on bpc194 and bpc198. Molecules 2017, 22, 1054. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Jeong, K.W.; Lee, J.; Shin, A.; Kim, J.K.; Lee, J.; Lee, D.G.; Kim, Y. Structure-activity relationships of cecropin-like peptides and their interactions with phospholipid membrane. BMB Rep. 2013, 46, 282–287. [Google Scholar] [CrossRef]
- Saiman, L.; Tabibi, S.; Starner, T.D.; San Gabriel, P.; Winokur, P.L.; Jia, H.P.; McCray, P.B., Jr.; Tack, B.F. Cathelicidin peptides inhibit multiply antibiotic-resistant pathogens from patients with cystic fibrosis. Antimicrob. Agents Chemother. 2001, 45, 2838–2844. [Google Scholar] [CrossRef] [Green Version]
- Tencza, S.B.; Douglass, J.P.; Creighton, D.J., Jr.; Montelaro, R.C.; Mietzner, T.A. Novel antimicrobial peptides derived from human immunodeficiency virus type 1 and other lentivirus transmembrane proteins. Antimicrob. Agents Chemother. 1997, 41, 2394–2398. [Google Scholar] [CrossRef] [Green Version]
- Lehrer, R.I.; Lichtenstein, A.K.; Ganz, T. Defensins: Antimicrobial and cytotoxic peptides of mammalian cells. Annu. Rev. Immunol. 1993, 11, 105–128. [Google Scholar] [CrossRef] [PubMed]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. Campr3: A database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Waring, A.J.; Ruchala, P.; Hong, M. Structures of beta-hairpin antimicrobial protegrin peptides in lipopolysaccharide membranes: Mechanism of gram selectivity obtained from solid-state nuclear magnetic resonance. Biochemistry 2011, 50, 2072–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitaram, N. Antimicrobial peptides with unusual amino acid compositions and unusual structures. Curr. Med. Chem. 2006, 13, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Dong, F.; Shi, C.; Liu, S.; Sun, J.; Chen, J.; Li, H.; Xu, H.; Lao, X.; Zheng, H. Dramp 2.0, an updated data repository of antimicrobial peptides. Sci. Data 2019, 6, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thennarasu, S.; Tan, A.; Penumatchu, R.; Shelburne, C.E.; Heyl, D.L.; Ramamoorthy, A. Antimicrobial and membrane disrupting activities of a peptide derived from the human cathelicidin antimicrobial peptide ll37. Biophys. J. 2010, 98, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Rozek, A.; Hancock, R.E. Interaction of cationic antimicrobial peptides with model membranes. J. Biol. Chem. 2001, 276, 35714–35722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, R.E.; O’Brien, L.M.; Thwaite, J.E.; Fox, M.A.; Atkins, H.; Ulaeto, D.O. A carpet-based mechanism for direct antimicrobial peptide activity against vaccinia virus membranes. Peptides 2010, 31, 1966–1972. [Google Scholar] [CrossRef]
- Sato, H.; Feix, J.B. Peptide-membrane interactions and mechanisms of membrane destruction by amphipathic alpha-helical antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Wohland, T.; Ho, B.; Ding, J.L. Perturbation of lipopolysaccharide (lps) micelles by sushi 3 (s3) antimicrobial peptide. The importance of an intermolecular disulfide bond in s3 dimer for binding, disruption, and neutralization of lps. J. Biol. Chem. 2004, 279, 50150–50156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, R.E.; Rozek, A. Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol. Lett. 2002, 206, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Chih, Y.H.; Wang, S.Y.; Yip, B.S.; Cheng, K.T.; Hsu, S.Y.; Wu, C.L.; Yu, H.Y.; Cheng, J.W. Dependence on size and shape of non-nature amino acids in the enhancement of lipopolysaccharide (lps) neutralizing activities of antimicrobial peptides. J. Colloid Interface Sci. 2019, 533, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Shang, D.; Zhang, Q.; Dong, W.; Liang, H.; Bi, X. The effects of lps on the activity of trp-containing antimicrobial peptides against gram-negative bacteria and endotoxin neutralization. Acta Biomater. 2016, 33, 153–165. [Google Scholar] [CrossRef]
- Deslouches, B.; Islam, K.; Craigo, J.K.; Paranjape, S.M.; Montelaro, R.C.; Mietzner, T.A. Activity of the de novo engineered antimicrobial peptide wlbu2 against pseudomonas aeruginosa in human serum and whole blood: Implications for systemic applications. Antimicrob. Agents Chemother. 2005, 49, 3208–3216. [Google Scholar] [CrossRef] [Green Version]
- Ryder, M.P.; Wu, X.; McKelvey, G.R.; McGuire, J.; Schilke, K.F. Binding interactions of bacterial lipopolysaccharide and the cationic amphiphilic peptides polymyxin b and wlbu2. Colloids Surf. B Biointerfaces 2014, 120, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Leung, L.M.; Cooper, V.S.; Rasko, D.A.; Guo, Q.; Pacey, M.P.; McElheny, C.L.; Mettus, R.T.; Yoon, S.H.; Goodlett, D.R.; Ernst, R.K.; et al. Structural modification of lps in colistin-resistant, kpc-producing klebsiella pneumoniae. J. Antimicrob. Chemother. 2017, 72, 3035–3042. [Google Scholar] [CrossRef]
