Key Challenges in Designing CHO Chassis Platforms
by
 , , , , and
, , , , and
Anis Hamdi
1,2,† ,
,
Diana Széliová
2,3,†,
David E. Ruckerbauer
2,3,
Isabel Rocha
1,4,
Nicole Borth
3 and
Jürgen Zanghellini
2,5,* 1
Centro de Engenharia Biológica, Universidade do Minho, 4710-057 Braga, Portugal
2
Acib GmbH– Austrian Centre of Industrial Biotechnology, 1190 Vienna, Austria
3
Department of Biotechnology, BOKU University of Natural Resources and Life Sciences, 1190 Vienna, Austria
4
ITQB-NOVA, Instituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, Av. da República, 2780-157 Oeiras, Portugal
5
Department of Analytical Chemistry, University of Vienna, 1090 Vienna, Austria
*
Author to whom correspondence should be addressed.
†
Both authors contributed equally.
Processes 2020, 8(6), 643; https://doi.org/10.3390/pr8060643
Submission received: 31 March 2020
/
Revised: 22 May 2020
/
Accepted: 25 May 2020
/
Published: 28 May 2020
(This article belongs to the Collection Principles of Modular Design and Control in Complex Systems)
Abstract
:Following the success of and the high demand for recombinant protein-based therapeutics during the last 25 years, the pharmaceutical industry has invested significantly in the development of novel treatments based on biologics. Mammalian cells are the major production systems for these complex biopharmaceuticals, with Chinese hamster ovary (CHO) cell lines as the most important players. Over the years, various engineering strategies and modeling approaches have been used to improve microbial production platforms, such as bacteria and yeasts, as well as to create pre-optimized chassis host strains. However, the complexity of mammalian cells curtailed the optimization of these host cells by metabolic engineering. Most of the improvements of titer and productivity were achieved by media optimization and large-scale screening of producer clones. The advances made in recent years now open the door to again consider the potential application of systems biology approaches and metabolic engineering also to CHO. The availability of a reference genome sequence, genome-scale metabolic models and the growing number of various “omics” datasets can help overcome the complexity of CHO cells and support design strategies to boost their production performance. Modular design approaches applied to engineer industrially relevant cell lines have evolved to reduce the time and effort needed for the generation of new producer cells and to allow the achievement of desired product titers and quality. Nevertheless, important steps to enable the design of a chassis platform similar to those in use in the microbial world are still missing. In this review, we highlight the importance of mammalian cellular platforms for the production of biopharmaceuticals and compare them to microbial platforms, with an emphasis on describing novel approaches and discussing still open questions that need to be resolved to reach the objective of designing enhanced modular chassis CHO cell lines.
1. Introduction
Following the approval of the first biopharmaceutical products by regulatory authorities in the early 1980s [1], innovation in the biopharmaceutical field has triggered the generation of many novel compounds that demonstrated great therapeutic potential towards the treatment of existing and emerging diseases [2]. Currently, 316 biopharmaceuticals are on the market, with a market value of $US 188 billion in total sales in 2017 [1]. This is projected to double by 2021 [3]. In parallel, the value of the global bioprocess technology market is expected to reach up to $US 71 billion by 2022 [4]. The biopharmaceutical market is currently dominated by monoclonal antibodies (mAbs), which generate two thirds of its global revenue [1] even though they make up only 25% of all approved biopharmaceuticals [3]. To illustrate, in 2018, eight of the top ten selling biopharmaceuticals were mAbs, and Humira represents the overall best-selling mAb-based pharmaceutical from 2013 to 2018 [1,5,6].
Biopharmaceuticals are mainly produced using heterologous expression in recombinant cell lines. The classic production host is Escherichia coli, which produced the first biopharmaceutical, insulin, already in the late 1970s [7]. Due to its rapid growth in cheap and simple media, its growth to high cell densities, and the ease with which it can be genetically manipulated, E. coli remains a prime production host for the biopharmaceutical and biotechnological industries [8,9,10,11]. For proteins that require the formation of disulfide bonds, including insulin as the best known example, several yeast species have been used and by now have well established platform technologies available [12]. However, for the production of high value biotherapeutics that require human-like glycosylation or other complex post-translational modifications (PTMs), higher host organisms, such as mammalian cells, are required [13,14].
Correct glycosylation is of key importance as it affects the efficacy of the therapeutic as well as its in vivo turnover rate and prevents immune responses which would be triggered by non-human glycans [15]. While all mammalian cells are natively able to generate proteins with species specific glycosylation patterns, there are some variants which may give rise to immunogenic reactions. This has been observed, for instance, in therapeutics produced in cell lines of mouse origin [16]. The predominant host used in today’s industry are Chinese hamster ovary (CHO) cells [13,14,17,18], which have the advantage of low susceptibility to viral infection and a species specific glycosylation pattern that differs only minimally from human glycosylation [19].
Due to their comparatively simple handling and their ability to grow in chemically defined media in suspension culture, CHO cell lines have proven extremely useful both for research and industrial applications [15]. However, despite several decades of research and process design, the productivity remains low compared to the theoretical maximum productivity predicted by a genome-scale metabolic model of CHO [20]. Furthermore, cell line development still heavily relies on laborious and time-consuming “trial and error” methods, such as random gene integration and amplification by a selection drug, which requires an extensive screening of thousands of clones. Even in well established and partially automated industrial platforms this takes at least 4 months [21]. Thus, along with the increasing demand for biopharmaceutical products, there is a growing need to optimize CHO’s production yield and to fast track the development of new production cell lines.
Over the last years, several new and powerful tools have been developed, including synthetic biology, systems biology and metabolic engineering. These tools have been used to optimize the production of several bio-based products, mainly focusing on strain optimization. The use of these tools is facilitated by the availability of the genetic information [22] as well as a genome-scale metabolic model of the target strain. While many of these tools are already widely applied in the field of recombinant protein production or strain engineering in microbial research, up to the level of design of chassis strains, their application to mammalian production hosts is still fragmentary and lagging far behind.
In this review, we therefore first address the systematic use and application of these tools for modular design and for the generation of stable chassis strains in microbial platforms. We then compare the size and complexity of microbial hosts such as E. coli and Saccharomyces cerevisiae with that of mammalian cell lines and investigate the current state of the art in mammalian cell engineering. Finally, we discuss the open challenges and required tools that need to be solved and established to transfer these approaches to mammalian systems.
2. Microbial Cell Factories: Current Engineering Paradigms for the Bio-based Production of Chemical Commodities
The sustainable and (bio-based) production of chemical commodities is a key challenge of the 21st century. Currently, only 2% of the global chemicals production are bio-based [23], the majority of this through microbial fermentation. As many microbes do not produce the product of interest naturally or, if they do, not at competitively high productivities and yields, cellular engineering is of uttermost importance.
2.1. Engineering Paradigms
The spectrum of common metabolic engineering strategies includes deletions, insertions, overexpression or downregulation of (multiple) genes. All of these interventions aim to modify the genetic circuits of cells and to engineer them to express phenotypes of interest at high yields. More specifically, this is achieved by reallocating growth resources towards production. Thus, growth-coupled production [24] emerged as the dominant engineering strategy. This approach aims to modify cellular metabolism such that the production of a product of interest is necessarily linked to cell growth [24]. Two “forms” can be distinguished: Strong and weak growth coupling [25]. In the former a non-zero amount of the product is synthesized at any growth rate, while in the latter this is only true for growth rates larger than a given threshold.
Growth-coupled production has been successfully implemented in several organisms, for example E. coli and S. cerevisiae, for the production of different compounds such as lactate, glycerol and succinate [26,27,28]. In fact, computational analysis indicates that (at least in A. niger, C. glutamicum, E. coli, S. cerevisiae and Synechocystis sp. PCC 6803 and under aerobic conditions) almost every (native) cellular metabolite can be produced in a strongly growth-coupled manner [29]. Typically, rationally designed growth-coupled production systems can be (easily) further optimized by relying on adaptive laboratory evolution experiments selecting for maximum growth, thereby improving product formation [30,31]. However, for optimal product yield, high growth is actually detrimental. Thus, continuous production processes at (near-)zero growth have recently gained interest [32]. Clearly, growth coupling is no longer feasible in such regimes. However, even at zero-growth, (maintenance) energy is still required. Thus, if production is linked to energy, adenosine triphosphate (ATP) synthesis rather than growth could drive product formation as has been shown recently [33]. This effect can be boosted if ATP consumption is increased by enforced ATP “wasting” in futile cycles. In fact, reinforcing the turnover of ATP has been previously successfully employed as a metabolic “engine” for the overproduction of key fermentation products [34,35,36,37].
2.2. Model-based Engineering Approaches
The availability of a full genome sequence enables researchers to reconstruct cellular metabolism in silico. This knowledge is captured in genome-scale metabolic models (GSMMs), which are essential for the rational identification of engineering targets [38,39]. It is, however, not obvious how to find a feasible implementation of the aforementioned strategies. Due to the size and complexity of the metabolic model, computational support is necessary in order to narrow down and select promising intervention strategies from the combinatorial universe of possible modifications. Computational strain design methods, many of which are based on constraint based reconstruction and analysis of cellular metabolism [40,41,42,43], are available towards this end [44] and are continuously being refined.
One of the earliest available methods was OptKnock [24]. It utilizes a bi-level optimization framework to find knockouts that maximize both product synthesis rate and growth rate. Since then, several improvements and alternatives evolved, summarised in Table 1 as a brief overview.
For example, in 2014, Campodonico et al. used an integrated approach through heterologous pathway integration combined with RobustKnock [46] and genetic design through local search (GDLS) frameworks [50,64] to predict gene deletion strategies that would achieve growth-coupled production for 20 non-native commodity chemicals (e.g., 1-Butanol, 1-Propanol, etc.). As a highlight, 15 out of 20 cases demonstrated a possible growth-coupling. For the other five cases (e.g., 2-phenylethanol and 4-hydroxybutyrate), it was not possible to determine any adequate interventions [64].
Failing to achieve growth-coupling may indicate a fundamental inability or the over-constraining of design choices [25]. However, growth-coupling strategies often introduce auxotrophies that need to be addressed by appropriate media supplements. For instance, growth-coupled production of itaconic acid in E. coli is infeasible on minimal M9 medium, but possible upon supplementation of methionine and glutamate, among other molecules [65,66]. OptCouple is a recent tool that aims to overcome such shortcomings by predicting more comprehensive intervention strategies consisting of, e.g., gene deletions, knock-ins and medium supplements [61].
2.3. Chassis Strains
In the last decades, numerous studies that aimed at improving product yield and productivity in a custom-tailored fashion emerged [67]. Thus, the question arises if there exists a common chassis cell that could act as host for generating many different cell factories [68,69]. Ideally, chassis cells are essentially pre-optimized microbes of minimal functionality that—in a “plug and play” fashion—can be turned into a full-scale cell factory by inserting any pathway that reliably achieves the desired product yield and productivity [70]. This would allow fast generation of new host cell lines, because the cells would have already been optimised before, and would then be reused, needing only minimal adaptation for the production of new products of interest [71]. As an example, the production of organic acids (e.g., succinic acid and malic acid) [72] and biofuels (e.g., n-butanol) [73] was implemented using potential chassis microbial strains.
A promising strategy for the generation of a chassis strain is the optimization of carbon metabolic flux towards the production of a desired product. This can be achieved by the removal of unnecessary genes and pathways from a genome, thus creating cells with high controllability and production efficiency compared to the original strains [74]. Consequently, a lower number of metabolites is produced in the cells, which helps to reroute the resources towards the synthesis of the industrial product of interest [75,76]. The production of a target compound is also often limited by the availability of specific metabolic precursors. To overcome this challenge, chassis strains can be engineered to overexpress biosynthetic pathways for these metabolites. Improvements in the availability of metabolic precursors such as malonyl-CoA, acetyl-CoA and aromatic amino acids were previously described [77,78,79,80].
2.4. Dynamic Control of Cell Metabolism
Cells are commonly engineered at the gene or pathway levels. Taking strain engineering one step further, metabolism can even be optimized to dynamically respond to the cell’s current state in a way that maximizes the synthesis of a target product. The expression of the product of interest can be timed to start at a certain point during the cultivation, for example when the cell concentration is sufficient [81,82]. The switch could be triggered by the addition of a small molecule or by engineering cells to respond to intracellular or extracellular signals [83]. Such a strategy was successfully applied to E. coli [84,85], where a toggle switch was used to redirect flux from the TCA cycle towards the synthesis of target products after reaching a sufficiently high cell concentration. Further, feedback loops could be put in place. These could activate biosynthetic pathways if precursors become limiting or degradation pathways if toxic byproducts accumulate [86]. If the synthesis of a product requires several steps, the expression of the individual enzymes might be fine-tuned such that no intermediates are accumulated [87]. Thus, by reducing the metabolic burden, additional resources for increasing the production of the product of interest can be freed.
2.5. Requirements for Such Strategies to Work
To apply the aforementioned strategies, it is necessary to better understand cells at different levels, such as genome sequence, RNA and protein expression, interactions between various macromolecules, signalling and metabolic pathways [88].
The field of genomics and transcriptomics witnessed a great advance in maturity as a result of the optimization of next generation sequencing (NGS) technologies, which are now routinely used to sequence whole genomes and to quantify gene expression using RNAseq [89]. Based on these information sets on the potential of an organism, GSMMs can be built in order to predict the best and most efficient interventions for metabolic engineering. Advances in other fields, such as proteomics, metabolomics or fluxomics will help to improve the predictive capabilities of these models. Based on these advances, the generation of accurate omics data and their analysis using meaningful models, helps to solve different aspects of the cellular “black box” and to improve our understanding of the genotype-phenotype interactions in cells.
In order to apply these predictions in vivo it is necessary to have robust tools for gene deletions, insertions and editing. The tools at our disposal have become efficient and more rapid with the discovery of the CRISPR/Cas9 system [90,91], the availability of cheap and easy-to-use DNA cloning techniques and the possibility to synthesize large numbers of genes of interest.
