Pan-Proteomic Analysis and Elucidation of Protein Abundance among the Closely Related Brucella Species, Brucella abortus and Brucella melitensis

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Brucella Culture
2.2. Whole-Cell Protein Extraction
2.3. In Solution Trypsin Digestion
2.4. Liquid Chromatography–Electrospray Ionization–Tandem Mass Spectrometry (LC–ESI–MS/MS)
2.5. Protein Identification
2.6. Data Analysis
2.7. Functional Categorization and Pathways Analysis
2.8. Screening for Virulence-Associated Proteins
2.9. Mass Spectrometry Data
3. Results and Discussion
3.1. Brucella Whole-Cell Protein Extraction
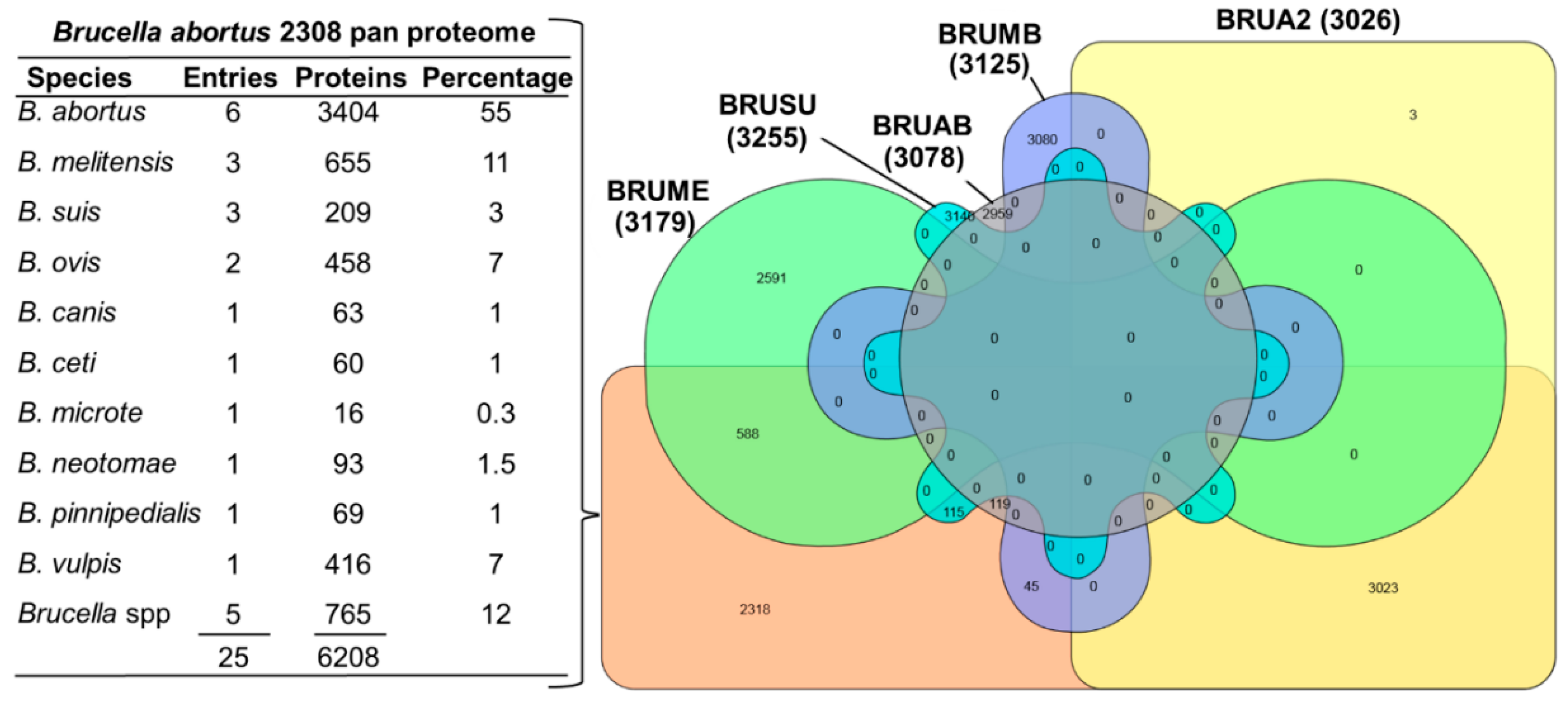
3.2. Databases and Protein Sequences
3.3. Protein Identification
3.4. Comparative Proteomics of B. abortus and B. melitensis
- Category I: B. melitensis 16M vs. B. abortus 544 (M vs. A);
- Category II: B. melitensis C vs. B. abortus T (M2 vs. A2);
- Category III: B. melitensis C vs. B. abortus 544 (M2 vs. A);
- Category IV: B. melitensis 16M vs. B. abortus T (M vs. A2);
- Category V: B. abortus T vs. B. abortus 544 (A2 vs. A);
- Category VI: B. melitensis C vs. B. melitensis 16M (M2 vs. M).