- Kamoshida, G.; Akaji, T.; Takemoto, N.; Suzuki, Y.; Sato, Y.; Kai, D.; Hibino, T.; Yamaguchi, D.; Kikuchi-Ueda, T.; Nishida, S.; et al. Lipopolysaccharide-deficient acinetobacter baumannii due to colistin resistance is killed by neutrophil-produced lysozyme. Front. Microbiol. 2020, 11, 573. [Google Scholar] [CrossRef]
- Li, B.; Yin, F.; Zhao, X.; Guo, Y.; Wang, W.; Wang, P.; Zhu, H.; Yin, Y.; Wang, X. Colistin resistance gene mcr-1 mediates cell permeability and resistance to hydrophobic antibiotics. Front. Microbiol. 2019, 10, 3015. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.R.; Bayer, A.S.; Arias, C.A. Mechanism of action and resistance to daptomycin in staphylococcus aureus and enterococci. Cold Spring Harb. Perspect Med. 2016, 6, a026997. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.D.; Palmer, M. The action mechanism of daptomycin. Bioorg. Med. Chem. 2016, 24, 6253–6268. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.T.; Munita, J.M.; Arias, C.A. Mechanisms of drug resistance: Daptomycin resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 32–53. [Google Scholar] [CrossRef] [Green Version]
- Hale, J.D.; Hancock, R.E. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev. Anti Infect. Ther. 2007, 5, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Ostorhazi, E.; Holub, M.C.; Rozgonyi, F.; Harmos, F.; Cassone, M.; Wade, J.D.; Otvos, L., Jr. Broad-spectrum antimicrobial efficacy of peptide a3-apo in mouse models of multidrug-resistant wound and lung infections cannot be explained by in vitro activity against the pathogens involved. Int. J. Antimicrob. Agents 2011, 37, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.T.; Lee, J.Y.; Kim, H.J.; Eu, Y.J.; Shin, S.Y.; Hahm, K.S.; Kim, J.I. Contribution of a central proline in model amphipathic alpha-helical peptides to self-association, interaction with phospholipids, and antimicrobial mode of action. FEBS J. 2006, 273, 4040–4054. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, C.L.; Rozek, A.; Patrzykat, A.; Hancock, R.E. Structure and mechanism of action of an indolicidin peptide derivative with improved activity against gram-positive bacteria. J. Biol. Chem. 2001, 276, 24015–24022. [Google Scholar] [CrossRef] [Green Version]
- Melchers, M.J.; Teague, J.; Warn, P.; Hansen, J.; Bernardini, F.; Wach, A.; Obrecht, D.; Dale, G.E.; Mouton, J.W. Pharmacokinetics and pharmacodynamics of murepavadin in neutropenic mouse models. Antimicrob. Agents Chemother. 2019, 63, e01699-18. [Google Scholar] [CrossRef] [Green Version]
- Scheinpflug, K.; Nikolenko, H.; Komarov, I.V.; Rautenbach, M.; Dathe, M. What goes around comes around-a comparative study of the influence of chemical modifications on the antimicrobial properties of small cyclic peptides. Pharmaceuticals 2013, 6, 1130–1144. [Google Scholar] [CrossRef]
- Bhopale, G.M. Antimicrobial peptides: A promising avenue for human healthcare. Curr. Pharm. Biotechnol. 2020, 21, 90–96. [Google Scholar] [CrossRef]
- Faya, M.; Kalhapure, R.S.; Dhumal, D.; Agrawal, N.; Omolo, C.; Akamanchi, K.G.; Govender, T. Antimicrobial cell penetrating peptides with bacterial cell specificity: Pharmacophore modelling, quantitative structure activity relationship and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2019, 37, 2370–2380. [Google Scholar] [CrossRef]
- Deslouches, B.; Hasek, M.L.; Craigo, J.K.; Steckbeck, J.D.; Montelaro, R.C. Comparative functional properties of engineered cationic antimicrobial peptides consisting exclusively of tryptophan and either lysine or arginine. J. Med. Microbiol. 2016, 65, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Hicks, R.P.; Abercrombie, J.J.; Wong, R.K.; Leung, K.P. Antimicrobial peptides containing unnatural amino acid exhibit potent bactericidal activity against eskape pathogens. Bioorg. Med. Chem. 2013, 21, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Deslouches, B.; Steckbeck, J.D.; Craigo, J.K.; Doi, Y.; Burns, J.L.; Montelaro, R.C. Engineered cationic antimicrobial peptides to overcome multidrug resistance by eskape pathogens. Antimicrob. Agents Chemother. 2015, 59, 1329–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steckbeck, J.D.; Deslouches, B.; Montelaro, R.C. Antimicrobial peptides: New drugs for bad bugs? Expert. Opin. Biol. Ther. 2014, 14, 11–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deslouches, B.; Phadke, S.M.; Lazarevic, V.; Cascio, M.; Islam, K.; Montelaro, R.C.; Mietzner, T.A. De novo generation of cationic antimicrobial peptides: Influence of length and tryptophan substitution on antimicrobial activity. Antimicrob. Agents Chemother. 