Finally, to succeed and to really take advantage of the full “design” space, it is necessary to have a collection of well-characterized synthetic biology parts, such as promoters, terminators, regulatory sequences, or genes encoding sensor proteins that can react to various stimuli. Any combination of these parts should result in predictable behaviour. Such a collection of parts is already available for bacteria http://parts.igem.org/Main_Page where one can choose from a wide selection of promoters, terminators and genes involved in various cellular processes, such as signalling, cell death, motility, recombination and many more. These parts can be combined into circuits with the use of logical gates, such as AND, OR, NOR or NAND. These circuits then can perform a wide variety of functions, such as oscillations, toggle switching, timekeeping or band-pass control [92].
3. Mammalian Expression Systems: CHO Cells
3.1. Importance of Mammalian Cells
Cultivated mammalian cell lines have emerged as a powerful tool to produce complex biopharmaceuticals, which is why the optimization of these platforms is a major objective of the biopharmaceutical industry. Within mammalian platforms, CHO cells are the most widely used cell line. While, as indicated by their name, these cells were derived from the ovary of the Chinese hamster, they are in fact mainly of epithelial phenotype [93]. One of the main advantages of mammalian cells is that they are able to perform complex PTMs. These are required to sustain optimal pharmacokinetic and pharmacodynamic properties for many biopharmaceuticals [94], where glycosylation represents one of the most important quality control attributes [95,96] and the most common structurally diversified modification in secreted proteins [97].
To satisfy the need for large scale production of complex biotherapeutics, various strategies have been employed to boost the titer of the product of interest, mostly based on media optimization and on high-throughput screening for good producers [98,99]. Alternative optimization strategies based on modular design, synthetic biology, and systems metabolic engineering, hold tremendous promise to further improve the productivity, yield and product quality, and thus to reduce the time and cost of cell line development. Nonetheless, applying such rational engineering tools to mammalian cells is more difficult compared to other platforms. This is due to the need to consider many additional factors, namely the large genome, sophisticated regulatory, signalling and metabolic networks, genome instability and epigenetic regulation.
3.2. Omics Landscape of CHO
CHO cells were originally established in the late 1950s by Theodore T. Puck [100]. Since then, the family of CHO cells has expanded, giving rise to various new lineages such as CHO-K1, CHO-S, CHO-GS-, CHO-DG44, etc. These lineages were developed to fulfill specific industrial requirements, such as suspension culture or specific gene deficiencies that enabled selection, and are the result of genetic modifications via chemical and radiation mutagenesis [101], targeted gene knockouts and adaptation to new culture conditions [102,103].
The CHO-K1 genome published in 2011 [104] was an important stepping stone towards the application of systems biology methods to CHO. However, in contrast to microbial systems, the genome of CHO cells often contains various chromosomal abnormalities caused by their genetic instability [105,106]. Thus, cells belonging to the same lineage within a CHO family can have distinct genetic information and phenotypes. Differences in phenotype can even be observed between cells that nominally belong to the same lineage, but were grown in different laboratories and under various culture conditions. These can in part be explained by the structural variations in the genome and the accumulated genetic changes such as single nucleotide polymorphisms (SNPs), transgene copy number variations and chromosomal rearrangements [107,108,109,110]. Vcelar et al. showed that chromosomal rearrangements within the genome of a population are observed during subcloning, adaption of the cells to a new medium or simply during long-term cultivation [111,112]. On top of this, variations in phenotypes that cannot be explained by genomic diversity and variation alone, are frequently observed, even in subclones of subclones [113]. These facts suggested that the genetic information of the CHO-K1 cells sequenced in 2011 [104] are not representative for all CHO cell lines and subclones, so a reliable and stable reference genome was needed. This was addressed by the generation of a common reference genome of the Chinese hamster Cricetulus griseus [19,114] which was further improved by a more complete genome assembly in 2018 [115]. These, along with the genomes of other cell lines sequenced in the meantime [19,109,110], serve as basic datasets for in silico studies of CHO via integrative analyses of omics data to support cell line and metabolic engineering studies.
3.3. Comparison of CHO to Bacteria and Yeast
From a systems biology perspective, mammalian cells are considered more complex than microbial systems as their genome is by far larger than that of E. coli and S. cerevisiae. The assembly of the sequenced CHO-K1 genome comprises 2.45 Gb with 24,383 predicted genes [104], while microbial cells used by the biotechnological industry have a smaller genome size by one to two orders of magnitude (Table 2). A comparison of E. coli, S. cerevisiae and CHO cell platforms is summarized in Table 2. Even though having a larger genome does not necessarily relate to the cell’s morphological complexity, it can be an indication of the intricacy of its proteome, fluxome, transcriptome and metabolome, and, in particular, of its regulatory capacities. Even from the viewpoint of the basic proteins that are encoded in these genomes, the proteins constituting prokaryotic cells are considered less complex than those of eukaryotic ones. The latter idea was claimed by different researchers such as Zhang et al. and Wang et al. [116,117], stating that the organism’s protein structural complexity (e.g., length) can directly affect the growth performance of cells. It was demonstrated that, when optimizing for growth, a higher growth rate was observed for cell types containing smaller proteins. This is due to the cells’ tendency to increase their mass-normalized kinetic efficiencies during growth [118].
In addition, cultivating mammalian cells is considered more demanding compared to microbial organisms, especially when focusing on their bioprocessing requirements. Due to the lack of a cell wall, there is significantly higher shear sensitivity and the cell culture medium must contain a higher number of essential nutrients compared to microbial systems. Bacteria (e.g., E. coli) and yeast (e.g., S. cerevisiae) can grow in a simple medium containing solely basic elements (e.g., glucose and salts) and usually only in specific cases a few amino acids or vitamins are supplemented. In contrast, mammalian cells require a larger and more complex set of nutrients, including amino acids, organic acids, vitamins, cofactors, carbohydrates and salts. This complexity of the growth medium reveals the strict nutritional demand of mammalian cells.
A major difference between microbial and mammalian cells is the fact that the genome of the latter actually encodes many different types of cells and developmental stages, namely more than 100 different types of tissue that are part of a mammalian body. To ensure correct expression of the required genes at the necessary level in each of these different tissue types, a much more complex regulatory network is required that includes highly sophisticated mechanisms such as epigenetics and chromatin remodeling that simply are not necessary for microbial cells and therefore are not present or only at immature levels of development [119]. Apart from these chromatin state and epigenetic mechanisms, other regulatory factors are abundant in mammalian cells, such as microRNAs or long-non-coding RNAs (lncRNAs), which are transcribed in large numbers [120,121]. While only 2–3% of the genome are regions coding for proteins, in total approximately 30% of the genome are actively transcribed in CHO cells (unpublished results), indicating the regulatory importance of these transcripts and the high level of sophistication.

{kind=link}
Table 2.
Comparison of the three major platforms for biopharmaceuticals production.
| Characteristic | E. coli | S. cerevisiae | CHO | References |
|---|---|---|---|---|
| Genome size (Mbp) | 4.6 | 12.1 | 2450 | [104,122,123] |
| Cell size (m) | <1 | 3–5 | 12–24 | [124,125,126] |
| Cell volume (m3) | 0.3–3 | 30–100 | 900–7200 | [125,126,127] |
| N-Linked Glycosylation | No | High Mannose | Complex | [128] |
| Cell culture medium complexity | Low | Low | High | [128] |
| Cost of cell culture medium | Low | Low | High | [128] |
| Doubling time (h) | Rapid (0.5–4) | Rapid (1.5–6) | Slow (18–48) | [75,129,130,131] |
| Number of protein coding genes | ∼4300 | ∼6600 | ∼24,000 | [115,132] |
| Gene length (bp) | ∼1000 | ∼1000 | ∼1300/18,000 † | [115,127] |
| Promoter length (bp) | ∼100 | ∼1000 | ∼104–105 * | [127,133] |
| Proteins per cell | ∼106 | ∼108 | ∼1010 * | [127,131] |
† Coding/transcript; * HeLa cell line.
4. Engineering CHO for Improved Performance
In view of the many different aspects of a well performing mammalian production cell line, several engineering approaches have been developed to address, typically individually, the many challenges encountered during cell line development and manufacturing of highly complex biotherapeutics. These include increasing cell specific productivity, maintaining a balance between the competing interests of growth and productivity, generating an efficient and targeted metabolism to enhance both, ensuring product quality and ensuring stability both of phenotype and of productivity.
4.1. Increasing Cell Specific Productivity and Process Performance
The larger part of the omics studies presented on CHO focuses on the identification of limiting pathways and genes and their subsequent engineering by overexpression or knock-out [134,135]. Most of these addressed the endoplasmic reticulum and the unfolded protein response, as cells producing high amounts of a “foreign” protein tend to have problems in processing and assembling such large cargos. Here, approaches to either overexpress specific helper proteins such as protein disulfide isomerase or to upregulate the entire ER were reported, as shown for example in [136,137,138].
The success of many of these studies was hampered by the fact that the differential regulation of a single gene is not likely to completely change the behaviour or phenotype of a cell line, so that these approaches were only successful if indeed the engineered gene was the main limiting factor due to its low expression level in the studied cell line [139]. Very little overlap in specific genes was observed in all of these studies, although frequently similar pathways were identified [140,141]. This led to the search for global engineering approaches where multiple genes and entire pathways could be controlled in a single step. One option that was extensively investigated, was the engineering of microRNAs (miRNAs), which globally regulate post-transcriptional processing of mRNAs and protein translation. With the annotation of miRNAs [142,143,144], their potential towards enhancing protein productivity by influencing various cellular pathways (e.g., cell cycle, apoptosis, metabolism, protein expression, etc.) was taken advantage of [145,146,147,148,149].
With respect to process performance, the main focus has been on the reduction of the process associated impurities such as host cell proteins (HCPs), which are considered a hurdle, especially for downstream processing. From this perspective, performing knockouts of pro-apoptotic genes (e.g., caspases), proteases, or overexpression of anti-apoptotic genes, chaperones or proteins involved in the secretory pathway has been previously described. Indeed, several studies demonstrated that overexpressing anti-apoptotic genes may improve the productivity of the cells through extending their lifespan [150,151]. It also results in a lower release of proteins from lysed cells, which were shown to make up a considerable amount of the total protein in culture supernatants [152,153]. Here, Fukuda et al. recently described a novel approach where they developed Anxa2- and Ctsd-knockout CHO cell lines aiming at minimizing the release of HCPs into the supernatant of cultures used for the production of biotherapeutics [154]. Subsequently, Kol et al. described a novel model-based approach to predict the effect of decreasing the secretion of HCPs on CHO and its impact on the cell productivity. These predictions helped directing the design of “clean” cells by knocking-out 14 genes (using multiplex CRISPR-Cas9) that were proven to be responsible for the production of HCPs [155]. The main outcomes of this study were the improvement of the production of recombinant proteins, in part due to the release of resources, and the reduction of impurities at the end of the culture. More targeted approaches were directed against specific HCPs that are known to pose problems in downstream processing [156].
An excellent summary of the different engineering approaches that have been applied to CHO so far is provided by Fischer et al. [157].
4.2. Maintaining the Balance between Growth and Productivity
As described earlier, growth-coupled production is a common design principle employed for the generation of several compounds using microbial cell factories. However, this approach is limited to simple metabolites which can be stoichiometrically coupled to growth, and cannot be applied to protein production, since it is competitive to growth. Hence, a contrasting solution is usually applied for the production of recombinant proteins in CHO cells, where growth and production phases are separated [158]. The switch from high-proliferation to high-production is commonly triggered by a reduction in cultivation temperature [159,160,161] or by treating the cells with certain chemicals, such as sodium butyrate, which promotes gene expression and growth suppression [162]. In addition, cell cycle arrest in the G1 phase was achieved by controlling the activity of cyclin-dependent kinase inhibitors (cdkis) and resulted in an increase in the specific productivity of the cells [163,164].
In fact, various approaches have already been employed to control the proliferation of the cells during cultivation. So far, however, mathematical modeling, although of great promise, has not been fully exploited to simulate bioprocesses and design better control of the switch from cell proliferation to increased heterologous protein production. One of the few examples is a study by Klamt et al., who computationally compared the volumetric productivities of two-stage fermentation strategies against the conventional one-stage production system [165].
4.3. Making Metabolism more Efficient
As already mentioned, cellular resources are limited and need to be shared between growth and recombinant protein production [166]. In the omics studies mentioned above, energy metabolism was the second most frequently found differentially regulated pathway, after unfolded protein response. Therefore, a deeper understanding of CHO metabolism was the main objective after the genomic sequence of CHO became available, which helped with the reconstruction of a genome-scale metabolic model of CHO cells [20]. This model provides the toolkit for in silico metabolic engineering and medium optimization [167]. Combined with different mathematical approaches and experimental data, the model can be used to identify possible metabolic bottlenecks and predict specific targets for metabolic engineering [51]. It allows the study of the cells by applying linear programming-based strategies such as FBA or flux variability analysis (FVA) along with other computational tools that are already commonly used for bacteria or yeast (see Table 1). Furthermore, feeding the GSMM with omics data (e.g., transcriptomics, proteomics, fluxomics and metabolomics) will further improve the in silico prediction accuracy and serve as a basis for cell line engineering [168], employing the tools summarized in Table 1.
Prediction tools based on metabolic modeling have only recently started to be applied to the design of engineering strategies in CHO cells, while also contributing to characterize and understand them better. One of the first examples of an application of the CHO GSMM to an industrial process is the work done by Calmels et al., who curated the genome-scale metabolic model [20] and tailored it to a CHO-DG44 producer cell line. They performed corrections, such as modifying 601 reactions (for example silencing of 537 amino acids transporters), which lead to an improvement of the growth rate and exometabolome predictions [169]. In addition, the secretory pathway was integrated into the GSMM of CHO by Gutierrez et al. to enable predictions of energetic and machinery demands of secreted proteins [170], which might lead to better predictions of engineering targets that aim at improving protein production, as shown for example in the work of Kol et al. [155].