3.5. Geno Ontology and Clusters of Orthologous Groups
3.6. Bioinformatics Annotation of Differentially Expressed Proteins
3.7. Predicted Virulence-Associated Proteins
3.8. Field Strains and Host Adaptability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moreno, E.; Stackebrandt, E.; Dorsch, M.; Wolters, J.; Busch, M.; Mayer, H. Brucella abortus 16S rRNA and lipid A reveal a phylogenetic relationship with members of the alpha-2 subdivision of the class Proteobacteria. J. Bacteriol. 1990, 172, 3569–3576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, P.G.; Macedo, G.C.; Azevedo, V.; Oliveira, S.C. Brucella spp noncanonical LPS: Structure, biosynthesis, and interaction with host immune system. Microb. Cell Fact. 2006, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christopher, S.; Umapathy, B.L.; Ravikumar, K.L. Brucellosis: Review on the recent trends in pathogenicity and laboratory diagnosis. J. Lab. Physicians 2010, 2, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Whatmore, A.M. Current understanding of the genetic diversity of Brucella, an expanding genus of zoonotic pathogens. Infect. Genet. Evol. 2009, 9, 1168–1184. [Google Scholar] [CrossRef] [PubMed]
- Verger, J.M.; Grimont, F.; Grimont, P.A.D.; Grayon, M. Brucella, a Monospecific Genus as Shown by Deoxyribonucleic-Acid Hybridization. Int. J. Syst. Bacteriol. 1985, 35, 292–295. [Google Scholar] [CrossRef]
- Brenner, D.; Staley, J.; Krieg, N. Classification of Procaryotic Organisms and the Concept of Bacterial Speciation. In Bergey’s Manual® of Systematic Bacteriology; Brenner, D., Krieg, N., Staley, J., Garrity, G., Eds.; Springer US: New York, NY, USA, 2005; pp. 27–32. [Google Scholar]
- Chain, P.S.; Comerci, D.J.; Tolmasky, M.E.; Larimer, F.W.; Malfatti, S.A.; Vergez, L.M.; Aguero, F.; Land, M.L.; Ugalde, R.A.; Garcia, E. Whole-genome analyses of speciation events in pathogenic Brucellae. Infect. Immun. 2005, 73, 8353–8361. [Google Scholar] [CrossRef] [Green Version]
- Halling, S.M.; Peterson-Burch, B.D.; Bricker, B.J.; Zuerner, R.L.; Qing, Z.; Li, L.L.; Kapur, V.; Alt, D.P.; Olsen, S.C. Completion of the genome sequence of Brucella abortus and comparison to the highly similar genomes of Brucella melitensis and Brucella suis. J. Bacteriol. 2005, 187, 2715–2726. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ke, Y.; Wang, Z.; Yuan, X.; Qiu, Y.; Zhen, Q.; Xu, J.; Li, T.; Wang, D.; Huang, L.; et al. Genome sequences of three live attenuated vaccine strains of Brucella species and implications for pathogenesis and differential diagnosis. J. Bacteriol. 2012, 194, 6012–6013. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.K.; Kumar, A.; Tiwari, A.K.; Sankarasubramanian, J.; Vishnu, U.S.; Sridhar, J.; Gunasekaran, P.; Rajendhran, J. Draft Genome Sequence of Brucella abortus Virulent Strain 544. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- DelVecchio, V.G.; Kapatral, V.; Redkar, R.J.; Patra, G.; Mujer, C.; Los, T.; Ivanova, N.; Anderson, I.; Bhattacharyya, A.; Lykidis, A.; et al. The genome sequence of the facultative intracellular pathogen Brucella melitensis. Proc. Natl. Acad. Sci. USA 2002, 99, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Rajashekara, G.; Glasner, J.D.; Glover, D.A.; Splitter, G.A. Comparative whole-genome hybridization reveals genomic islands in Brucella species. J. Bacteriol. 