2005, 49, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirel, L.; Jayol, A.; Nordmann, P. Polymyxins: Antibacterial activity, susceptibility testing, and resistance mechanisms encoded by plasmids or chromosomes. Clin. Microbiol. Rev. 2017, 30, 557–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deslouches, B.; Steckbeck, J.D.; Craigo, J.K.; Doi, Y.; Mietzner, T.A.; Montelaro, R.C. Rational design of engineered cationic antimicrobial peptides consisting exclusively of arginine and tryptophan: Wr ecap activity against multidrug-resistant pathogens. Antimicrob. Agents Chemother. 2013, 57, 2511–2521. [Google Scholar] [CrossRef] [Green Version]
- Doi, Y.; van Duin, D. Polymyxin resistance in klebsiella pneumoniae: Complexity at every level. Clin. Infect. Dis. 2020, 70, 2092–2094. [Google Scholar] [CrossRef]
- Barker, W.T.; Martin, S.E.; Chandler, C.E.; Nguyen, T.V.; Harris, T.L.; Goodell, C.; Melander, R.J.; Doi, Y.; Ernst, R.K.; Melander, C. Small molecule adjuvants that suppress both chromosomal and mcr-1 encoded colistin-resistance and amplify colistin efficacy in polymyxin-susceptible bacteria. Bioorg. Med. Chem. 2017, 25, 5749–5753. [Google Scholar] [CrossRef]
- Goldner, N.K.; Bulow, C.; Cho, K.; Wallace, M.; Hsu, F.F.; Patti, G.J.; Burnham, C.A.; Schlesinger, P.; Dantas, G. Mechanism of high-level daptomycin resistance in corynebacterium striatum. mSphere 2018, 3, e00371-18. [Google Scholar] [CrossRef] [Green Version]
- Kelesidis, T. Transport of daptomycin resistance genes between animals and humans as a possible mechanism for development of de novo daptomycin resistance in enterococci. Epidemiol. Infect. 2013, 141, 2185–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Deslouches, B.; Montelaro, R.C.; Di, Y.P. Prevention of eskape pathogen biofilm formation by antimicrobial peptides wlbu2 and ll37. Int. J. Antimicrob. Agents 2018, 52, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Baindara, P.; Ghosh, A.K.; Mandal, S.M. Coevolution of resistance against antimicrobial peptides. Microb. Drug Resist. 2020. [Google Scholar] [CrossRef]
- El Shazely, B.; Yu, G.; Johnston, P.R.; Rolff, J. Resistance evolution against antimicrobial peptides in staphylococcus aureus alters pharmacodynamics beyond the mic. Front. Microbiol. 2020, 11, 103. [Google Scholar] [CrossRef] [PubMed]
- Duperthuy, M. Antimicrobial peptides: Virulence and resistance modulation in gram-negative bacteria. Microorganisms 2020, 8, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosikowska, P.; Lesner, A. Antimicrobial peptides (amps) as drug candidates: A patent review (2003–2015). Expert. Opin. Ther. Pat. 2016, 26, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Tonk, M.; Vilcinskas, A.; Rahnamaeian, M. Insect antimicrobial peptides: Potential tools for the prevention of skin cancer. Appl. Microbiol. Biotechnol. 2016, 100, 7397–7405. [Google Scholar] [CrossRef] [Green Version]
- Tanphaichitr, N.; Srakaew, N.; Alonzi, R.; Kiattiburut, W.; Kongmanas, K.; Zhi, R.; Li, W.; Baker, M.; Wang, G.; Hickling, D. Potential use of antimicrobial peptides as vaginal spermicides/microbicides. Pharmaceuticals 2016, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Stensen, W.; Turner, R.; Brown, M.; Kondori, N.; Svendsen, J.S.; Svenson, J. Short cationic antimicrobial peptides display superior antifungal activities toward candidiasis and onychomycosis in comparison with terbinafine and amorolfine. Mol. Pharm. 2016, 13, 3595–3600. [Google Scholar] [CrossRef]
- Ramesh, S.; Govender, T.; Kruger, H.G.; de la Torre, B.G.; Albericio, F. Short antimicrobial peptides (samps) as a class of extraordinary promising therapeutic agents. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2016, 22, 438–451. [Google Scholar] [CrossRef]