Metabolism is an important network of chemical reactions involved in every feature of cellular function. In the case of CHO cells, the metabolism is considered suboptimal. Due to the complexity of the metabolic network compared to microbial systems, it is still unclear how the cells react to nutrient availability and why certain pathways are used by the cells and not others. A major focus for many years was on the inefficient utilisation of glucose and the rapid accumulation of, in part toxic, waste products.
Usually, CHO cells consume high amounts of glucose, which leads to the production of lactate, despite sufficient oxygen supply (the so called “Warburg effect”) [171]. This is a result of the inefficient use of glucose by the cells. Efforts trying to decrease lactate dehydrogenase A (LDHA) activity using small interfering RNAs (siRNAs) vectors showed promise in reducing the levels of lactate in the culture without influencing the process productivity [172]. Additionally, the metabolism of amino acids leads to the production of toxic by-products which can impact cell growth or productivity. Ammonia represents one of the major byproducts of amino acids metabolism (e.g., glutamine catabolism) [173]. Furthermore, several other byproducts of the amino acid metabolism (e.g., formate, homocysteine or indolelactate) as well as metabolites originating from lipid metabolism were determined to be detrimental to CHO growth [173,174]. To overcome these issues, numerous successful interventions, such as engineering amino acid catabolism, were performed to reduce the accumulation of toxic metabolites and to improve growth and product titers of CHO cell cultures [157,175,176,177].
CHO cells are auxotrophic for several amino acids and therefore need to take them up from the medium to support growth and recombinant protein production. However, the uptake of these amino acids is quite low, even though the amount in the medium is high [131], which might be due to insufficient capacity of the transporters. Increasing the transport capacity of the essential amino acids or inserting the complete pathways for their synthesis might be favourable for cell growth and protein production. Geoghegan et al. identified amino acid transporters that are likely upregulated in producer cell lines and suggested that overexpression of one or more amino acid transporters might be beneficial for growth and productivity [178].
4.4. Ensuring Product Quality
An additional area of focus, apart from improving growth and final titers, is controlling the PTMs, especially glycosylation. Glycosylation patterns are often heterogeneous and they are heavily influenced by the culture status [179,180]. It has been described that glycosylation patterns within CHO platforms can be modulated by varying the culture conditions (e.g., culture medium supplements) [181]. Identifying the role of the individual glycosylation enzymes as well as the limiting steps of glycosylation would help to engineer CHO cells to consistently produce completely glycosylated proteins with desired glycosylation patterns. In particular for monoclonal antibodies, the specific glycan structure plays an important role in the immunoactivity of the product [182] which led to the development of production cell lines that lack, for instance, fucosyltransferase FUT8, or that overexpress enzymes to generate more complex glycan structures [183,184]. Coats et al. showed that with increased productivity, the quality of N-glycosylation of EpoFc decreases [185]. The next step would be to identify the rate limiting step(s) and overexpress the necessary glycosylation enzymes or pathways for precursor synthesis. Fisher et al. described several approaches for modulating post-translational modifications of recombinant proteins by genome editing in CHO [157].
Steps towards custom and consistent glycosylation have already been taken. For example, a panel of cell lines with custom glycosylation patterns was created with the use of CRISPR/Cas9 technology [186]. In another study, the level of galactosylation was manipulated based on predictions from a kinetic model, leading to a reduction in glycan heterogeneity [187]. While for monoclonal antibodies with their relatively simple glycosylation pattern, work on detailed control has already been initiated, the field is still open for more complex proteins bearing multiple glycosylation structures with high prevalence of tetra-antennary structure and the need for full terminal sialylation [188].
4.5. Maintaining Stability
The high variations of behaviour observed between different CHO cell lines and subclones are two sides of Yin and Yang: On the one hand, this high variation contributes largely to their adaptability and ease of cultivation and also usually enables the isolation of a subclone that is able to produce the product of interest in large amounts (even if thousands of clones need to be screened to find such). On the other hand, once such a subclone is identified, there is the danger of phenotypic drift and instability, where the term is usually applied to instability in specific productivity of the transgene rather than other types of phenotypic drift that relate to other cellular properties. While these two types of instability should be separated, there are some common features that underlie both types [189].
For phenotypic instability the main causes are likely a mixture of genetic changes as discussed above and changes in the expression pattern of genes, where changes in expression levels are more frequent than ON/OFF types of differential expression. Traditionally, studies of genetic variation have focused on the coding genes, where of course mutations may cause loss of function or enhanced activity. However, mutations in regulatory regions have been largely ignored so far, mostly because of our limited understanding of their underlying mechanisms, but they could conceivably contribute to the differences in the transcriptome of the coding genes, even if the coding genes themselves are not directly affected. On top of such variations that are connected to changes in the genome sequence, there are the complex epigenetic mechanisms that determine the precise level of expression of all transcripts in a cell [190]. In fact, a recent study of the impact of genome variation and altered DNA-methylation patterns of the genes involved in the oxidative phosphorylation of cells, including the mitochondrial genome, revealed that for cells with specific phenotypes there is an accumulation of both genomic variants and changes in DNA-methylation in genes that are likely to be associated with that phenotype [108].
Analysis of these epigenetic regulatory mechanisms is still in its infancy in the field of CHO cell research, even though some basic mechanisms have been explored. Typically, when discussing DNA-methylation, one only thinks of the fact that the status of methylation of CpG islands in the promoter will effectively determine whether a gene is expressed or not. However, of all Cs in a genome, between 60% and 80% are methylated, including 100% of the Cs in transcribed regions and most of the Cs in inactive heterochromatin regions. The regions of the genome that show the highest variation in DNA methylation levels under different culture conditions or between subclones are those in chromatin states that have regulatory function, such as the areas around the transcription start sites and both genic and other enhancers [109]. Another level of regulation is the presence or absence of activating or repressing histone marks which can lead to constitutive expression or to moderate upregulation or downregulation of genes, in particular in response to short term changes in the environment, such as nutrient depletion or the accumulation of waste molecules [191]. High amounts of histone methylation (at certain amino acid positions) and low amounts of acetylation in the promoter’s environment can also decrease the expression of recombinant proteins [107]. Recent developments now allow the controlled manipulation of such epigenetic marks at specific sites using CRISPR/dCas9 tools [192,193]. Finally, it was recently shown that random changes in a population’s DNA-methylation pattern induce higher phenotypic diversity and thus enable a more efficient isolation of “outliers” with enhanced performance [194]. A more detailed understanding of these mechanisms is still required, but the new tools recently developed will contribute to generate such understanding while at the same time providing the possibility of more sophisticated and reliable control to researchers.
As mentioned before, the main focus of all “stability” related research so far has been on the stability of productivity. Here, several factors come into play that work both on the level of genome and epigenome. Traditionally, recombinant cell lines were generated by random integration of the transgene(s) into the host cell genome. Thus, as random integration usually occurs at adventitious double strand breaks that just happened to be present in the cell at the time of transfection, these integration sites could reside in all types of chromatin regions, including regions of high transcriptional activity and such of low activity or even silenced chromatin states. Consequently, the resulting productivity may vary, as it was shown that adjacent chromatin states can spread into newly integrated sequences. To avoid this, genetic sequences that maintain an open chromatin configuration have been used [195,196]. Direct silencing of the CMV promoter used for most transgenes, either by DNA-methylation or by changes in the histone modifications has also been shown [197,198,199,200]. These different strategies to silence the gene of interest are most likely due to the cell’s drive to remove the stress of high productivity [201,202,203], by which ever means possible. Therefore, a frequent cause of loss of productivity is also the complete deletion of the recombinant genes from the genome [204,205].
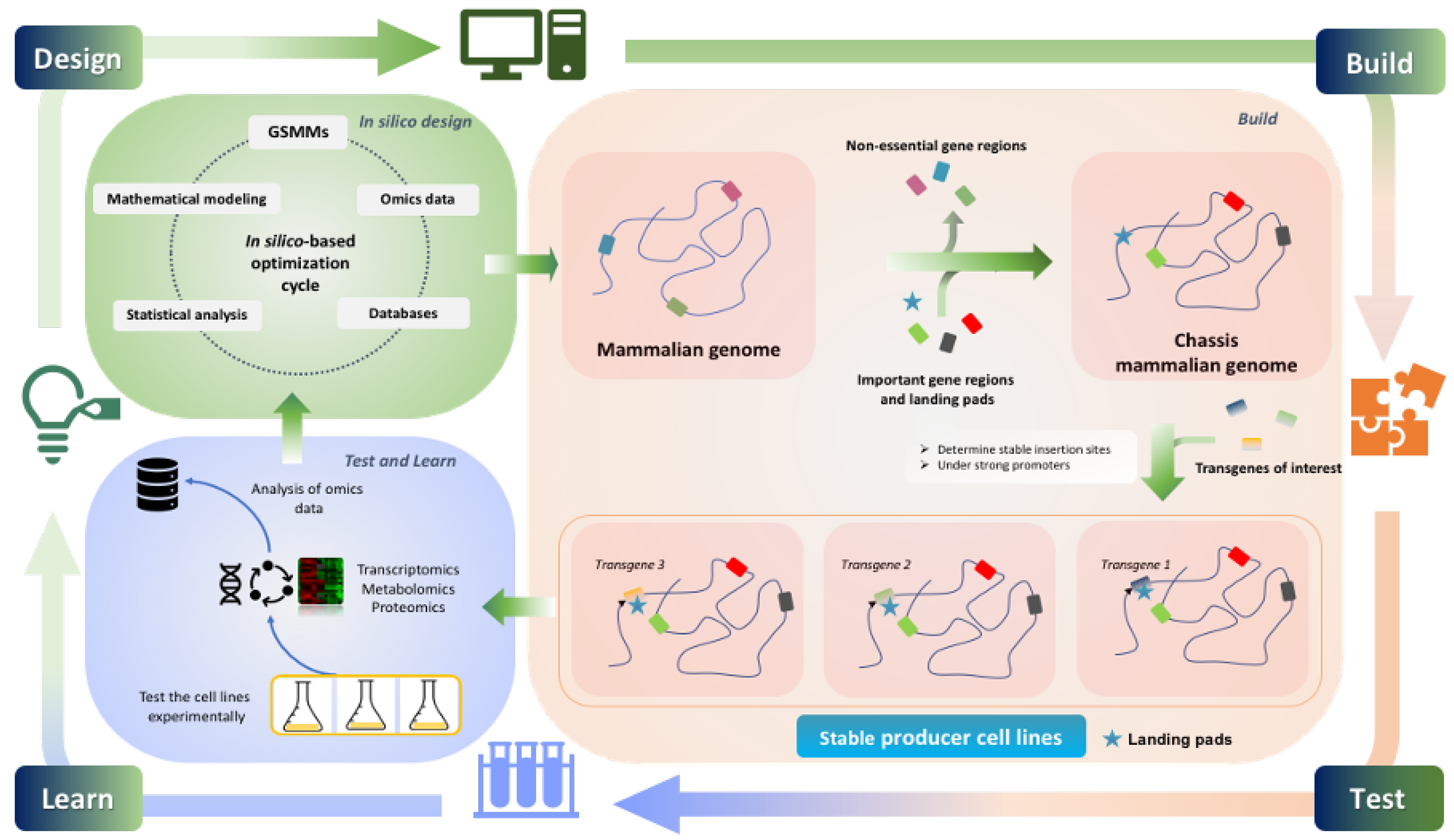
One way of controlling most of the above that has been extensively investigated over the last years, is targeted integration into safe harbours. For this purpose, well-characterized and transcriptionally active regions are identified that enable high stability and continuous expression of the transgenes over long periods of time [206]. Such sites can be identified either computationally, based on transcriptome data, by random selection [207,208,209] or using recombinase pseudosites [210] or viral vectors [208]. For reuse, "landing-pads" containing recombination sites and selection markers [208,210] are inserted into these stable sites, allowing easy integration of heterologous DNA of interest and thereby facilitating the development of stable producer cell lines with a known genomic environment [208]. Some approaches target already known open chromatin areas of the genome [211,212,213]. Another example is the Hipp11 gene locus [214]. The main advantage of these approaches is the efficiency and reduced time required for establishing a new production clone.
5. What Is Missing Towards the Construction of a CHO Chassis Cell?
Developing strong modular mammalian expression platforms is an important R&D goal of the biopharmaceutical industry. As the development of stable CHO production cell lines is still a challenging and slow process, the establishment of pre-optimised chassis cell lines with plug-and-play modules that can be combined at will and as needed is a promising approach [215], even if it is more challenging than in simpler microbial systems.
The most important obstacle here is our lack of knowledge on the many regulatory mechanisms that are in place in cells to enable them to respond to different conditions and challenges. As an example, the regulatory function of long-non-coding RNAs was already discussed. Currently, for lncRNAs, there are 127,802 transcripts and 56,946 genes annotated, however, only for 1867 is their function understood or at least known to be associated with a given phenotype in 3762 cases [216,217]. Similarly, for coding genes, many of these, while annotated, are still labeled as EST (expressed sequence tag), with no understanding of their function or at best a prediction. In many cases, the annotation is derived from studies of embryogenesis and development or disease association, which does not properly capture their everyday function in in vitro cultivated cells. This is why frequently the analysis of differentially expressed genes results in pathway enrichments such as “Neural Development” which are hard to interpret in the context of a recombinant production cell line derived from the ovary and grown in protein free suspension culture. In fact, the development of the human proteome atlas revealed that there are very few proteins that are expressed exclusively in only one tissue type, while the majority is expressed in multiple ones, albeit at different levels and in some cases even in different intracellular locations [218]. As long as no precise functional annotation for all the genes and transcripts that are expressed in CHO is available for the context of cells grown in a bioprocessing environment, our understanding of CHO metabolism will remain perforce incomplete, making the full design of a chassis cell line, as already done for microbial cells [69], difficult.