2004, 186, 5040–5051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloeckaert, A.; Verger, J.M.; Grayon, M.; Paquet, J.Y.; Garin-Bastuji, B.; Foster, G.; Godfroid, J. Classification of Brucella spp. isolated from marine mammals by DNA polymorphism at the omp2 locus. Microbes. Infect. Inst. Pasteur 2001, 3, 729–738. [Google Scholar] [CrossRef]
- Cloeckaert, A.; Vizcaino, N.; Paquet, J.Y.; Bowden, R.A.; Elzer, P.H. Major outer membrane proteins of Brucella spp.: Past, present and future. Vet. Microbiol. 2002, 90, 229–247. [Google Scholar] [CrossRef]
- Adone, R.; Muscillo, M.; La Rosa, G.; Francia, M.; Tarantino, M. Antigenic, immunologic and genetic characterization of rough strains B. abortus RB51, B. melitensis B115 and B. melitensis B18. PLoS ONE 2011, 6, e24073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Wang, L.; Yin, J.; Wang, X.; Cheng, S.; Lang, X.; Wang, X.; Qu, H.; Sun, C.; Wang, J.; et al. Immunoproteomic analysis of Brucella melitensis and identification of a new immunogenic candidate protein for the development of brucellosis subunit vaccine. Mol. Immunol. 2011, 49, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Al Dahouk, S.; Nöckler, K.; Scholz, H.C.; Tomaso, H.; Bogumil, R.; Neubauer, H. Immunoproteomic characterization of Brucella abortus 1119-3 preparations used for the serodiagnosis of Brucella infections. J. Immunol. Methods 2006, 309, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Tabynov, K.; Ryskeldinova, S.; Sansyzbay, A. An influenza viral vector Brucella abortus vaccine induces good cross-protection against Brucella melitensis infection in pregnant heifers. Vaccine 2015, 33, 3619–3623. [Google Scholar] [CrossRef]
- Dorneles, E.M.; Sriranganathan, N.; Lage, A.P. Recent advances in Brucella abortus vaccines. Vet. Res. 2015, 46, 76. [Google Scholar] [CrossRef] [Green Version]
- Al Dahouk, S.; Tomaso, H.; Nockler, K.; Neubauer, H.; Frangoulidis, D. Laboratory-based diagnosis of brucellosis—A review of the literature. Part II: Serological tests for brucellosis. Clin. Lab. 2003, 49, 577–589. [Google Scholar]
- Al Dahouk, S.; Scholz, H.C.; Tomaso, H.; Bahn, P.; Gollner, C.; Karges, W.; Appel, B.; Hensel, A.; Neubauer, H.; Nockler, K. Differential phenotyping of Brucella species using a newly developed semi-automated metabolic system. BMC Microbiol. 2010, 10, 269. [Google Scholar] [CrossRef] [Green Version]
- Alton, G.G.; Forsyth, J.R.L. Brucella. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Wareth, G.; Eravci, M.; Weise, C.; Roesler, U.; Melzer, F.; Sprague, L.D.; Neubauer, H.; Murugaiyan, J. Comprehensive identification of immunodominant proteins of Brucella abortus and Brucella melitensis using antibodies in the sera from naturally infected hosts. Int. J. Mol. Sci. 2016, 17, 659. [Google Scholar] [CrossRef] [Green Version]