- Melvin, J.A.; Lashua, L.P.; Kiedrowski, M.R.; Yang, G.; Deslouches, B.; Montelaro, R.C.; Bomberger, J.M. Simultaneous antibiofilm and antiviral activities of an engineered antimicrobial peptide during virus-bacterium coinfection. mSphere 2016, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelbaqi, S.; Deslouches, B.; Steckbeck, J.; Montelaro, R.; Reed, D.S. Novel engineered cationic antimicrobial peptides display broad-spectrum activity against francisella tularensis, yersinia pestis and burkholderia pseudomallei. J. Med. Microbiol. 2016, 65, 188–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelhaus, C.; Jacobs, T.; Andra, J.; Leippe, M. The antimicrobial peptide nk-2, the core region of mammalian nk-lysin, kills intraerythrocytic plasmodium falciparum. Antimicrob. Agents Chemother. 2008, 52, 1713–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, C.K.; Rodrigues, F.G.; Ghosh, A.; Varotti Fde, P.; Miranda, A.; Daffre, S.; Jacobs-Lorena, M.; Moreira, L.A. Effect of the antimicrobial peptide gomesin against different life stages of plasmodium spp. Exp. Parasitol. 2007, 116, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.N.; Pan, C.Y.; Chen, J.Y. Grouper (epinephelus coioides) antimicrobial peptide epinecidin-1 exhibits antiviral activity against foot-and-mouth disease virus in vitro. Peptides 2018, 106, 91–95. [Google Scholar] [CrossRef]
- Brandenburg, K.; Garidel, P.; Fukuoka, S.; Howe, J.; Koch, M.H.; Gutsmann, T.; Andra, J. Molecular basis for endotoxin neutralization by amphipathic peptides derived from the alpha-helical cationic core-region of nk-lysin. Biophys. Chem. 2010, 150, 80–87. [Google Scholar] [CrossRef]
- Lin, Q.P.; Zhou, L.F.; Li, N.N.; Chen, Y.Q.; Li, B.C.; Cai, Y.F.; Zhang, S.Q. Lipopolysaccharide neutralization by the antibacterial peptide cm4. Eur. J. Pharmacol. 2008, 596, 160–165. [Google Scholar] [CrossRef]
- Paranjape, S.M.; Lauer, T.W.; Montelaro, R.C.; Mietzner, T.A.; Vij, N. Modulation of proinflammatory activity by the engineered cationic antimicrobial peptide wlbu-2. F1000Research 2013, 2, 36. [Google Scholar] [CrossRef]
- Pfalzgraff, A.; Heinbockel, L.; Su, Q.; Gutsmann, T.; Brandenburg, K.; Weindl, G. Synthetic antimicrobial and lps-neutralising peptides suppress inflammatory and immune responses in skin cells and promote keratinocyte migration. Sci. Rep. 2016, 6, 31577. [Google Scholar] [CrossRef] [Green Version]
- Di, Y.P.; Lin, Q.; Chen, C.; Montelaro, R.C.; Doi, Y.; Deslouches, B. Enhanced therapeutic index of an antimicrobial peptide in mice by increasing safety and activity against multidrug-resistant bacteria. Sci. Adv. 2020, 6, eaay6817. [Google Scholar] [CrossRef]
- Chen, C.; Deslouches, B.; Montelaro, R.C.; Di, Y.P. Enhanced efficacy of the engineered antimicrobial peptide wlbu2 via direct airway delivery in a murine model of pseudomonas aeruginosa pneumonia. Clin. Microbiol. Infect. 2018, 24, 547.e1–547.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandell, J.B.; Deslouches, B.; Montelaro, R.C.; Shanks, R.M.Q.; Doi, Y.; Urish, K.L. Elimination of antibiotic resistant surgical implant biofilms using an engineered cationic amphipathic peptide wlbu2. Sci. Rep. 2017, 7, 18098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deslouches, B.; Di, Y.P. Antimicrobial peptides: A potential therapeutic option for surgical site infections. Clin. Surg. 2017, 2, 1740. [Google Scholar] [PubMed]
- Lashua, L.P.; Melvin, J.A.; Deslouches, B.; Pilewski, J.M.; Montelaro, R.C.; Bomberger, J.M. Engineered cationic antimicrobial peptide (ecap) prevents pseudomonas aeruginosa biofilm growth on airway epithelial cells. J. Antimicrob. Chemother. 2016, 71, 2200–2207. [Google Scholar] [CrossRef] [Green Version]
- Deslouches, B.; Di, Y.P. Antimicrobial peptides with selective antitumor mechanisms: Prospect for anticancer applications. Oncotarget 2017, 8, 46635–46651. [Google Scholar] [CrossRef] [Green Version]
- Sertcelik, A.; Baran, I.; Akinci, E.; Mumcuoglu, I.; Bodur, H. Synergistic activities of colistin combinations with meropenem, sulbactam, minocycline, disodium fosfomycin, or vancomycin against different clones of carbapenem-resistant acinetobacter baumannii strains. Microb. Drug Resist. 2020, 26, 429–433. [Google Scholar] [CrossRef]
- Pollini, S.; Brunetti, J.; Sennati, S.; Rossolini, G.M.; Bracci, L.; Pini, A.; Falciani, C. Synergistic activity profile of an antimicrobial peptide against multidrug-resistant and extensively drug-resistant strains of gram-negative bacterial pathogens. J. Pept. Sci. 2017, 23, 329–333. [Google Scholar] [CrossRef]
- Gomes, D.; Santos, R.; Soares, R.S.; Reis, S.; Carvalho, S.; Rego, P.; M., C.P.; Tavares, L.; Oliveira, M. Pexiganan in combination with nisin to control polymicrobial diabetic foot infections. Antibiotics 2020, 9, 128. [Google Scholar] [CrossRef] [Green Version]
- Cote, C.K.; Blanco, I.I.; Hunter, M.; Shoe, J.L.; Klimko, C.P.; Panchal, R.G.; Welkos, S.L. Combinations of early generation antibiotics and antimicrobial peptides are effective against a broad spectrum of bacterial biothreat agents. Microb. Pathog. 2020, 142, 104050. [Google Scholar] [CrossRef]