In addition, such design strategies would require the availability of parts and modules for different functionality that can be combined at will and as required for the production of a given product (Figure 1). Due to the complexity of mammalian cells and the large number of coding genes and apparently required non-coding transcripts, a full design of such a chassis cell line seems quite a challenge in the near future. However, both the selection of pre-adapted cell lines [219,220] and the development of pre-optimised and pre-engineered cell lines with specific properties have already been addressed to good purpose [155,175,176,207,221]. With the recently emerged and still emerging tools, such model-based designed engineering approaches become more and more feasible, where multiple genes are targeted to achieve a specific purpose. New circuits and tools from synthetic biology [222,223,224], including as an example a panel of both endogenous promoters with defined properties [133,225] as well as artificial ones that can be adapted and modified as needed [226,227], will contribute and will be more and more required. Such engineering approaches will require a diverse set of such control units that are based on different mechanisms, as more and more genes may have to be controlled at different expression levels. Such expression levels may be ON/OFF, which is easily achieved by CRISPR/Cas9 strategies of deletion or knock-in, but fine-tuning the expression level and integrating it into endogenous cellular circuits so as to make them responsive to the cellular state, is still a challenge. Here, again, recently developed tools based on dCas9 linked to different modulators of expression or of epigenetic marks, will come in useful [192,193]. Finally, apart from deletion and overexpression and modulation of transcription, in the future other control strategies that target mRNA processing or translation will be needed to enlarge the range and number of possible interventions that can be used for this purpose [228]. As the load of cargo to be engineered increases, artificial chromosomes are a promising alternative that could be used to integrate an entire circuit and multiple genes in predefined combinations into cells in a single step [229].
As the genomic instability and large number of genomic variation of CHO cells is associated with their rapid growth and high division rate, as in other rapidly growing cell lines such as cancer cells, engineering them for more precise DNA replication and a lower rate of faulty chromosome segregation bears the risk of reducing growth rate to a level that is industrially unfeasible. This may be the price we have to pay for high growth rate and good process performance. Nevertheless, with respect to production stability, many successful approaches have been described that can be combined and used for an integrated designer cell line, including insulators [230], scaffold attachment regions [231] and CpG island regions [232]. In addition, removal of transposons or of chromosomal hotspots for rearrangements such as translocations may promote a higher stability of the entire genome [233], similarly as it was performed for E. coli [75]. Finally, simply speeding up cell line development by new and more efficient selection mechanisms or by the above mentioned use of targeted integration and of pre-optimised cell lines, will reduce the number of divisions that a cell line will undergo until ready for manufacturing, thus also reducing the accumulation of variants or mutations. Finally, in all these approaches the impact of the product gene and its specific requirements for translation, processing and secretion will have to be taken into account [215,234].
In the attempt to reach a fully designed chassis cell line—rather than an optimised or engineered one—model-based predictions and computational strategies for the identification of the most useful engineering strategies will play a key role. A challenge that still needs to be addressed here is the availability of tools to combine and correlate the different omics data sets in a comprehensive and automated way. Traditionally, most of these are analysed separately [235]. A proper connection should link different omics layers in order to obtain a complete picture of their interrelationship and its combined influence on the system [236]. So far, several algorithms have been developed to integrate omics data, such as transcriptomics, proteomics or metabolomics into GSMMs [237,238,239,240,241,242,243,244,245], but no truly comprehensive solution is yet available. Thus, with the rapid advances that have been achieved over the last years, both in our basic understanding of cellular mechanisms and regulatory circuits, and with the new tools that have emerged, we are today in a much better position to aim for the design of mammalian chassis cell lines with defined characteristics, even though a number of challenges still need to be resolved.
Author Contributions
Conceptualization, J.Z.; writing—original draft preparation, A.H. and D.S.; writing—review and editing, all; visualization, A.H.; supervision, J.Z., N.B., I.R. and D.E.R.; funding acquisition, A.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been supported the Federal Ministry for Digital and Economic Affairs (bmwd), the Federal Ministry for Transport, Innovation and Technology (bmvit), the Styrian Business Promotion Agency SFG, the Standortagentur Tirol, Government of Lower Austria and ZIT - Technology Agency of the City of Vienna through the COMET-Funding Program managed by the Austrian Research Promotion Agency FFG. A.H. has been supported by the Portuguese NORTE-08-5369-FSE-000053 operation. Additional funding came from the PhD program BioToP (Biomolecular Technology of Proteins) of the Austrian Science Fund (FWF Project W1224) and MIT-Portugal PhD program (Bioengineering Systems). The funding agencies had no influence on the conduct of this research. Open Access Funding by the University of Vienna.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Walsh, G. Biopharmaceutical benchmarks 2018. Nat. Biotechnol. 2018, 36, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Chan, G.; Hu, Y.; Hu, H.; Ouyang, D. A comprehensive map of FDA-approved pharmaceutical products. Pharmaceutics 2018, 10, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohlscheidt, M.; Kiss, R.; Gottschalk, U. An Introduction to Recent Trends in the Biotechnology Industry: Development and Manufacturing of Recombinant Antibodies and Proteins. In New Bioprocessing Strategies: Development and Manufacturing of Recombinant Antibodies and Proteins; Springer: Cham, Switzerland, 2018; pp. 1–8. [Google Scholar]
- Market Research Future Bio process Technology Market to Expand in Size by Highest Revenue up to 2022. Available online: https://www.medgadget.com/2018/08/bio-process-technology-market-to-expand-in-size-by-highest-revenue-up-to-2022-market-research-future.html (accessed on 9 January 2020).
- Bandaranayake, A.D.; Almo, S.C. Recent advances in mammalian protein production. FEBS Lett. 2014, 588, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harcum, S.W.; Lee, K.H. CHO Cells Can Make More Protein. Cell Syst. 2016, 3, 412–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goeddel, D.V.; Kleid, D.G.; Bolivar, F.; Heyneker, H.L.; Yansura, D.G.; Crea, R.; Hirose, T.; Kraszewski, A.; Itakura, K.; Riggs, A.D. Expression in Escherichia coli of chemically synthesized genes for human insulin. Proc. Natl. Acad. Sci. USA 1979, 76, 106–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Li, H.; Sourjik, V. Assembly and stability of flagellar motor in Escherichia coli. Mol. Microbiol. 2011, 80, 886–899. [Google Scholar] [CrossRef]
- Sivashanmugam, A.; Murray, V.; Cui, C.; Zhang, Y.; Wang, J.; Li, Q. Practical protocols for production of very high yields of recombinant proteins using Escherichia coli. Protein Sci. 2009, 18, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Keasling, J.D. Manufacturing molecules through metabolic engineering. Science 2010, 330, 1355–1358. [Google Scholar] [CrossRef]
- Nielsen, J. Production of biopharmaceutical proteins by yeast: advances through metabolic engineering. Bioengineered 2013, 4, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Paddon, C.J.; Keasling, J.D. Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Microbiol. 2014, 12, 355. [Google Scholar] [CrossRef]
- Wohlgemuth, R. Biocatalysis—Key to sustainable industrial chemistry. Curr. Opin. Biotechnol. 2010, 21, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Cummings, R.; Esko, J.; Stanley, P.; Hart, G.; Aebi, M.; Darvill, A.; Kinoshita, T.; Packer, N.; Prestegard, J.; et al. Glycan-Recognizing Probes as Tools–Essentials of Glycobiology. In Essentials of Glycobiology [Internet], 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017. [Google Scholar]
- Liu, L. Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J. Pharm. Sci. 2015, 104, 1866–1884. [Google Scholar] [CrossRef] [PubMed]
- Aslankoohi, E.; Herrera-Malaver, B.; Rezaei, M.N.; Steensels, J.; Courtin, C.M.; Verstrepen, K.J. Non-conventional yeast strains increase the aroma complexity of bread. PLoS ONE 2016, 11, e0165126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, M.; Lin, L.J.; Chiang, C.J.; Chao, Y.P. Synthetic consortium of Escherichia coli for n-butanol production by fermentation of the glucose–xylose mixture. J. Agric. Food Chem. 2017, 65, 10040–10047. [Google Scholar] [CrossRef]
- Lewis, N.E.; Liu, X.; Li, Y.; Nagarajan, H.; Yerganian, G.; O’brien, E.; Bordbar, A.; Roth, A.M.; Rosenbloom, J.; Bian, C.; et al. Genomic landscapes of Chinese hamster ovary cell lines as revealed by the Cricetulus griseus draft genome. Nat. Biotechnol. 2013, 31, 759. [Google Scholar] [CrossRef] [Green Version]
- Hefzi, H.; Ang, K.S.; Hanscho, M.; Bordbar, A.; Ruckerbauer, D.; Lakshmanan, M.; Orellana, C.A.; Baycin-Hizal, D.; Huang, Y.; Ley, D.; et al. A Consensus Genome-scale Reconstruction of Chinese Hamster Ovary Cell Metabolism. Cell Syst. 2016, 3, 434–443.e8. [Google Scholar] [CrossRef] [Green Version]
- Lai, T.; Yang, Y.; Ng, S.K. Advances in mammalian cell line development technologies for recombinant protein production. Pharmaceuticals 2013, 6, 579–603. [Google Scholar] [CrossRef] [Green Version]
- Long, M.R.; Ong, W.K.; Reed, J.L. Computational methods in metabolic engineering for strain design. Curr. Opin. Biotechnol. 2015, 34, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Ögmundarson, Ó.; Herrgård, M.J.; Forster, J.; Hauschild, M.Z.; Fantke, P. Addressing environmental sustainability of biochemicals. Nat. Sustain. 2020, 1–8. [Google Scholar] [CrossRef]
- Burgard, A.P.; Pharkya, P.; Maranas, C.D. Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 2003, 84, 647–657. [Google Scholar] [CrossRef]
- Klamt, S.; Mahadevan, R. On the feasibility of growth-coupled product synthesis in microbial strains. Metab. Eng. 2015, 30, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Fong, S.S.; Burgard, A.P.; Herring, C.D.; Knight, E.M.; Blattner, F.R.; Maranas, C.D.; Palsson, B.O. In silico design and adaptive evolution of Escherichia coli for production of lactic acid. Biotechnol. Bioeng. 2005, 91, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Trinh, C.T.; Srienc, F. Metabolic engineering of Escherichia coli for efficient conversion of glycerol to ethanol. Appl. Environ. Microbiol. 2009, 75, 6696–6705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otero, J.M.; Cimini, D.; Patil, K.R.; Poulsen, S.G.; Olsson, L.; Nielsen, J. Industrial systems biology of Saccharomyces cerevisiae enables novel succinic acid cell factory. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Von Kamp, A.; Klamt, S. Growth-coupled overproduction is feasible for almost all metabolites in five major production organisms. Nat. Commun. 2017, 8, 15956. [Google Scholar] [CrossRef]
- Dinh, H.V.; King, Z.A.; Palsson, B.O.; Feist, A.M. Identification of growth-coupled production strains considering protein costs and kinetic variability. Metab. Eng. Commun. 2018, 7, e00080. [Google Scholar] [CrossRef]
- Portnoy, V.A.; Bezdan, D.; Zengler, K. Adaptive laboratory evolution–harnessing the power of biology for metabolic engineering. Curr. Opin. Biotechnol. 2011, 22, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Van Mastrigt, O.; Abee, T.; Lillevang, S.K.; Smid, E.J. Quantitative physiology and aroma formation of a dairy Lactococcus lactis at near-zero growth rates. Food Microbiol. 2018, 73, 216–226. [Google Scholar] [CrossRef]
- Boecker, S.; Zahoor, A.; Schramm, T.; Link, H.; Klamt, S. Broadening the Scope of Enforced ATP Wasting as a Tool for Metabolic Engineering in Escherichia coli. Biotechnol. J. 2019, 1800438. [Google Scholar] [CrossRef] [Green Version]
- Hädicke, O.; Bettenbrock, K.; Klamt, S. Enforced ATP futile cycling increases specific productivity and yield of anaerobic lactate production in Escherichia coli. Biotechnol. Bioeng. 2015, 112, 2195–2199. [Google Scholar] [CrossRef]
- Semkiv, M.V.; Dmytruk, K.V.; Abbas, C.A.; Sibirny, A.A. Activation of futile cycles as an approach to increase ethanol yield during glucose fermentation in Saccharomyces cerevisiae. Bioengineered 2016, 7, 106–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Kandasamy, V.; Würtz, A.; Jensen, P.R.; Solem, C. Stimulation of acetoin production in metabolically engineered Lactococcus lactis by increasing ATP demand. Appl. Microbiol. Biotechnol. 2016, 100, 9509–9517. [Google Scholar] [CrossRef] [PubMed]
- Holm, A.K.; Blank, L.M.; Oldiges, M.; Schmid, A.; Solem, C.; Jensen, P.R.; Vemuri, G.N. Metabolic and transcriptional response to cofactor perturbations in Escherichia coli. J. Biol. Chem. 2010, 285, 17498–17506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feist, A.M.; Palsson, B.Ø. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat. Biotechnol. 2008, 26, 659. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef] [Green Version]