- Wareth, G.; Melzer, F.; Weise, C.; Neubauer, H.; Roesler, U.; Murugaiyan, J. Proteomics-based identification of immunodominant proteins of Brucellae using sera from infected hosts points towards enhanced pathogen survival during the infection. Biochem. Biophys. Res. Commun. 2015, 456, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Connolly, J.P.; Comerci, D.; Alefantis, T.G.; Walz, A.; Quan, M.; Chafin, R.; Grewal, P.; Mujer, C.V.; Ugalde, R.A.; DelVecchio, V.G. Proteomic analysis of Brucella abortus cell envelope and identification of immunogenic candidate proteins for vaccine development. Proteomics 2006, 6, 3767–3780. [Google Scholar] [CrossRef] [PubMed]
- DelVecchio, V.G.; Wagner, M.A.; Eschenbrenner, M.; Horn, T.A.; Kraycer, J.A.; Estock, F.; Elzer, P.; Mujer, C.V. Brucella proteomes—A review. Vet. Microbiol. 2002, 90, 593–603. [Google Scholar] [CrossRef]
- Eschenbrenner, M.; Horn, T.A.; Wagner, M.A.; Mujer, C.V.; Miller-Scandle, T.L.; DelVecchio, V.G. Comparative proteome analysis of laboratory grown Brucella abortus 2308 and Brucella melitensis 16M. J. Proteome Res. 2006, 5, 1731–1740. [Google Scholar] [CrossRef]
- Eschenbrenner, M.; Wagner, M.A.; Horn, T.A.; Kraycer, J.A.; Mujer, C.V.; Hagius, S.; Elzer, P.; DelVecchio, V.G. Comparative proteome analysis of Brucella melitensis vaccine strain Rev 1 and a virulent strain, 16M. J. Bacteriol. 2002, 184, 4962–4970. [Google Scholar] [CrossRef] [Green Version]
- Lamontagne, J.; Beland, M.; Forest, A.; Cote-Martin, A.; Nassif, N.; Tomaki, F.; Moriyon, I.; Moreno, E.; Paramithiotis, E. Proteomics-based confirmation of protein expression and correction of annotation errors in the Brucella abortus genome. BMC Genom. 2010, 11, 300. [Google Scholar] [CrossRef] [Green Version]
- Mujer, C.V.; Wagner, M.A.; Eschenbrenner, M.; Horn, T.; Kraycer, J.A.; Redkar, R.; Hagius, S.; Elzer, P.; Delvecchio, V.G. Global analysis of Brucella melitensis proteomes. Ann. N. Y. Acad. Sci. 2002, 969, 97–101. [Google Scholar] [CrossRef]
- Paredes-Cervantes, V.; Flores-Mejia, R.; Moreno-Lafont, M.C.; Lanz-Mendoza, H.; Tello-Lopez, A.T.; Castillo-Vera, J.; Pando-Robles, V.; Hurtado-Sil, G.; Gonzalez-Gonzalez, E.; Rodriguez-Cortes, O.; et al. Comparative proteome analysis of Brucella abortus 2308 and its virB type IV secretion system mutant reveals new T4SS-related candidate proteins. J. Proteom. 2011, 74, 2959–2971. [Google Scholar] [CrossRef]
- Wagner, M.A.; Eschenbrenner, M.; Horn, T.A.; Kraycer, J.A.; Mujer, C.V.; Hagius, S.; Elzer, P.; DelVecchio, V.G. Global analysis of the Brucella melitensis proteome: Identification of proteins expressed in laboratory-grown culture. Proteomics 2002, 2, 1047–1060. [Google Scholar] [CrossRef]
- Zai, X.; Yang, Q.; Yin, Y.; Li, R.; Qian, M.; Zhao, T.; Li, Y.; Zhang, J.; Fu, L.; Xu, J.; et al. Relative Quantitative Proteomic Analysis of Brucella abortus Reveals Metabolic Adaptation to Multiple Environmental Stresses. Front. Microbiol. 2017, 8, 2347. [Google Scholar] [CrossRef] [PubMed]
- Zai, X.; Yang, Q.; Liu, K.; Li, R.; Qian, M.; Zhao, T.; Li, Y.; Yin, Y.; Dong, D.; Fu, L.; et al. A comprehensive proteogenomic study of the human Brucella vaccine strain 104 M. BMC Genom. 2017, 18, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Candiano, G.; Bruschi, M.; Musante, L.; Santucci, L.; Ghiggeri, G.M.; Carnemolla, B.; Orecchia, P.; Zardi, L.; Righetti, P.G. Blue silver: A very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 2004, 25, 1327–1333. [Google Scholar] [CrossRef]