- Bengtsson, T.; Selegard, R.; Musa, A.; Hultenby, K.; Utterstrom, J.; Sivler, P.; Skog, M.; Nayeri, F.; Hellmark, B.; Soderquist, B.; et al. Plantaricin nc8 alphabeta exerts potent antimicrobial activity against staphylococcus spp. And enhances the effects of antibiotics. Sci. Rep. 2020, 10, 3580. [Google Scholar] [CrossRef]
- Falagas, M.E.; Kasiakou, S.K. Colistin: The revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirt, H.; Hall, J.W.; Larson, E.; Gorr, S.U. A d-enantiomer of the antimicrobial peptide gl13k evades antimicrobial resistance in the gram positive bacteria enterococcus faecalis and streptococcus gordonii. PLoS ONE 2018, 13, e0194900. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, H.; Matsumoto, H.; Hashimoto, K.; Teraguchi, S.; Takase, M.; Hayasawa, H. N-acylated and d enantiomer derivatives of a nonamer core peptide of lactoferricin b showing improved antimicrobial activity. Antimicrob. Agents Chemother. 1999, 43, 1267–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, I.; Hansen, J.E.; Jana, B.; Molchanova, N.; Oddo, A.; Thulstrup, P.W.; Damborg, P.; Guardabassi, L.; Hansen, P.R. Structure(-)activity study, characterization, and mechanism of action of an antimicrobial peptoid d2 and its d- and l-peptide analogues. Molecules 2019, 24, 1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molchanova, N.; Wang, H.; Hansen, P.R.; Hoiby, N.; Nielsen, H.M.; Franzyk, H. Antimicrobial activity of alpha-peptide/beta-peptoid lysine-based peptidomimetics against colistin-resistant pseudomonas aeruginosa isolated from cystic fibrosis patients. Front. Microbiol. 2019, 10, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molchanova, N.; Hansen, P.R.; Damborg, P.; Nielsen, H.M.; Franzyk, H. Lysine-based alpha-peptide/beta-peptoid peptidomimetics: Influence of hydrophobicity, fluorination, and distribution of cationic charge on antimicrobial activity and cytotoxicity. ChemMedChem 2017, 12, 312–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, A.; Lee, E.; Jeon, D.; Park, Y.G.; Bang, J.K.; Park, Y.S.; Shin, S.Y.; Kim, Y. Peptoid-substituted hybrid antimicrobial peptide derived from papiliocin and magainin 2 with enhanced bacterial selectivity and anti-inflammatory activity. Biochemistry 2015, 54, 3921–3931. [Google Scholar] [CrossRef]
- Deslouches, B.; Gonzalez, I.A.; DeAlmeida, D.; Islam, K.; Steele, C.; Montelaro, R.C.; Mietzner, T.A. De novo-derived cationic antimicrobial peptide activity in a murine model of pseudomonas aeruginosa bacteraemia. J. Antimicrob. Chemother. 2007, 60, 669–672. [Google Scholar] [CrossRef]
- Mwangi, J.; Yin, Y.; Wang, G.; Yang, M.; Li, Y.; Zhang, Z.; Lai, R. The antimicrobial peptide zy4 combats multidrug-resistant pseudomonas aeruginosa and acinetobacter baumannii infection. Proc. Natl. Acad. Sci. USA 2019, 116, 26516–26522. [Google Scholar] [CrossRef] [Green Version]
- de Breij, A.; Riool, M.; Cordfunke, R.A.; Malanovic, N.; de Boer, L.; Koning, R.I.; Ravensbergen, E.; Franken, M.; van der Heijde, T.; Boekema, B.K.; et al. The antimicrobial peptide saap-148 combats drug-resistant bacteria and biofilms. Sci. Transl. Med. 2018, 10, eaan4044. [Google Scholar] [CrossRef] [Green Version]
- Bals, R.; Wang, X.; Zasloff, M.; Wilson, J.M. The peptide antibiotic ll-37/hcap-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc. Natl. Acad. Sci. USA 1998, 95, 9541–9546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, D.S.; Tomassini, N.; Zasloff, M. Expression of magainin antimicrobial peptide genes in the developing granular glands of xenopus skin and induction by thyroid hormone. Dev. Biol. 1994, 162, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Phadke, S.M.; Lazarevic, V.; Bahr, C.C.; Islam, K.; Stolz, D.B.; Watkins, S.; Tencza, S.B.; Vogel, H.J.; Montelaro, R.C.; Mietzner, T.A. Lentivirus lytic peptide 1 perturbs both outer and inner membranes of serratia marcescens. Antimicrob. Agents Chemother. 2002, 46, 2041–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phadke, S.M.; Islam, K.; Deslouches, B.; Kapoor, S.A.; Beer Stolz, D.; Watkins, S.C.; Montelaro, R.C.; Pilewski, J.M.; Mietzner, T.A. Selective toxicity of engineered lentivirus lytic peptides in a cf airway cell model. Peptides 2003, 24, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Shi, C.; Kalia, V.; Tencza, S.B.; Montelaro, R.C.; Gupta, P. Hiv gp120 v(1)/v(2) and c(2)-v(3) domains glycoprotein compatibility is required for viral replication. Virus Res. 2001, 79, 91–101. [Google Scholar] [CrossRef]
- Tencza, S.B.; Creighton, D.J.; Yuan, T.; Vogel, H.J.; Montelaro, R.C.; Mietzner, T.A. Lentivirus-derived antimicrobial peptides: Increased potency by sequence engineering and dimerization. J. Antimicrob. Chemother. 1999, 44, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Javadpour, M.M.; Juban, M.M.; Lo, W.C.; Bishop, S.M.; Alberty, J.B.; Cowell, S.M.; Becker, C.L.; McLaughlin, M.L. De novo antimicrobial peptides with low mammalian cell toxicity. J. Med. Chem. 1996, 39, 3107–3113. [Google Scholar] [CrossRef]