- Lewis, N.E.; Nagarajan, H.; Palsson, B.O. Constraining the metabolic genotype–phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 2012, 10, 291–305. [Google Scholar] [CrossRef] [Green Version]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245. [Google Scholar] [CrossRef]
- Zanghellini, J.; Ruckerbauer, D.E.; Hanscho, M.; Jungreuthmayer, C. Elementary flux modes in a nutshell: properties, calculation and applications. Biotechnol. J. 2013, 8, 1009–1016. [Google Scholar] [CrossRef]
- Gagneur, J.; Klamt, S. Computation of elementary modes: a unifying framework and the new binary approach. BMC Bioinform. 2004, 5, 175. [Google Scholar] [CrossRef] [Green Version]
- Maia, P.; Rocha, M.; Rocha, I. In Silico Constraint-Based Strain Optimization Methods: The Quest for Optimal Cell Factories. Microbiol. Mol. Biol. Rev. 2016, 80, 45–67. [Google Scholar] [CrossRef] [Green Version]
- Alter, T.B.; Ebert, B.E. Determination of growth-coupling strategies and their underlying principles. BMC Bioinform. 2019, 20, 447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tepper, N.; Shlomi, T. Predicting metabolic engineering knockout strategies for chemical production: accounting for competing pathways. Bioinformatics 2009, 26, 536–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pharkya, P.; Burgard, A.P.; Maranas, C.D. OptStrain: a computational framework for redesign of microbial production systems. Genome Res. 2004, 14, 2367–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pharkya, P.; Maranas, C.D. An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab. Eng. 2006, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Patil, K.R.; Rocha, I.; Förster, J.; Nielsen, J. Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinform. 2005, 6, 308. [Google Scholar]
- Lun, D.S.; Rockwell, G.; Guido, N.J.; Baym, M.; Kelner, J.A.; Berger, B.; Galagan, J.E.; Church, G.M. Large-scale identification of genetic design strategies using local search. Mol. Syst. Biol. 2009, 5, 296. [Google Scholar] [CrossRef] [Green Version]
- Rocha, I.; Maia, P.; Evangelista, P.; Vilaça, P.; Soares, S.; Pinto, J.P.; Nielsen, J.; Patil, K.R.; Ferreira, E.C.; Rocha, M. OptFlux: An open-source software platform for in silico metabolic engineering. BMC Syst. Biol. 2010, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Hamdi, A.; Santos, S.T.; Rocha, I. Towards metabolic optimization of CHO cells: in silico improvement of culture medium. In Proceedings of the 8th IFAC Conference on Foundations of Systems Biology in Engineering, Valencia, Spain, 15–18 October 2019. [Google Scholar]
- Kim, J.; Reed, J.L. OptORF: Optimal metabolic and regulatory perturbations for metabolic engineering of microbial strains. BMC Syst. Biol. 2010, 4, 53. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Reed, J.L.; Maravelias, C.T. Large-scale bi-level strain design approaches and mixed-integer programming solution techniques. PLoS ONE 2011, 6, e24162. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Zheng, P.; Sun, J.; Ma, Y. ReacKnock: identifying reaction deletion strategies for microbial strain optimization based on genome-scale metabolic network. PLoS ONE 2013, 8, e72150. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Zeng, B.; Qian, X. Adaptive bi-level programming for optimal gene knockouts for targeted overproduction under phenotypic constraints. BMC Bioinform. 2013, 14, S17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Ji, B.; Mardinoglu, A.; Nielsen, J.; Hua, Q. Logical transformation of genome-scale metabolic models for gene level applications and analysis. Bioinformatics 2015, 31, 2324–2331. [Google Scholar] [CrossRef] [Green Version]
- Jian, X.; Zhou, S.; Zhang, C.; Hua, Q. In silico identification of gene amplification targets based on analysis of production and growth coupling. Biosystems 2016, 145, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Zhang, C.; Zhou, S.; Wei, L.; Hua, Q. IdealKnock: a framework for efficiently identifying knockout strategies leading to targeted overproduction. Comput. Biol. Chem. 2016, 61, 229–237. [Google Scholar] [CrossRef]
- Hartmann, A.; Vila-Santa, A.; Kallscheuer, N.; Vogt, M.; Julien-Laferrière, A.; Sagot, M.F.; Marienhagen, J.; Vinga, S. OptPipe-a pipeline for optimizing metabolic engineering targets. BMC Syst. Biol. 2017, 11, 143. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.; Broeken, V.; Hansen, A.S.L.; Sonnenschein, N.; Herrgård, M.J. OptCouple: Joint simulation of gene knockouts, insertions and medium modifications for prediction of growth-coupled strain designs. Metab. Eng. Commun. 2019, 8, e00087. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Sun, R.; Yao, J.; Li, J.; Liu, Q.; Price, N.D.; Liu, C.; Wang, Z. OptRAM: In-silico strain design via integrative regulatory-metabolic network modeling. PLoS Comput. Biol. 2019, 15, e1006835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z. egKnock: Identifying direct gene knockout strategies for microbial strain optimization based on metabolic network with gene-protein-reaction relationships. bioRxiv 2019, 514653. [Google Scholar]
- Campodonico, M.A.; Andrews, B.A.; Asenjo, J.A.; Palsson, B.O.; Feist, A.M. Generation of an atlas for commodity chemical production in Escherichia coli and a novel pathway prediction algorithm, GEM-Path. Metab. Eng. 2014, 25, 140–158. [Google Scholar] [CrossRef]
- Shepelin, D.; Hansen, A.S.L.; Lennen, R.; Luo, H.; Herrgård, M.J. Selecting the best: evolutionary engineering of chemical production in microbes. Genes 2018, 9, 249. [Google Scholar] [CrossRef] [Green Version]
- Harder, B.J.; Bettenbrock, K.; Klamt, S. Model-based metabolic engineering enables high yield itaconic acid production by Escherichia coli. Metab. Eng. 2016, 38, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Klamt, S. Characterizing and ranking computed metabolic engineering strategies. Bioinformatics 2019, 35, 3063–3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, S.; Trinh, C.T. Harnessing natural modularity of cellular metabolism to design a modular chassis cell for a diverse class of products by using goal attainment optimization. bioRxiv 2019, 748350. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Tanaka, T.; Kondo, A. Engineering metabolic pathways in Escherichia coli for constructing a “microbial chassis” for biochemical production. Bioresour. Technol. 2017, 245, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Jouhten, P.; Boruta, T.; Andrejev, S.; Pereira, F.; Rocha, I.; Patil, K.R. Yeast metabolic chassis designs for diverse biotechnological products. Sci. Rep. 2016, 6, 29694. [Google Scholar] [CrossRef] [Green Version]
- Trinh, C.T.; Mendoza, B. Modular cell design for rapid, efficient strain engineering toward industrialization of biology. Curr. Opin. Chem. Eng. 2016, 14, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Pereira, F.; Lopes, H.; Maia, P.; Meyer, B.; Konstantinidis, D.; Kafkia, E.; Kötter, P.; Rocha, I.; Patil, K. Yeast chassis design for production of dicarboxylic acids. New Biotechnol. 2018, 44, S8. [Google Scholar] [CrossRef]
- Wen, Z.; Ledesma-Amaro, R.; Lin, J.; Jiang, Y.; Yang, S. Improved n-butanol production from Clostridium cellulovorans by integrated metabolic and evolutionary engineering. Appl. Environ. Microbiol. 2019, 85, e02560-18. [Google Scholar] [CrossRef] [Green Version]
- Xavier, J.C.; Patil, K.R.; Rocha, I. Systems biology perspectives on minimal and simpler cells. Microbiol. Mol. Biol. Rev. 2014, 78, 487–509. [Google Scholar] [CrossRef] [Green Version]
- Pósfai, G.; Plunkett, G.; Fehér, T.; Frisch, D.; Keil, G.M.; Umenhoffer, K.; Kolisnychenko, V.; Stahl, B.; Sharma, S.S.; De Arruda, M.; et al. Emergent properties of reduced-genome Escherichia coli. Science 2006, 312, 1044–1046. [Google Scholar] [CrossRef] [Green Version]
- Couto, J.M.; McGarrity, A.; Russell, J.; Sloan, W.T. The effect of metabolic stress on genome stability of a synthetic biology chassis Escherichia coli K12 strain. Microb. Cell Factories 2018, 17, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, A.; Kildegaard, K.R.; Li, M.; Borodina, I.; Nielsen, J. Establishment of a yeast platform strain for production of p-coumaric acid through metabolic engineering of aromatic amino acid biosynthesis. Metab. Eng. 2015, 31, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.; Chen, Y.; Siewers, V.; Nielsen, J. Improving production of malonyl coenzyme A-derived metabolites by abolishing Snf1-dependent regulation of Acc1. MBio 2014, 5, e01130-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, J.; Si, T.; Nair, N.U.; Zhao, H. Design and construction of acetyl-CoA overproducing Saccharomyces cerevisiae strains. Metab. Eng. 2014, 24, 139–149. [Google Scholar] [CrossRef]
- Kozak, B.U.; van Rossum, H.M.; Luttik, M.A.; Akeroyd, M.; Benjamin, K.R.; Wu, L.; de Vries, S.; Daran, J.M.; Pronk, J.T.; van Maris, A.J. Engineering acetyl coenzyme A supply: functional expression of a bacterial pyruvate dehydrogenase complex in the cytosol of Saccharomyces cerevisiae. MBio 2014, 5, e01696-14. [Google Scholar] [CrossRef] [Green Version]
- Soma, Y.; Hanai, T. Self-induced metabolic state switching by a tunable cell density sensor for microbial isopropanol production. Metab. Eng. 2015, 30, 7–15. [Google Scholar] [CrossRef]
- Soma, Y.; Fujiwara, Y.; Nakagawa, T.; Tsuruno, K.; Hanai, T. Reconstruction of a metabolic regulatory network in Escherichia coli for purposeful switching from cell growth mode to production mode in direct GABA fermentation from glucose. Metab. Eng. 2017, 43, 54–63. [Google Scholar] [CrossRef]
- Brockman, I.M.; Prather, K.L. Dynamic metabolic engineering: new strategies for developing responsive cell factories. Biotechnol. J. 2015, 10, 1360–1369. [Google Scholar] [CrossRef] [Green Version]
- Soma, Y.; Tsuruno, K.; Wada, M.; Yokota, A.; Hanai, T. Metabolic flux redirection from a central metabolic pathway toward a synthetic pathway using a metabolic toggle switch. Metab. Eng. 2014, 23, 175–184. [Google Scholar] [CrossRef]
- Tsuruno, K.; Honjo, H.; Hanai, T. Enhancement of 3-hydroxypropionic acid production from glycerol by using a metabolic toggle switch. Microb. Cell Factories 2015, 14, 155. [Google Scholar] [CrossRef] [Green Version]
- Moser, F.; Borujeni, A.E.; Ghodasara, A.N.; Cameron, E.; Park, Y.; Voigt, C.A. Dynamic control of endogenous metabolism with combinatorial logic circuits. Mol. Syst. Biol. 2018, 14, e8605. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Breitling, R.; Bovenberg, R.; Takano, E. Exploiting plug-and-play synthetic biology for drug discovery and production in microorganisms. Nat. Rev. Microbiol. 2011, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Aderem, A. Systems biology: its practice and challenges. Cell 2005, 121, 511–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCombie, W.R.; McPherson, J.D.; Mardis, E.R. Next-generation sequencing technologies. Cold Spring Harb. Perspect. Med. 2019, 9, a036798. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230. [Google Scholar] [CrossRef]
- Khalil, A.S.; Collins, J.J. Synthetic biology: Applications come of age. Nat. Rev. Genet. 2010, 11, 367. [Google Scholar] [CrossRef]
- Walsh, G.; Jefferis, R. Post-translational modifications in the context of therapeutic proteins. Nat. Biotechnol. 2006, 24, 1241. [Google Scholar] [CrossRef]
- Meehl, M.A.; Stadheim, T.A. Biopharmaceutical discovery and production in yeast. Curr. Opin. Biotechnol. 2014, 30, 120–127. [Google Scholar] [CrossRef]
- Anurag, A.J.R.A.W.; Rathore, S. Defining critical quality attributes for monoclonal antibody therapeutic products. BioPharm Int. 2014, 27, 34–43. [Google Scholar]
- Batra, J.; Rathore, A.S. Glycosylation of monoclonal antibody products: Current status and future prospects. Biotechnol. Prog. 2016, 32, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Higgins, E. Carbohydrate analysis throughout the development of a protein therapeutic. Glycoconj. J. 2010, 27, 211–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gifford, J.; Albee, A.; Deeds, Z.; Delong, B.; Kao, K.; Ross, S.; Caple, M. An Efficient Approach to Cell Culture Medium Optimization—A statistical method to medium mixing. In Animal Cell Technology Meets Genomics; Springer: Dordrecht, The Netherlands, 2005; pp. 549–553. [Google Scholar]
- Puente-Massaguer, E.; Badiella, L.; Gutiérrez-Granados, S.; Cervera, L.; Gòdia, F. A statistical approach to improve compound screening in cell culture media. Eng. Life Sci. 2019, 19, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Puck, T.T.; Cieciura, S.J.; Robinson, A. Genetics of somatic mammalian cells: III. Long-term cultivation of euploid cells from human and animal subjects. J. Exp. Med. 1958, 108, 945–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urlaub, G.; Käs, E.; Carothers, A.M.; Chasin, L.A. Deletion of the diploid dihydrofolate reductase locus from cultured mammalian cells. Cell 1983, 33, 405–412. [Google Scholar] [CrossRef]
- Fan, L.; Kadura, I.; Krebs, L.E.; Hatfield, C.C.; Shaw, M.M.; Frye, C.C. Improving the efficiency of CHO cell line generation using glutamine synthetase gene knockout cells. Biotechnol. Bioeng. 2012, 109, 1007–1015. [Google Scholar] [CrossRef]
- Bort, J.A.H.; Stern, B.; Borth, N. CHO-K1 host cells adapted to growth in glutamine-free medium by FACS-assisted evolution. Biotechnol. J. 2010, 5, 1090–1097. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Nagarajan, H.; Lewis, N.E.; Pan, S.; Cai, Z.; Liu, X.; Chen, W.; Xie, M.; Wang, W.; Hammond, S.; et al. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat. Biotechnol. 2011, 29, 735. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Kimura, S.; Itoi, T.; Honda, K.; Ohtake, H.; Omasa, T. Construction of BAC-based physical map and analysis of chromosome rearrangement in Chinese hamster ovary cell lines. Biotechnol. Bioeng. 2012, 109, 1357–1367. [Google Scholar] [CrossRef]