- Murugaiyan, J.; Eravci, M.; Weise, C.; Roesler, U. Label-free quantitative proteomic analysis of harmless and pathogenic strains ofinfectious microalgae, Prototheca spp. Int. J. Mol. Sci. 2016, 18, 59. [Google Scholar] [CrossRef]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Matic, I.; Hilger, M.; Nagaraj, N.; Selbach, M.; Olsen, J.V.; Mann, M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 2009, 4, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Luber, C.A.; Cox, J.; Lauterbach, H.; Fancke, B.; Selbach, M.; Tschopp, J.; Akira, S.; Wiegand, M.; Hochrein, H.; O’Keeffe, M.; et al. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity 2010, 32, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. R. Stat. Soc. B Met. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, 11033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Gupta, D. VirulentPred: A SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinform. 2008, 9, 62. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Meyer, M.E.; Morgan, W.J. Metabolic characterization of Brucella strains that show conflicting identity by biochemical and serological methods. Bull. World Health Organ. 1962, 26, 823–827. [Google Scholar]
- He, Y. Analyses of Brucella pathogenesis, host immunity, and vaccine targets using systems biology and bioinformatics. Front. Cell. Infect. Microbiol. 2012, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Teixeira-Gomes, A.P.; Cloeckaert, A.; Bezard, G.; Dubray, G.; Zygmunt, M.S. Mapping and identification of Brucella melitensis proteins by two-dimensional electrophoresis and microsequencing. Electrophoresis 1997, 18, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Crasta, O.R.; Folkerts, O.; Fei, Z.; Mane, S.P.; Evans, C.; Martino-Catt, S.; Bricker, B.; Yu, G.; Du, L.; Sobral, B.W. Genome sequence of Brucella abortus vaccine strain S19 compared to virulent strains yields candidate virulence genes. PLoS ONE 2008, 3, e2193. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, J.; Forest, A.; Marazzo, E.; Denis, F.; Butler, H.; Michaud, J.F.; Boucher, L.; Pedro, I.; Villeneuve, A.; Sitnikov, D.; et al. Intracellular adaptation of Brucella abortus. J. Proteome Res. 2009, 8, 1594–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoktouridis, G.; Merz, C.A.; Manning, S.P.; Giovagnoli-Kurtz, R.; Williams, L.E.; Mujer, C.V.; Hagius, S.; Elzer, P.; Redkar, R.J.; Patra, G.; et al. Molecular characterization of Brucella abortus chromosome II recombination. J. Bacteriol. 2003, 185, 6130–6136. [Google Scholar] [CrossRef] [Green Version]
- Teixeira-Gomes, A.P.; Cloeckaert, A.; Zygmunt, M.S. Characterization of heat, oxidative, and acid stress responses in Brucella melitensis. Infect. Immun. 2000, 68, 2954–2961. [Google Scholar] [CrossRef] [Green Version]
- UniProt. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16. [Google Scholar] [CrossRef]