- Tew, G.N.; Liu, D.; Chen, B.; Doerksen, R.J.; Kaplan, J.; Carroll, P.J.; Klein, M.L.; DeGrado, W.F. De novo design of biomimetic antimicrobial polymers. Proc. Natl. Acad. Sci. USA 2002, 99, 5110–5114. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; DeGrado, W.F. De novo design, synthesis, and characterization of antimicrobial beta-peptides. J. Am. Chem. Soc. 2001, 123, 7553–7559. [Google Scholar] [CrossRef]
- Ahmad, S.; Gilmore, R.C.; Alexis, N.E.; Tarran, R. Splunc1 loses its antimicrobial activity in acidic cystic fibrosis airway secretions. Am. J. Respir. Crit. Care Med. 2019, 200, 633–636. [Google Scholar] [CrossRef]
- Goldman, M.J.; Anderson, G.M.; Stolzenberg, E.D.; Kari, U.P.; Zasloff, M.; Wilson, J.M. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 1997, 88, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Waghu, F.H.; Idicula-Thomas, S. Collection of antimicrobial peptides database and its derivatives: Applications and beyond. Protein Sci. 2020, 29, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. The antimicrobial peptide database provides a platform for decoding the design principles of naturally occurring antimicrobial peptides. Protein Sci. 2020, 29, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, R.; Tong, A.; Li, X.; Gao, X.; Mei, L.; Zhou, L.; Zhang, X.; You, C.; Guo, G. Enhanced antitumor effects by docetaxel/ll37-loaded thermosensitive hydrogel nanoparticles in peritoneal carcinomatosis of colorectal cancer. Int. J. Nanomed. 2015, 10, 7291–7305. [Google Scholar]
- Steinstraesser, L.; Ring, A.; Bals, R.; Steinau, H.U.; Langer, S. The human host defense peptide ll37/hcap accelerates angiogenesis in pegt/pbt biopolymers. Ann. Plast. Surg. 2006, 56, 93–98. [Google Scholar] [CrossRef]
- Kittaka, M.; Shiba, H.; Kajiya, M.; Fujita, T.; Iwata, T.; Rathvisal, K.; Ouhara, K.; Takeda, K.; Fujita, T.; Komatsuzawa, H.; et al. The antimicrobial peptide ll37 promotes bone regeneration in a rat calvarial bone defect. Peptides 2013, 46, 136–142. [Google Scholar] [CrossRef]
- Ramos, R.; Silva, J.P.; Rodrigues, A.C.; Costa, R.; Guardao, L.; Schmitt, F.; Soares, R.; Vilanova, M.; Domingues, L.; Gama, M. Wound healing activity of the human antimicrobial peptide ll37. Peptides 2011, 32, 1469–1476. [Google Scholar] [CrossRef] [Green Version]
- Kajiya, M.; Shiba, H.; Komatsuzawa, H.; Ouhara, K.; Fujita, T.; Takeda, K.; Uchida, Y.; Mizuno, N.; Kawaguchi, H.; Kurihara, H. The antimicrobial peptide ll37 induces the migration of human pulp cells: A possible adjunct for regenerative endodontics. J. Endod. 2010, 36, 1009–1013. [Google Scholar] [CrossRef]
- Chang, H.T.; Tsai, P.W.; Huang, H.H.; Liu, Y.S.; Chien, T.S.; Lan, C.Y. Ll37 and hbd-3 elevate the beta-1,3-exoglucanase activity of candida albicans xog1p, resulting in reduced fungal adhesion to plastic. Biochem. J. 2012, 441, 963–970. [Google Scholar] [CrossRef]
- Casanova, V.; Sousa, F.H.; Shakamuri, P.; Svoboda, P.; Buch, C.; D’Acremont, M.; Christophorou, M.A.; Pohl, J.; Stevens, C.; Barlow, P.G. Citrullination alters the antiviral and immunomodulatory activities of the human cathelicidin ll-37 during rhinovirus infection. Front. Immunol. 2020, 11, 85. [Google Scholar] [CrossRef]
- Gunn, J.S. Bacterial modification of lps and resistance to antimicrobial peptides. J. Endotoxin Res. 2001, 7, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Collins, L.V. Staphylococcal resistance to antimicrobial peptides of mammalian and bacterial origin. Peptides 2001, 22, 1651–1659. [Google Scholar] [CrossRef]
- Abdi, M.; Mirkalantari, S.; Amirmozafari, N. Bacterial resistance to antimicrobial peptides. J. Pept. Sci. 2019, 25, e3210. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.M.; Holden, B.S.; Coburn, J.; Taylor, M.F.; Weber, S.; Hilton, B.; Zaugg, A.L.; McEwan, C.; Carson, R.; Andersen, J.L.; et al. Proteomic analysis of resistance of gram-negative bacteria to chlorhexidine and impacts on susceptibility to colistin, antimicrobial peptides, and ceragenins. Front. Microbiol. 2019, 10, 210. [Google Scholar] [CrossRef] [PubMed]