- Derouazi, M.; Martinet, D.; Schmutz, N.B.; Flaction, R.; Wicht, M.; Bertschinger, M.; Hacker, D.; Beckmann, J.; Wurm, F. Genetic characterization of CHO production host DG44 and derivative recombinant cell lines. Biochem. Biophys. Res. Commun. 2006, 340, 1069–1077. [Google Scholar] [CrossRef]
- Lee, J.S.; Kildegaard, H.F.; Lewis, N.E.; Lee, G.M. Mitigating Clonal Variation in Recombinant Mammalian Cell Lines. Trends Biotechnol. 2019, 37, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, H.; Gerstl, M.P.; Ruckerbauer, D.; Hanscho, M.; Himmelbauer, H.; Clarke, C.; Barron, N.; Zanghellini, J.; Borth, N. Genetic and epigenetic variation across genes involved in energy metabolism and mitochondria of Chinese Hamster Ovary cell lines. Biotechnol. J. 2019, 14, 1800681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feichtinger, J.; Hernández, I.; Fischer, C.; Hanscho, M.; Auer, N.; Hackl, M.; Jadhav, V.; Baumann, M.; Krempl, P.M.; Schmidl, C.; et al. Comprehensive genome and epigenome characterization of CHO cells in response to evolutionary pressures and over time. Biotechnol. Bioeng. 2016, 113, 2241–2253. [Google Scholar] [CrossRef] [Green Version]
- Kaas, C.S.; Kristensen, C.; Betenbaugh, M.J.; Andersen, M.R. Sequencing the CHO DXB11 genome reveals regional variations in genomic stability and haploidy. BMC Genom. 2015, 16, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vcelar, S.; Melcher, M.; Auer, N.; Hrdina, A.; Puklowski, A.; Leisch, F.; Jadhav, V.; Wenger, T.; Baumann, M.; Borth, N. Changes in chromosome counts and patterns in CHO cell lines upon generation of recombinant cell lines and subcloning. Biotechnol. J. 2018, 13, 1700495. [Google Scholar] [CrossRef] [PubMed]
- Vcelar, S.; Jadhav, V.; Melcher, M.; Auer, N.; Hrdina, A.; Sagmeister, R.; Heffner, K.; Puklowski, A.; Betenbaugh, M.; Wenger, T.; et al. Karyotype variation of CHO host cell lines over time in culture characterized by chromosome counting and chromosome painting. Biotechnol. Bioeng. 2018, 115, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Pilbrough, W.; Munro, T.P.; Gray, P. Intraclonal protein expression heterogeneity in recombinant CHO cells. PLoS ONE 2009, 4, e8432. [Google Scholar] [CrossRef] [Green Version]
- Brinkrolf, K.; Rupp, O.; Laux, H.; Kollin, F.; Ernst, W.; Linke, B.; Kofler, R.; Romand, S.; Hesse, F.; Budach, W.E.; et al. Chinese hamster genome sequenced from sorted chromosomes. Nat. Biotechnol. 2013, 31, 694. [Google Scholar] [CrossRef]
- Rupp, O.; MacDonald, M.L.; Li, S.; Dhiman, H.; Polson, S.; Griep, S.; Heffner, K.; Hernandez, I.; Brinkrolf, K.; Jadhav, V.; et al. A reference genome of the Chinese hamster based on a hybrid assembly strategy. Biotechnol. Bioeng. 2018, 115, 2087–2100. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J. Protein-length distributions for the three domains of life. Trends Genet. 2000, 16, 107–109. [Google Scholar] [CrossRef]
- Wang, M.; Kurland, C.G.; Caetano-Anollés, G. Reductive evolution of proteomes and protein structures. Proc. Natl. Acad. Sci. USA 2011, 108, 11954–11958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurland, C.G.; Canbäck, B.; Berg, O.G. The origins of modern proteomes. Biochimie 2007, 89, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K.; Moqtaderi, Z. The TAFs in the HAT. Cell 1998, 94, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Jadhav, V.; Hackl, M.; Druz, A.; Shridhar, S.; Chung, C.Y.; Heffner, K.M.; Kreil, D.P.; Betenbaugh, M.; Shiloach, J.; Barron, N.; et al. CHO microRNA engineering is growing up: Recent successes and future challenges. Biotechnol. Adv. 2013, 31, 1501–1513. [Google Scholar] [CrossRef] [Green Version]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017, 18, 206. [Google Scholar] [CrossRef] [Green Version]
- Blattner, F.R.; Plunkett, G.; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [Green Version]
- Cherry, J.M.; Ball, C.; Weng, S.; Juvik, G.; Schmidt, R.; Adler, C.; Dunn, B.; Dwight, S.; Riles, L.; Mortimer, R.K.; et al. Genetic and physical maps of Saccharomyces cerevisiae. Nature 1997, 387, 67. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, N. On the way to commercializing plant cell culture platform for biopharmaceuticals: present status and prospect. Pharm. Bioprocess. 2014, 2, 499. [Google Scholar] [CrossRef] [Green Version]
- Martínez, V.S.; Buchsteiner, M.; Gray, P.; Nielsen, L.K.; Quek, L.E. Dynamic metabolic flux analysis using B-splines to study the effects of temperature shift on CHO cell metabolism. Metab. Eng. Commun. 2015, 2, 46–57. [Google Scholar] [CrossRef]
- Pan, X.; Dalm, C.; Wijffels, R.H.; Martens, D.E. Metabolic characterization of a CHO cell size increase phase in fed-batch cultures. Appl. Microbiol. Biotechnol. 2017, 101, 8101–8113. [Google Scholar] [CrossRef] [Green Version]
- Milo, R.; Phillips, R. Cell Biology by the Numbers; Garland Science: New York, NY, USA; Abingdon, UK, 2015. [Google Scholar]
- Fernandez, J.M.; Hoeffler, J.P. Gene Expression Systems: Using Nature for the Art of Expression; Academic Press: San Diego, CA, USA, 1998. [Google Scholar]
- Bitter, G.A.; Egan, K.M.; Burnette, W.N.; Samal, B.; Fieschko, J.C.; Peterson, D.L.; Downing, M.R.; Wypych, J.; Langley, K.E. Hepatitis B vaccine produced in yeast. J. Med. Virol. 1988, 25, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Salari, R.; Salari, R. Investigation of the best Saccharomyces cerevisiae growth condition. Electron. Phys. 2017, 9, 3592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Széliová, D.; E Ruckerbauer, D.; N Galleguillos, S.; B Petersen, L.; Natter, K.; Hanscho, M.; Troyer, C.; Causon, T.; Schoeny, H.; B Christensen, H.; et al. What CHO is made of: Variations in the biomass composition of Chinese hamster ovary cell lines. Metab. Eng. 2020. Accepted manuscript. [Google Scholar]
- Rocha, M.; Ferreira, P.G. Bioinformatics Algorithms: Design and Implementation in Python; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Nguyen, L.N.; Novak, N.; Baumann, M.; Koehn, J.; Borth, N. Bioinformatic Identification of Chinese Hamster Ovary (CHO) Cold-Shock Genes and Biological Evidence of their Cold-Inducible Promoters. Biotechnol. J. 2020, 15, 1900359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monger, C.; Kelly, P.S.; Gallagher, C.; Clynes, M.; Barron, N.; Clarke, C. Towards next generation CHO cell biology: Bioinformatics methods for RNA-Seq-based expression profiling. Biotechnol. J. 2015, 10, 950–966. [Google Scholar] [CrossRef] [PubMed]
- Kildegaard, H.F.; Baycin-Hizal, D.; Lewis, N.E.; Betenbaugh, M.J. The emerging CHO systems biology era: Harnessing the ‘omics revolution for biotechnology. Curr. Opin. Biotechnol. 2013, 24, 1102–1107. [Google Scholar] [CrossRef]
- Borth, N.; Mattanovich, D.; Kunert, R.; Katinger, H. Effect of increased expression of protein disulfide isomerase and heavy chain binding protein on antibody secretion in a recombinant CHO cell line. Biotechnol. Prog. 2005, 21, 106–111. [Google Scholar] [CrossRef]
- Mohan, C.; Park, S.H.; Chung, J.Y.; Lee, G.M. Effect of doxycycline-regulated protein disulfide isomerase expression on the specific productivity of recombinant CHO cells: thrombopoietin and antibody. Biotechnol. Bioeng. 2007, 98, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Tigges, M.; Fussenegger, M. Xbp1-based engineering of secretory capacity enhances the productivity of Chinese hamster ovary cells. Metab. Eng. 2006, 8, 264–272. [Google Scholar] [CrossRef]
- Harreither, E.; Hackl, M.; Pichler, J.; Shridhar, S.; Auer, N.; Łabaj, P.P.; Scheideler, M.; Karbiener, M.; Grillari, J.; Kreil, D.P.; et al. Microarray profiling of preselected CHO host cell subclones identifies gene expression patterns associated with in-creased production capacity. Biotechnol. J. 2015, 10, 1625–1638. [Google Scholar] [CrossRef] [Green Version]
- Vishwanathan, N.; Yongky, A.; Johnson, K.C.; Fu, H.Y.; Jacob, N.M.; Le, H.; Yusufi, F.N.; Lee, D.Y.; Hu, W.S. Global insights into the Chinese hamster and CHO cell transcriptomes. Biotechnol. Bioeng. 2015, 112, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Fu, H.Y.; Raju, R.; Vishwanathan, N.; Hu, W.S. Unveiling gene trait relationship by cross-platform meta-analysis on Chinese hamster ovary cell transcriptome. Biotechnol. Bioeng. 2017, 114, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Diendorfer, A.B.; Hackl, M.; Klanert, G.; Jadhav, V.; Reithofer, M.; Stiefel, F.; Hesse, F.; Grillari, J.; Borth, N. Annotation of additional evolutionary conserved microRNAs in CHO cells from updated genomic data. Biotechnol. Bioeng. 2015, 112, 1488–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackl, M.; Jadhav, V.; Jakobi, T.; Rupp, O.; Brinkrolf, K.; Goesmann, A.; Pühler, A.; Noll, T.; Borth, N.; Grillari, J. Computational identification of microRNA gene loci and precursor microRNA sequences in CHO cell lines. J. Biotechnol. 2012, 158, 151–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackl, M.; Jakobi, T.; Blom, J.; Doppmeier, D.; Brinkrolf, K.; Szczepanowski, R.; Bernhart, S.H.; zu Siederdissen, C.H.; Bort, J.A.H.; Wieser, M.; et al. Next-generation sequencing of the Chinese hamster ovary microRNA transcriptome: Identification, annotation and profiling of microRNAs as targets for cellular engineering. J. Biotechnol. 2011, 153, 62–75. [Google Scholar] [CrossRef] [Green Version]
- Loh, W.P.; Loo, B.; Zhou, L.; Zhang, P.; Lee, D.Y.; Yang, Y.; Lam, K.P. Overexpression of microRNAs enhances recombinant protein production in Chinese hamster ovary cells. Biotechnol. J. 2014, 9, 1140–1151. [Google Scholar] [CrossRef]
- Druz, A.; Son, Y.J.; Betenbaugh, M.; Shiloach, J. Stable inhibition of mmu-miR-466h-5p improves apoptosis resistance and protein production in CHO cells. Metab. Eng. 2013, 16, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Jadhav, V.; Hackl, M.; Klanert, G.; Bort, J.A.H.; Kunert, R.; Grillari, J.; Borth, N. Stable overexpression of miR-17 enhances recombinant protein production of CHO cells. J. Biotechnol. 2014, 175, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Kelly, P.S.; Breen, L.; Gallagher, C.; Kelly, S.; Henry, M.; Lao, N.T.; Meleady, P.; O’Gorman, D.; Clynes, M.; Barron, N. Re-programming CHO cell metabolism using miR-23 tips the balance towards a highly productive phenotype. Biotechnol. J. 2015, 10, 1029–1040. [Google Scholar] [CrossRef]
- Fischer, S.; Marquart, K.F.; Pieper, L.A.; Fieder, J.; Gamer, M.; Gorr, I.; Schulz, P.; Bradl, H. miRNA engineering of CHO cells facilitates production of difficult-to-express proteins and increases success in cell line development. Biotechnol. Bioeng. 2017, 114, 1495–1510. [Google Scholar] [CrossRef] [Green Version]
- Baek, E.; Noh, S.M.; Lee, G.M. Anti-apoptosis engineering for improved protein production from CHO cells. In Heterologous Protein Production in CHO Cells; Springer: New York, NY, USA, 2017; pp. 71–85. [Google Scholar]
- Lee, S.; Lee, G.M. Bcl-2 overexpression in CHO cells improves polyethylenimine-mediated gene transfection. Process. Biochem. 2013, 48, 1436–1440. [Google Scholar] [CrossRef]
- Kroll, P.; Eilers, K.; Fricke, J.; Herwig, C. Impact of cell lysis on the description of cell growth and death in cell culture. Eng. Life Sci. 2017, 17, 440–447. [Google Scholar] [CrossRef]
- Klein, T.; Heinzel, N.; Kroll, P.; Brunner, M.; Herwig, C.; Neutsch, L. Quantification of cell lysis during CHO bioprocesses: Impact on cell count, growth kinetics and productivity. J. Biotechnol. 2015, 207, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, N.; Senga, Y.; Honda, S. Anxa2-and Ctsd-knockout CHO cell lines to diminish the risk of contamination with host cell proteins. Biotechnol. Prog. 2019, 35, e2820. [Google Scholar] [CrossRef] [PubMed]
- Kol, S.; Ley, D.; Wulff, T.; Decker, M.; Arnsdorf, J.; Schoffelen, S.; Hansen, A.H.; Jensen, T.L.; Gutierrez, J.M.; Chiang, A.W.; et al. Multiplex secretome engineering enhances recombinant protein production and purity. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chiu, J.; Valente, K.N.; Levy, N.E.; Min, L.; Lenhoff, A.M.; Lee, K.H. Knockout of a difficult-to-remove CHO host cell protein, lipoprotein lipase, for improved polysorbate stability in monoclonal antibody formulations. Biotechnol. Bioeng. 2017, 114, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.; Handrick, R.; Otte, K. The art of CHO cell engineering: a comprehensive retrospect and future perspectives. Biotechnol. Adv. 2015, 33, 1878–1896. [Google Scholar] [CrossRef]
- Simpson, N.; Singh, R.; Emery, A.; Al-Rubeai, M. Bcl-2 over-expression reduces growth rate and prolongs G1 phase in continuous chemostat cultures of hybridoma cells. Biotechnol. Bioeng. 1999, 64, 174–186. [Google Scholar] [CrossRef]
- Kaufmann, H.; Mazur, X.; Fussenegger, M.; Bailey, J.E. Influence of low temperature on productivity, proteome and protein phosphorylation of CHO cells. Biotechnol. Bioeng. 1999, 63, 573–582. [Google Scholar] [CrossRef]
- Xu, J.; Tang, P.; Yongky, A.; Drew, B.; Borys, M.C.; Liu, S.; Li, Z.J. Systematic Development of Temperature Shift Strategies for Chinese Hamster Ovary Cells Based on Short Duration Cultures and Kinetic Modeling. mAbs 2019, 11, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Bedoya-López, A.; Estrada, K.; Sanchez-Flores, A.; Ramirez, O.T.; Altamirano, C.; Segovia, L.; Miranda-Rios, J.; Trujillo-Roldan, M.A.; Valdez-Cruz, N.A. Effect of temperature downshift on the transcriptomic responses of Chinese hamster ovary cells using recombinant human tissue plasminogen activator production culture. PLoS ONE 2016, 11, e0151529. [Google Scholar] [CrossRef]