- Silva, T.M.A.; Mol, J.P.S.; Winter, M.G.; Atluri, V.; Xavier, M.N.; Pires, S.F.; Paixao, T.A.; Andrade, H.M.; Santos, R.L.; Tsolis, R.M. The Predicted ABC Transporter AbcEDCBA Is Required for Type IV Secretion System Expression and Lysosomal Evasion by Brucella ovis. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Jenner, D.C.; Dassa, E.; Whatmore, A.M.; Atkins, H.S. ATP-Binding Cassette Systems of Brucella. Comp. Funct. Genom. 2009, 354649. [Google Scholar] [CrossRef] [Green Version]
- Smircich, P.; Eastman, G.; Bispo, S.; Duhagon, M.A.; Guerra-Slompo, E.P.; Garat, B.; Goldenberg, S.; Munroe, D.J.; Dallagiovanna, B.; Holetz, F.; et al. Ribosome profiling reveals translation control as a key mechanism generating differential gene expression in Trypanosoma cruzi. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [Green Version]
- Caswell, C.C.; Elhassanny, A.E.; Planchin, E.E.; Roux, C.M.; Weeks-Gorospe, J.N.; Ficht, T.A.; Dunman, P.M.; Roop, R.M., II. Diverse genetic regulon of the virulence-associated transcriptional regulator MucR in Brucella abortus 2308. Infect. Immun. 2013, 81, 1040–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibor, A.; Wansard, V.; Bielartz, V.; Delrue, R.M.; Danese, I.; Michel, P.; Walravens, K.; Godfroid, J.; Letesson, J.J. Effect of omp10 or omp19 deletion on Brucella abortus outer membrane properties and virulence in mice. Infect. Immun. 2002, 70, 5540–5546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goolab, S.; Roth, R.L.; van Heerden, H.; Crampton, M.C. Analyzing the molecular mechanism of lipoprotein localization in Brucella. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simborio, H.L.T.; Lee, J.J.; Reyes, A.W.B.; Hop, H.T.; Arayan, L.T.; Min, W.; Lee, H.J.; Yoo, H.S.; Kim, S. Evaluation of the combined use of the recombinant Brucella abortus Omp10, Omp19 and Omp28 proteins for the clinical diagnosis of bovine brucellosis. Microb. Pathog. 2015, 83–84, 41–46. [Google Scholar] [CrossRef]
- Gee, J.M.; Valderas, M.W.; Kovach, M.E.; Grippe, V.K.; Robertson, G.T.; Ng, W.L.; Richardson, J.M.; Winkler, M.E.; Roop, R.M. The Brucella abortus Cu,Zn superoxide dismutase is required for optimal resistance to oxidative killing by murine macrophages an wild-type virulence in experimentally infected mice. Infect. Immun. 2005, 73, 2873–2880. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Joo, Y.J.; Kim, J. Asc1p, a ribosomal protein, plays a pivotal role in cellular adhesion and virulence in Candida albicans. J. Microbiol. 2010, 48, 842–848. [Google Scholar] [CrossRef]
- Du, Z.Q.; Li, X.; Wang, J.Y. Immunogenicity analysis of a novel subunit vaccine candidate molecule-recombinant L7/L12 ribosomal protein of Brucella suis. Appl. Biochem. Biotechnol. 2016, 179, 1445–1455. [Google Scholar] [CrossRef]
- Jain, S.; Kumar, S.; Dohre, S.; Afley, P.; Sengupta, N.; Alam, S.I. Identification of a protective protein from stationary-phase exoproteome of Brucella abortus. Pathog. Dis. 2014, 70, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Atluri, V.L.; Xavier, M.N.; de Jong, M.F.; den Hartigh, A.B.; Tsolis, R.M. Interactions of the human pathogenic Brucella Species with their hosts. Annu. Rev. Microbiol. 2011, 65, 523–541. [Google Scholar] [CrossRef]