- Lutsar, I.; Telling, K.; Metsvaht, T. Treatment option for sepsis in children in the era of antibiotic resistance. Expert Rev. Anti Infect. Ther. 2014, 12, 1237–1252. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.R.; Bastug, A.T.; But, A.; Yildiz, S.; Yetkin, M.A.; Kanyilmaz, D.; Akinci, E.; Bodur, H. Clinical and microbiological efficacy and toxicity of colistin in patients infected with multidrug-resistant gram-negative pathogens. J. Infect. Chemother. Off. J. Jpn. Soc. Chemother. 2013, 19, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the eskape pathogens. Expert Rev. Anti Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- Ho, J.Y.; Cira, N.J.; Crooks, J.A.; Baeza, J.; Weibel, D.B. Rapid identification of eskape bacterial strains using an autonomous microfluidic device. PLoS ONE 2012, 7, e41245. [Google Scholar] [CrossRef]
- Skinner, M.C.; Kiselev, A.O.; Isaacs, C.E.; Mietzner, T.A.; Montelaro, R.C.; Lampe, M.F. Evaluation of wlbu2 peptide and 3-o-octyl-sn-glycerol lipid as active ingredients for a topical microbicide formulation targeting chlamydia trachomatis. Antimicrob. Agents Chemother. 2010, 54, 627–636. [Google Scholar] [CrossRef] [Green Version]
- Flamm, R.K.; Rhomberg, P.R.; Farrell, D.J.; Jones, R.N. In vitro spectrum of pexiganan activity; bactericidal action and resistance selection tested against pathogens with elevated mic values to topical agents. Diagn. Microbiol. Infect. Dis. 2016, 86, 66–69. [Google Scholar] [CrossRef]
- Kulkarni, M.M.; Karafova, A.; Kamysz, W.; McGwire, B.S. Design of protease-resistant pexiganan enhances antileishmanial activity. Parasitol. Res. 2014, 113, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Usan Council. List no. 419. New names. Pexiganan acetate. Clin. Pharmacol. Ther. 1999, 66, 330. [Google Scholar]
- Lamb, H.M.; Wiseman, L.R. Pexiganan acetate. Drugs 1998, 56, 1047–1052; discussion 1053–1044. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, J.; Falciani, C.; Roscia, G.; Pollini, S.; Bindi, S.; Scali, S.; Arrieta, U.C.; Gomez-Vallejo, V.; Quercini, L.; Ibba, E.; et al. In vitro and in vivo efficacy, toxicity, bio-distribution and resistance selection of a novel antibacterial drug candidate. Sci. Rep. 2016, 6, 26077. [Google Scholar] [CrossRef]
- Shah, N.B.; Hersh, B.L.; Kreger, A.; Sayeed, A.; Bullock, A.G.; Rothenberger, S.D.; Klatt, B.; Hamlin, B.; Urish, K.L. Benefits and adverse events associated with extended antibiotic use in total knee arthroplasty periprosthetic joint infection. Clin. Infect. Dis. 2020, 70, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Mandell, J.B.; Orr, S.; Koch, J.; Nourie, B.; Ma, D.; Bonar, D.D.; Shah, N.; Urish, K.L. Large variations in clinical antibiotic activity against staphylococcus aureus biofilms of periprosthetic joint infection isolates. J. Orthop. Res. 2019, 37, 1604–1609. [Google Scholar] [CrossRef]
- Ma, D.; Shanks, R.M.Q.; Davis, C.M., 3rd; Craft, D.W.; Wood, T.K.; Hamlin, B.R.; Urish, K.L. Viable bacteria persist on antibiotic spacers following two-stage revision for periprosthetic joint infection. J. Orthop. Res. 2018, 36, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Hersh, B.L.; Shah, N.B.; Rothenberger, S.D.; Zlotnicki, J.P.; Klatt, B.A.; Urish, K.L. Do culture negative periprosthetic joint infections remain culture negative? J. Arthroplast. 2019, 34, 2757–2762. [Google Scholar] [CrossRef]
- Urish, K.L.; Bullock, A.G.; Kreger, A.M.; Shah, N.B.; Jeong, K.; Rothenberger, S.D.; Infected Implant, C. A multicenter study of irrigation and debridement in total knee arthroplasty periprosthetic joint infection: Treatment failure is high. J. Arthroplast. 2018, 33, 1154–1159. [Google Scholar] [CrossRef]
- Urish, K.L.; DeMuth, P.W.; Kwan, B.W.; Craft, D.W.; Ma, D.; Haider, H.; Tuan, R.S.; Wood, T.K.; Davis, C.M., 3rd. Antibiotic-tolerant staphylococcus aureus biofilm persists on arthroplasty materials. Clin. Orthop. Relat. Res. 2016, 474, 1649–1656. [Google Scholar] [CrossRef]
- Greber, K.E.; Dawgul, M. Antimicrobial peptides under clinical trials. Curr. Top. Med. Chem. 2017, 17, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Migon, D.; Neubauer, D.; Kamysz, W. Hydrocarbon stapled antimicrobial peptides. Protein J. 2018, 37, 2–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luong, H.X.; Kim, D.H.; Lee, B.J.; Kim, Y.W. Antimicrobial activity and stability of stapled helices of polybia-mp1. Arch. Pharm. Res. 2017, 40, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- Caroline Rufo, P. Carb-x Award Worth Up to $12.17 m USD to Develop Peptilogics’ Lead Compound in Prosthetic Joint Infection and Accelerate Development of Additional Novel Antibiotics. Available online: https://www.peptilogics.com/single-post/2020/02/24/Peptilogics-Announces-CARB-X-Funding-to-Accelerate-Development-of-First-in-Class-Antibiotics-to-Address-the-Global-Threat-of-Multi-drug-Resistant-Bacteria (accessed on 25 February 2020).