- Jiang, Z.; Sharfstein, S.T. Sodium butyrate stimulates monoclonal antibody over-expression in CHO cells by improving gene accessibility. Biotechnol. Bioeng. 2008, 100, 189–194. [Google Scholar] [CrossRef]
- Bi, J.X.; Shuttleworth, J.; Al-Rubeai, M. Uncoupling of cell growth and proliferation results in enhancement of productivity in p21CIP1-arrested CHO cells. Biotechnol. Bioeng. 2004, 85, 741–749. [Google Scholar] [CrossRef]
- Fussenegger, M.; Schlatter, S.; Dätwyler, D.; Mazur, X.; Bailey, J.E. Controlled proliferation by multigene metabolic engineering enhances the productivity of Chinese hamster ovary cells. Nat. Biotechnol. 1998, 16, 468. [Google Scholar] [CrossRef]
- Klamt, S.; Mahadevan, R.; Hädicke, O. When do two-stage processes outperform one-stage processes? Biotechnol. J. 2018, 13, 1700539. [Google Scholar] [CrossRef] [Green Version]
- Kallehauge, T.B.; Li, S.; Pedersen, L.E.; Ha, T.K.; Ley, D.; Andersen, M.R.; Kildegaard, H.F.; Lee, G.M.; Lewis, N.E. Ribosome profiling-guided depletion of an mRNA increases cell growth rate and protein secretion. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, E.J.; Monk, J.M.; Palsson, B.O. Using genome-scale models to predict biological capabilities. Cell 2015, 161, 971–987. [Google Scholar] [CrossRef] [Green Version]
- Galleguillos, S.N.; Ruckerbauer, D.; Gerstl, M.P.; Borth, N.; Hanscho, M.; Zanghellini, J. What can mathematical modelling say about CHO metabolism and protein glycosylation? Comput. Struct. Biotechnol. J. 2017, 15, 212–221. [Google Scholar] [CrossRef]
- Calmels, C.; McCann, A.; Malphettes, L.; Andersen, M.R. Application of a curated genome-scale metabolic model of CHO DG44 to an industrial fed-batch process. Metab. Eng. 2019, 51, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, J.M.; Feizi, A.; Li, S.; Kallehauge, T.B.; Hefzi, H.; Grav, L.M.; Ley, D.; Hizal, D.B.; Betenbaugh, M.J.; Voldborg, B.; et al. Genome-scale reconstructions of the mammalian secretory pathway predict metabolic costs and limitations of protein secretion. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, G.M. Down-regulation of lactate dehydrogenase-A by siRNAs for reduced lactic acid formation of Chinese hamster ovary cells producing thrombopoietin. Appl. Microbiol. Biotechnol. 2007, 74, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.; Kildegaard, H.F.; Andersen, M.R. Impact of CHO metabolism on cell growth and protein production: an overview of toxic and inhibiting metabolites and nutrients. Biotechnol. J. 2018, 13, 1700499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulukutla, B.C.; Kale, J.; Kalomeris, T.; Jacobs, M.; Hiller, G.W. Identification and control of novel growth inhibitors in fed-batch cultures of Chinese hamster ovary cells. Biotechnol. Bioeng. 2017, 114, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Mulukutla, B.C.; Mitchell, J.; Geoffroy, P.; Harrington, C.; Krishnan, M.; Kalomeris, T.; Morris, C.; Zhang, L.; Pegman, P.; Hiller, G.W. Metabolic engineering of Chinese hamster ovary cells towards reduced biosynthesis and accumulation of novel growth inhibitors in fed-batch cultures. Metab. Eng. 2019, 54, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Ley, D.; Pereira, S.; Pedersen, L.E.; Arnsdorf, J.; Hefzi, H.; Davy, A.M.; Ha, T.K.; Wulff, T.; Kildegaard, H.F.; Andersen, M.R. Reprogramming AA catabolism in CHO cells with CRISPR/Cas9 genome editing improves cell growth and reduces byproduct secretion. Metab. Eng. 2019, 56, 120–129. [Google Scholar] [CrossRef]
- O’Brien, C.; Allman, A.; Daoutidis, P.; Hu, W.S. Kinetic model optimization and its application to mitigating the Warburg effect through multiple enzyme alterations. Metab. Eng. 2019, 56, 154–164. [Google Scholar] [CrossRef]
- Geoghegan, D.; Arnall, C.; Hatton, D.; Noble-Longster, J.; Sellick, C.; Senussi, T.; James, D.C. Control of amino acid transport into Chinese hamster ovary cells. Biotechnol. Bioeng. 2018, 115, 2908–2929. [Google Scholar] [CrossRef]
- Butler, M.; Spearman, M. The choice of mammalian cell host and possibilities for glycosylation engineering. Curr. Opin. Biotechnol. 2014, 30, 107–112. [Google Scholar] [CrossRef]
- Spearman, M.; Butler, M. Glycosylation in cell culture. In Animal Cell Culture; Springer: Cham, Switzerland, 2015; pp. 237–258. [Google Scholar]
- Ehret, J.; Zimmermann, M.; Eichhorn, T.; Zimmer, A. Impact of cell culture media additives on IgG glycosylation produced in Chinese hamster ovary cells. Biotechnol. Bioeng. 2019, 116, 816–830. [Google Scholar]
- Jefferis, R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat. Rev. Drug Discov. 2009, 8, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Malphettes, L.; Freyvert, Y.; Chang, J.; Liu, P.Q.; Chan, E.; Miller, J.C.; Zhou, Z.; Nguyen, T.; Tsai, C.; Snowden, A.W.; et al. Highly efficient deletion of FUT8 in CHO cell lines using zinc-finger nucleases yields cells that produce completely nonfucosylated antibodies. Biotechnol. Bioeng. 2010, 106, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Mascarenhas, J.; Sealover, N.R.; George, H.J.; Brooks, J.; Kayser, K.J.; Gau, B.; Yasa, I.; Azadi, P.; Archer-Hartmann, S. Chinese hamster ovary (CHO) host cell engineering to increase sialylation of recombinant therapeutic proteins by modulating sialyltransferase expression. Biotechnol. Prog. 2015, 31, 334–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coats, M.T.; Bydlinski, N.; Maresch, D.; Diendorfer, A.; Klanert, G.; Borth, N. mRNA Transfection into CHO-Cells Reveals Production Bottlenecks. Biotechnol. J. 2019, e1900198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Ye, Z.; Wang, S.; Schulz, M.A.; Van Coillie, J.; Sun, L.; Chen, Y.H.; Narimatsu, Y.; Hansen, L.; Kristensen, C.; et al. The glycosylation design space for recombinant lysosomal replacement enzymes produced in CHO cells. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stach, C.S.; McCann, M.G.; O’Brien, C.M.; Le, T.S.; Somia, N.; Chen, X.; Lee, K.; Fu, H.Y.; Daoutidis, P.; Zhao, L.; et al. Model-driven engineering of N-linked glycosylation in Chinese Hamster Ovary cells. ACS Synth. Biol. 2019, 8, 2524–2535. [Google Scholar] [CrossRef]
- Kotidis, P.; Jedrzejewski, P.; Sou, S.N.; Sellick, C.; Polizzi, K.; del Val, I.J.; Kontoravdi, C. Model-based optimization of antibody galactosylation in CHO cell culture. Biotechnol. Bioeng. 2019, 116, 1612–1626. [Google Scholar] [CrossRef]
- Dahodwala, H.; Lee, K.H. The fickle CHO: A review of the causes, implications, and potential alleviation of the CHO cell line instability problem. Curr. Opin. Biotechnol. 2019, 60, 128–137. [Google Scholar] [CrossRef]
- Willbanks, A.; Leary, M.; Greenshields, M.; Tyminski, C.; Heerboth, S.; Lapinska, K.; Haskins, K.; Sarkar, S. The evolution of epigenetics: From prokaryotes to humans and its biological consequences. Genet. Epigenet. 2016, 8, GEG–S31863. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, I.; Dhiman, H.; Klanert, G.; Jadhav, V.; Auer, N.; Hanscho, M.; Baumann, M.; Esteve-Codina, A.; Dabad, M.; Gómez, J.; et al. Epigenetic regulation of gene expression in Chinese Hamster Ovary cells in response to the changing environment of a batch culture. Biotechnol. Bioeng. 2019, 116, 677–692. [Google Scholar] [CrossRef] [Green Version]
- Marx, N.; Grünwald-Gruber, C.; Bydlinski, N.; Dhiman, H.; Ngoc Nguyen, L.; Klanert, G.; Borth, N. CRISPR-Based Targeted Epigenetic Editing Enables Gene Expression Modulation of the Silenced Beta-Galactoside Alpha-2, 6-Sialyltransferase 1 in CHO Cells. Biotechnol. J. 2018, 13, 1700217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karottki, K.J.l.C.; Hefzi, H.; Xiong, K.; Shamie, I.; Hansen, A.H.; Li, S.; Pedersen, L.E.; Li, S.; Lee, J.S.; Lee, G.M.; et al. Awakening dormant glycosyltransferases in CHO cells with CRISPRa. Biotechnol. Bioeng. 2019, 117, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Weinguny, M.; Eisenhut, P.; Klanert, G.; Virgolini, N.; Johnson, A.; Ivansson, D.; Lövgren, A.; Borth, N. Random epigenetic modulation of CHO cells by repeated knock-down of DNA-methyltransferases increases population diversity and enables sorting of cells with higher production capacities. Biotechnol. Bioeng. 2020. Submitted manuscript. [Google Scholar]
- Kostyrko, K.; Neuenschwander, S.; Junier, T.; Regamey, A.; Iseli, C.; Schmid-Siegert, E.; Bosshard, S.; Majocchi, S.; Le Fourn, V.; Girod, P.A.; et al. MAR-mediated transgene integration into permissive chromatin and increased expression by recombination pathway engineering. Biotechnol. Bioeng. 2017, 114, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Betts, Z.; Dickson, A.J. Assessment of UCOE on recombinant EPO production and expression stability in amplified Chinese hamster ovary cells. Mol. Biotechnol. 2015, 57, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chusainow, J.; Yap, M.G. DNA methylation contributes to loss in productivity of monoclonal antibody-producing CHO cell lines. J. Biotechnol. 2010, 147, 180–185. [Google Scholar] [CrossRef]
- Moritz, B.; Woltering, L.; Becker, P.B.; Göpfert, U. High levels of histone H 3 acetylation at the CMV promoter are predictive of stable expression in Chinese hamster ovary cells. Biotechnol. Prog. 2016, 32, 776–786. [Google Scholar] [CrossRef]
- Soo, B.P.; Tay, J.; Ng, S.; Ho, S.C.; Yang, Y.; Chao, S.H. Correlation between expression of recombinant proteins and abundance of H3K4Me3 on the enhancer of human cytomegalovirus major immediate-early promoter. Mol. Biotechnol. 2017, 59, 315–322. [Google Scholar] [CrossRef]
- Spencer, S.; Gugliotta, A.; Koenitzer, J.; Hauser, H.; Wirth, D. Stability of single copy transgene expression in CHOK1 cells is affected by histone modifications but not by DNA methylation. J. Biotechnol. 2015, 195, 15–29. [Google Scholar] [CrossRef]
- Zelensky, A.N.; Schimmel, J.; Kool, H.; Kanaar, R.; Tijsterman, M. Inactivation of Pol θ and C-NHEJ eliminates off-target integration of exogenous DNA. Nat. Commun. 2017, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.; Maeda, R.; Adachi, N. Dual loss of human POLQ and LIG4 abolishes random integration. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Wang, J.; Luo, M.; Luo, H.; Zhao, M.; Han, L.; Zhang, M.; Yang, H.; Xie, Y.; Jiang, H.; et al. Rapid development of stable transgene CHO cell lines by CRISPR/Cas9-mediated site-specific integration into C12orf35. Appl. Microbiol. Biotechnol. 2018, 102, 6105–6117. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; O’Callaghan, P.M.; Droms, K.A.; James, D.C. A mechanistic understanding of production instability in CHO cell lines expressing recombinant monoclonal antibodies. Biotechnol. Bioeng. 2011, 108, 2434–2446. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.A.; O’Brien, S.A.; Zhao, L.; Fu, H.Y.; Vishwanathan, N.; Hu, W.S. Recurring genomic structural variation leads to clonal instability and loss of productivity. Biotechnol. Bioeng. 2019, 116, 41–53. [Google Scholar] [CrossRef]
- O’Brien, S.A.; Lee, K.; Fu, H.Y.; Lee, Z.; Le, T.S.; Stach, C.S.; McCann, M.G.; Zhang, A.Q.; Smanski, M.J.; Somia, N.V.; et al. Single Copy Transgene Integration in a Transcriptionally Active Site for Recombinant Protein Synthesis. Biotechnol. J. 2018, 13, 1800226. [Google Scholar] [CrossRef]
- Baumann, M.; Gludovacz, E.; Sealover, N.; Bahr, S.; George, H.; Lin, N.; Kayser, K.; Borth, N. Preselection of recombinant gene integration sites enabling high transcription rates in CHO cells using alternate start codons and recombinase mediated cassette exchange. Biotechnol. Bioeng. 2017, 114, 2616–2627. [Google Scholar] [CrossRef]
- Gaidukov, L.; Wroblewska, L.; Teague, B.; Nelson, T.; Zhang, X.; Liu, Y.; Jagtap, K.; Mamo, S.; Tseng, W.A.; Lowe, A.; et al. A multi-landing pad DNA integration platform for mammalian cell engineering. Nucleic Acids Res. 2018, 46, 4072–4086. [Google Scholar] [CrossRef] [Green Version]
- Pristovšek, N.; Nallapareddy, S.; Grav, L.M.; Hefzi, H.; Lewis, N.E.; Rugbjerg, P.; Hansen, H.G.; Lee, G.M.; Andersen, M.R.; Kildegaard, H.F. Systematic Evaluation of Site-Specific Recombinant Gene Expression for Programmable Mammalian Cell Engineering. ACS Synth. Biol. 2019, 8, 758–774. [Google Scholar] [CrossRef]
- Crawford, Y.; Zhou, M.; Hu, Z.; Joly, J.; Snedecor, B.; Shen, A.; Gao, A. Fast identification of reliable hosts for targeted cell line development from a limited-genome screening using combined φC31 integrase and CRE-Lox technologies. Biotechnol. Prog. 2013, 29, 1307–1315. [Google Scholar] [CrossRef]
- Irion, S.; Luche, H.; Gadue, P.; Fehling, H.J.; Kennedy, M.; Keller, G. Identification and targeting of the ROSA26 locus in human embryonic stem cells. Nat. Biotechnol. 2007, 25, 1477. [Google Scholar] [CrossRef]
- Zambrowicz, B.P.; Imamoto, A.; Fiering, S.; Herzenberg, L.A.; Kerr, W.G.; Soriano, P. Disruption of overlapping transcripts in the ROSA βgeo 26 gene trap strain leads to widespread expression of β-galactosidase in mouse embryos and hematopoietic cells. Proc. Natl. Acad. Sci. USA 1997, 94, 3789–3794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zboray, K.; Sommeregger, W.; Bogner, E.; Gili, A.; Sterovsky, T.; Fauland, K.; Grabner, B.; Stiedl, P.; Moll, H.P.; Bauer, A.; et al. Heterologous protein production using euchromatin-containing expression vectors in mammalian cells. Nucleic Acids Res. 2015, 43, e102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, X.; Zheng, Q.; Jiang, R.; Chen-Tsai, R.Y.; Kong, L.J. A system for site-specific integration of transgenes in mammalian cells. PLoS ONE 2019, 14. [Google Scholar] [CrossRef]
- Brown, A.J.; Gibson, S.J.; Hatton, D.; Arnall, C.L.; James, D.C. Whole synthetic pathway engineering of recombinant protein production. Biotechnol. Bioeng. 2019, 116, 375–387. [Google Scholar] [CrossRef] [PubMed]
- LncBook a Curated Knowledgebase of Human Long Non-coding RNAs. Available online: http://bigd.big.ac.cn/lncbook/index (accessed on 1 May 2020).