- Saenz, H.L.; Engel, P.; Stoeckli, M.C.; Lanz, C.; Raddatz, G.; Vayssier-Taussat, M.; Birtles, R.; Schuster, S.C.; Dehio, C. Genomic analysis of Bartonella identifies type IV secretion systems as host adaptability factors. Nat. Genet. 2007, 39, 1469–1476. [Google Scholar] [CrossRef]
- Boschiroli, M.L.; Ouahrani-Bettache, S.; Foulongne, V.; Michaux-Charachon, S.; Bourg, G.; Allardet-Servent, A.; Cazevieille, C.; Lavigne, J.P.; Liautard, J.P.; Ramuz, M.; et al. Type IV secretion and Brucella virulence. Vet. Microbiol. 2002, 90, 341–348. [Google Scholar] [CrossRef]
- Wattam, A.R.; Williams, K.P.; Snyder, E.E.; Almeida, N.F.; Shukla, M.; Dickerman, A.W.; Crasta, O.R.; Kenyon, R.; Lu, J.; Shallom, J.M.; et al. Analysis of ten Brucella genomes reveals evidence for horizontal gene transfer despite a preferred intracellular lifestyle. J. Bacteriol. 2009, 191, 3569–3579. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Designation/ID/Number | Host | Geographical Region | ||
|---|---|---|---|---|---|
| FAO | ATCC | NCTC | |||
| B. melitensisT | 16M | 23456 | 10094 | Goat | USA |
| B. abortusT | 544 | 23448 | 10093 | Cattle | UK |
| B. melitensis | C * | Sheep | China | ||
| B. abortus | T * | Cattle | Turkey | ||
| Regulation | Category | |||||
|---|---|---|---|---|---|---|
| I M vs. A | II M2 vs. A2 | III M2 vs. A | IV M vs. A2 | V A2 vs. A | VI M2 vs. M | |
| Down- | 173 | 142 | 166 | 223 | 83 | 96 |
| Up- | 216 | 199 | 221 | 248 | 97 | 129 |
| KEGG Pathways | Category | |||||
|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | |
| Metabolic pathways | 34 ↑ | |||||
| Carbon metabolism | 6 ↑ | |||||
| Glycolysis/gluconeogenesis | 7 ↓ | |||||
| Pentose and glucuronate interconversions | 3 ↓ | |||||
| Pyruvate metabolism | 4 ↑ | |||||
| 2-Oxocarboxylic acid metabolism | 4 ↓ | |||||
| Microbial metabolism in diverse environments | 14 ↑ | 10 ↑ | ||||
| Biosynthesis of amino acids | 11 ↓ | 14 ↓ | ||||
| Histidine metabolism | 5 ↓ | 5 ↓ | 5 ↓ | 6 ↓ | ||
| Valine, leucine and isoleucine biosynthesis | 4 ↓ | |||||
| Purine metabolism | 7 ↑ | |||||
| Pyrimidine metabolism | 5 ↑ | 6 ↑ | 7 ↑ | |||
| Biosynthesis of secondary metabolites | 19 ↓ | 25 ↓ | ||||
| RNA polymerase | 3 ↑ | |||||
| Ribosome | 17 ↑ | 18 ↓ | 14 ↓ | 19 ↑ | 22 ↓ | |
| Bacterial secretion system | 5 ↑ | 5 ↑ | 4 ↑ | 5 ↑ | ||
| ABC transporters | 14 ↑ | |||||
| Acc. | Protein Description | Reg | Significance | Reference |
|---|---|---|---|---|
| D0B8I3* | Antifreeze protein | (+) | Associated with MucR, a transcriptional regulator linked to Brucella virulence | [63] |
| Q8YIA9 | Antifreeze protein | (-) | ||
| Q2YM39 | Antifreeze protein type I | (-) | ||
| D0B248 * | LipA family protein | (-) | ||
| Q2YIP8 | Lipoprotein Omp10 | (-) | Reduced virulence in B. abortus with gene deletion and used in diagnostics | [64,65,66] |
| Q2YKV9 | Superoxide dismutase [Cu-Zn] | (-) | intracellular survival and used as antigens for subunit vaccines | [19,67] |
| Q2YRA8 | 30S ribosomal protein S14 | (-) | role in cellular adhesion and virulence in Candida albicans | [68] |
| ribosomal protein L7/L12 based subunit vaccines | [69,70] |
| UniProt ID | Protein |
|---|---|
| Downregulated in Field Isolates | |
| Q8YBH7 | Bacterial extracellular solute-binding protein family 1 |