Figure 1.
The human antimicrobial peptide (AMP) LL37. Composed of 14 different amino acids arranged to form a partial amphipathic helix (helical wheel) of 37 residues, LL37 (left) has evolved to display multiple functions (right); µH, hydrophobic moment, a measure of amphipathicity.
Figure 1.
The human antimicrobial peptide (AMP) LL37. Composed of 14 different amino acids arranged to form a partial amphipathic helix (helical wheel) of 37 residues, LL37 (left) has evolved to display multiple functions (right); µH, hydrophobic moment, a measure of amphipathicity.

Figure 2.
Structures of lentivirus lytic peptides (LLPs). The intracellular domains of the gp41 protein were identified as potential antimicrobial peptides because of their characteristic cationic amphipathic motif, with the helical wheel of (A) the more potent antibacterial LLP-1 (charge of +5) and (B) that of LLP-2 (charge of +3). These LLP domains set the foundation for the engineered AMPs, which we termed engineered cationic antimicrobial peptides (eCAPs) (Figure 3). The number on the left and the number on the right of each sequence indicate the beginning and the end of the primary sequence, respectively, in the protein gp41; µH, hydrophobic moment; H, hydrophobicity derived from the online software heliquest.fr.
Figure 2.
Structures of lentivirus lytic peptides (LLPs). The intracellular domains of the gp41 protein were identified as potential antimicrobial peptides because of their characteristic cationic amphipathic motif, with the helical wheel of (A) the more potent antibacterial LLP-1 (charge of +5) and (B) that of LLP-2 (charge of +3). These LLP domains set the foundation for the engineered AMPs, which we termed engineered cationic antimicrobial peptides (eCAPs) (Figure 3). The number on the left and the number on the right of each sequence indicate the beginning and the end of the primary sequence, respectively, in the protein gp41; µH, hydrophobic moment; H, hydrophobicity derived from the online software heliquest.fr.

Figure 3.
Progression of eCAP structures to overcome limitations of natural AMPs. The helical wheel of LL37 shows remarkable diversity in amino acid composition compared to eCAP LBU2 (A), which displays similar activity to LL37 (B). Sequence optimization of eCAPs (C) using Trp (W) resulted in resistance to saline (D) when the peptides were tested against methicillin-resistant S. aureus (MRSA, B and D). This figure is a summarized adaptation from previously published work [55,71,75,77].
Figure 3.
Progression of eCAP structures to overcome limitations of natural AMPs. The helical wheel of LL37 shows remarkable diversity in amino acid composition compared to eCAP LBU2 (A), which displays similar activity to LL37 (B). Sequence optimization of eCAPs (C) using Trp (W) resulted in resistance to saline (D) when the peptides were tested against methicillin-resistant S. aureus (MRSA, B and D). This figure is a summarized adaptation from previously published work [55,71,75,77].

Figure 4.
Comparison of the design of eCAP WLBU2 to that of recent AMPs with high potency. Both SAAP-148 and ZY4 are based on the modification of a cathelicidin with incorporation of Trp (W) and reduction in the number of amino acid residues to form a more ideal cationic amphipathic structure. ZY4 has a disulfide bridge indicated by a broken line in red.
Figure 4.
Comparison of the design of eCAP WLBU2 to that of recent AMPs with high potency. Both SAAP-148 and ZY4 are based on the modification of a cathelicidin with incorporation of Trp (W) and reduction in the number of amino acid residues to form a more ideal cationic amphipathic structure. ZY4 has a disulfide bridge indicated by a broken line in red.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Deslouches, B.; Montelaro, R.C.; Urish, K.L.; Di, Y.P. Engineered Cationic Antimicrobial Peptides (eCAPs) to Combat Multidrug-Resistant Bacteria. Pharmaceutics 2020, 12, 501. https://doi.org/10.3390/pharmaceutics12060501
AMA Style
Deslouches B, Montelaro RC, Urish KL, Di YP. Engineered Cationic Antimicrobial Peptides (eCAPs) to Combat Multidrug-Resistant Bacteria. Pharmaceutics. 2020; 12(6):501. https://doi.org/10.3390/pharmaceutics12060501
Chicago/Turabian StyleDeslouches, Berthony, Ronald C. Montelaro, Ken L. Urish, and Yuanpu P. Di. 2020. "Engineered Cationic Antimicrobial Peptides (eCAPs) to Combat Multidrug-Resistant Bacteria" Pharmaceutics 12, no. 6: 501. https://doi.org/10.3390/pharmaceutics12060501
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.