| Q8YIX9 | Glyceraldehyde-3-phosphate dehydrogenase |
| Q2YMI0 | Phosphatidylserine decarboxylase proenzyme |
| Q2YLU2 | Peptidylprolyl isomerase |
| Q2YLF8 | Leu/Ile/Val-binding protein homolog 1 |
| Q2YLG0 | Leu/Ile/Val-binding protein homolog 2 |
| Q2YJA9 | Leu/Ile/Val-binding protein homolog 5 |
| Q2YQQ6 | Glutelin:Lipoprotein YaeC family:NLPA lipoprotein |
| Q2YRP7 | Ribosome-recycling factor |
| Q2YR20 | Uncharacterized protein |
| Upregulated in Field Isolates | |
| Q2YJ78 | Type IV secretion system protein virB8 |
| Q2YJ79 | Type IV secretion system protein virB9 |
| Q2YJ81 | Type IV secretion system protein virB10 |
| Q2YJ83 | Type IV secretion system-outer membrane lipoprotein |
| Q2YJ77 | Type IV secretion system putative lipoprotein virB7 |
| Q8YB25 | Alpha-methylacyl-CoA racemase |
| Q2YIG7 | NADH:flavin oxidoreductase/NADH oxidase |
| Q2YKS6 | Aminotransferase class IV |
| Q8YCZ2 | N-acetylglucosamine-6-phosphate deacetylase |
| Q8YI04 | ATP-dependent DNA ligase |
| Q2YK32 | Catalase |
| Q2YLX9 | Lipoprotein putative |
| Q2YN45 | Probable cytosol aminopeptidase |
| Q2YRJ0 | ATP/GTP-binding site motif A (P-loop) |
| Q2YRN7 | Uncharacterized protein |
| Q8YBU9 | Putative uroporphyrin-iii c-methyltransferase |
| Q2YKY2 | Uncharacterized protein |
| Q2YL40 | Uncharacterized protein |
| Q2YLM7 | Uncharacterized protein |
| Downregulated in B. abortus and Upregulated in B. melitensis | |
| Q2YME1 | Threonylcarbamoyl-AMP synthase |
| Q2YNW9 | Dihydroxy-acid dehydratase |
| Q8YHA5 | Glutaryl-CoA dehydrogenase |
| Q2YP66 | Zinc-containing alcohol dehydrogenase |
| Q2YQE3 | Periplasmic binding protein |
| Q2YMW1 | Uncharacterized protein |
| Q2YPK6 | Uncharacterized protein |
| Upregulated in B. abortus and Downregulated in B. melitensis | |
| Q2YQV7 | 50S ribosomal protein L20 |
| Q2YR56 | 50S ribosomal protein L28 |
| Q2YRY0 | ABC-type glycine betaine transport system |
| Q8YBF5 | Maltose-binding periplasmic protein (Sugar ABC transporter) |
| Q8YCD1 | Cystine-binding periplasmic protein |
| Q8YGE8 | Cationic amino acid ABC transporter |
| Q2YKN7 | Uncharacterized protein |
| Q2YKN9 | Uncharacterized protein |
| Q2YQM2 | Uncharacterized protein |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murugaiyan, J.; Eravci, M.; Weise, C.; Roesler, U.; Sprague, L.D.; Neubauer, H.; Wareth, G. Pan-Proteomic Analysis and Elucidation of Protein Abundance among the Closely Related Brucella Species, Brucella abortus and Brucella melitensis. Biomolecules 2020, 10, 836. https://doi.org/10.3390/biom10060836
Murugaiyan J, Eravci M, Weise C, Roesler U, Sprague LD, Neubauer H, Wareth G. Pan-Proteomic Analysis and Elucidation of Protein Abundance among the Closely Related Brucella Species, Brucella abortus and Brucella melitensis. Biomolecules. 2020; 10(6):836. https://doi.org/10.3390/biom10060836
Chicago/Turabian StyleMurugaiyan, Jayaseelan, Murat Eravci, Christoph Weise, Uwe Roesler, Lisa D. Sprague, Heinrich Neubauer, and Gamal Wareth. 2020. "Pan-Proteomic Analysis and Elucidation of Protein Abundance among the Closely Related Brucella Species, Brucella abortus and Brucella melitensis" Biomolecules 10, no. 6: 836. https://doi.org/10.3390/biom10060836