- A Comprehensive Compendium of Human Long Non-coding RNAs. Available online: https://lncipedia.org/ (accessed on 1 May 2020).
- Uhlén, M.; Björling, E.; Agaton, C.; Szigyarto, C.A.K.; Amini, B.; Andersen, E.; Andersson, A.C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteom. 2005, 4, 1920–1932. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.L.; Lovelady, C.S.; Grainger, R.K.; Racher, A.J.; Young, R.J.; James, D.C. Functional heterogeneity and heritability in CHO cell populations. Biotechnol. Bioeng. 2013, 110, 260–274. [Google Scholar] [CrossRef]
- Fernandez-Martell, A.; Johari, Y.B.; James, D.C. Metabolic phenotyping of CHO cells varying in cellular biomass accumulation and maintenance during fed-batch culture. Biotechnol. Bioeng. 2018, 115, 645–660. [Google Scholar] [CrossRef]
- Pichler, J.; Galosy, S.; Mott, J.; Borth, N. Selection of CHO host cell subclones with increased specific antibody production rates by repeated cycles of transient transfection and cell sorting. Biotechnol. Bioeng. 2011, 108, 386–394. [Google Scholar] [CrossRef]
- Ausländer, S.; Ausländer, D.; Müller, M.; Wieland, M.; Fussenegger, M. Programmable single-cell mammalian biocomputers. Nature 2012, 487, 123. [Google Scholar] [CrossRef]
- Guinn, M.; Bleris, L. Biological 2-input decoder circuit in human cells. ACS Synth. Biol. 2014, 3, 627–633. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, B.H.; Pham, N.H.; Caraballo, L.D.; Lozanoski, T.; Engel, A.; Bhatia, S.; Wong, W.W. Large-scale design of robust genetic circuits with multiple inputs and outputs for mammalian cells. Nat. Biotechnol. 2017, 35, 453. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.N.; Baumann, M.; Dhiman, H.; Marx, N.; Schmieder, V.; Hussein, M.; Eisenhut, P.; Hernandez, I.; Koehn, J.; Borth, N. Novel promoters derived from Chinese Hamster Ovary cells via in silico and in vitro analysis. Biotechnol. J. 2019, 14, 1900125. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Gibson, S.J.; Hatton, D.; James, D.C. In silico design of context-responsive mammalian promoters with user-defined functionality. Nucleic Acids Res. 2017, 45, 10906–10919. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Sweeney, B.; Mainwaring, D.O.; James, D.C. Synthetic promoters for CHO cell engineering. Biotechnol. Bioeng. 2014, 111, 1638–1647. [Google Scholar] [CrossRef]
- Zucchelli, S.; Patrucco, L.; Persichetti, F.; Gustincich, S.; Cotella, D. Engineering translation in mammalian cell factories to increase protein yield: the unexpected use of long non-coding SINEUP RNAs. Comput. Struct. Biotechnol. J. 2016, 14, 404–410. [Google Scholar] [CrossRef] [Green Version]
- Martella, A.; Pollard, S.M.; Dai, J.; Cai, Y. Mammalian synthetic biology: time for big MACs. ACS Synth. Biol. 2016, 5, 1040–1049. [Google Scholar] [CrossRef]
- Pikaart, M.J.; Recillas-Targa, F.; Felsenfeld, G. Loss of transcriptional activity of a transgene is accompanied by DNA methylation and histone deacetylation and is prevented by insulators. Genes Dev. 1998, 12, 2852–2862. [Google Scholar] [CrossRef] [Green Version]
- Dang, Q.; Auten, J.; Plavec, I. Human beta interferon scaffold attachment region inhibits de novo methylation and confers long-term, copy number-dependent expression to a retroviral vector. J. Virol. 2000, 74, 2671–2678. [Google Scholar] [CrossRef] [Green Version]
- Hejnar, J.; Hájková, P.; Plachỳ, J.; Elleder, D.; Stepanets, V.; Svoboda, J. CpG island protects Rous sarcoma virus-derived vectors integrated into nonpermissive cells from DNA methylation and transcriptional suppression. Proc. Natl. Acad. Sci. USA 2001, 98, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Choe, D.; Cho, S.; Kim, S.C.; Cho, B.K. Minimal genome: worthwhile or worthless efforts toward being smaller? Biotechnol. J. 2016, 11, 199–211. [Google Scholar] [CrossRef]
- Cartwright, J.F.; Arnall, C.L.; Patel, Y.D.; Barber, N.O.; Lovelady, C.S.; Rosignoli, G.; Harris, C.L.; Dunn, S.; Field, R.P.; Dean, G.; et al. A platform for context-specific genetic engineering of recombinant protein production by CHO cells. J. Biotechnol. 2020, 312, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Mias, G.I.; Li-Pook-Than, J.; Jiang, L.; Lam, H.Y.; Chen, R.; Miriami, E.; Karczewski, K.J.; Hariharan, M.; Dewey, F.E.; et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 2012, 148, 1293–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinu, F.R.; Beale, D.J.; Paten, A.M.; Kouremenos, K.; Swarup, S.; Schirra, H.J.; Wishart, D. Systems biology and multi-omics integration: Viewpoints from the metabolomics research community. Metabolites 2019, 9, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, S.A.; Palsson, B.O. Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 2008, 4, e1000082. [Google Scholar] [CrossRef]
- Colijn, C.; Brandes, A.; Zucker, J.; Lun, D.S.; Weiner, B.; Farhat, M.R.; Cheng, T.Y.; Moody, D.B.; Murray, M.; Galagan, J.E. Interpreting expression data with metabolic flux models: predicting Mycobacterium tuberculosis mycolic acid production. PLoS Comput. Biol. 2009, 5, e1000489. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Price, N.D. Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 17845–17850. [Google Scholar] [CrossRef] [Green Version]
- Zur, H.; Ruppin, E.; Shlomi, T. iMAT: An integrative metabolic analysis tool. Bioinformatics 2010, 26, 3140–3142. [Google Scholar] [CrossRef]
- Schmidt, B.J.; Ebrahim, A.; Metz, T.O.; Adkins, J.N.; Palsson, B.Ø.; Hyduke, D.R. GIM3E: Condition-specific models of cellular metabolism developed from metabolomics and expression data. Bioinformatics 2013, 29, 2900–2908. [Google Scholar] [CrossRef] [Green Version]
- Song, H.S.; Reifman, J.; Wallqvist, A. Prediction of metabolic flux distribution from gene expression data based on the flux minimization principle. PLoS ONE 2014, 9, e112524. [Google Scholar] [CrossRef]
- Motamedian, E.; Mohammadi, M.; Shojaosadati, S.A.; Heydari, M. TRFBA: An algorithm to integrate genome-scale metabolic and transcriptional regulatory networks with incorporation of expression data. Bioinformatics 2016, 33, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Banos, D.T.; Trébulle, P.; Elati, M. Integrating transcriptional activity in genome-scale models of metabolism. BMC Syst. Biol. 2017, 11, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, M.; Reed, J.L. Integrating proteomic or transcriptomic data into metabolic models using linear bound flux balance analysis. Bioinformatics 2018, 34, 3882–3888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Steps towards creating a Chinese hamster ovary (CHO) chassis cell line. A combination of various in silico tools is used to predict intervention targets (e.g., for deletion or overexpression). A chassis cell line is constructed by performing genetic changes that boost growth or productivity and ensure desired product quality and stability. Various transgenes of interest can be inserted into a pre-defined landing-pad in the chassis cell line. Afterwards, the performance of the producer cell lines is validated experimentally and the results can be used to further optimize the chassis cell line.
Figure 1.
Steps towards creating a Chinese hamster ovary (CHO) chassis cell line. A combination of various in silico tools is used to predict intervention targets (e.g., for deletion or overexpression). A chassis cell line is constructed by performing genetic changes that boost growth or productivity and ensure desired product quality and stability. Various transgenes of interest can be inserted into a pre-defined landing-pad in the chassis cell line. Afterwards, the performance of the producer cell lines is validated experimentally and the results can be used to further optimize the chassis cell line.

Table 1.
Computational frameworks used for metabolic engineering.
| Tool | Year | Predictions | Application (Strains) | References |
|---|---|---|---|---|
| OptKnock | 2003 | Possible knockouts that can improve production yield. Maximize the target compound production using Bi-Level linear programming | E. coli, G. sulfurreducens, S. cerevisiae | [24,45,46] |
| OptStrain | 2004 | Reaction addition/deletion Possible knockouts. Possible insertions | E. coli, C. acetobutylicum, M. extorquens | [47] |
| OptReg | 2005 | Up and downregulation of reactions for strain design | E. coli | [48] |
| OptGene | 2008 | Optimization of non-linear objective functions Faster predictions compared to OptKnock | S. cerevisiae | [49] |
| RobustKnock | 2009 | Triple level optimization pipeline The framework targets the maximization of minimal target fluxes of industrial targets based on FBA and FVA Production of chemicals of interest is an obligatory by-product of growth rate maximization | E. coli | [45,46] |
| GDLS | 2009 | Employs MILP search approach Finding direct gene-deletion targets Low-complexity search of the space of genetic manipulations ignoring Gene-Protein-Reaction relationships | E. coli | [50] |
| Optflux | 2010 | Bi-level Optimization (OptGene/OptKnock) Multiobjective Optimization | E. coli, yeast, mammalian cells | [51,52] |
| OptORF | 2010 | Depict metabolic engineering strategies based on gene deletion/overexpression | E. coli | [53] |
| SimOptstrain | 2011 | Concurrent gene insertions and knockouts Non-native reaction addition. Bi-level strain design | E. coli | [54] |
| BiMoMa | 2011 | Knockout predictions based on mixed-integer programming solution techniques GPR association | E. coli | [54] |
| ReacKnock | 2013 | Predictions using Bi-Level FBA Improved computation time compared to OptKnock | E. coli | [55] |
| MOMAKnock | 2013 | Relies on MOMA assumption to restrict constraints to steady-state fluxes Identification of robust knockout strategies | E. coli core model | [56] |
| OptGeneKnock | 2015 | Incorporates logic transformation of model (LTM) with a bilevel mixed integer linear programming (MILP)-based knockout method Designing fast genetic intervention strategies | E. coli | [57] |
| APCG | 2016 | Analysis of production and growth coupling Identification of gene targets for improving production of the desired metabolite | E. coli | [58] |
| IdealKnock | 2016 | Predict knockout strategies for overproduction of compounds of interest | Y. lipolytica | [59] |
| SelFI | 2017 | Identification of selection pathways for directed enzyme evolution | ||
| OptPipe | 2017 | Knockout prediction procedures and rank the suggested mutants according to the expected growth rate, production and a new adaptability measure | C. glutamicum | [60] |
| gcOPT | 2018 | Prediction of possible strong growth-coupling combinations and intervention strategies | E. coli | [45] |
| OptCouple | 2019 | Determination of knockouts, insertions and also medium supplements | E. coli | [61] |
| OptRAM | 2019 | Overexpression, knockdown or knockout of both metabolic genes and transcription factors | Saccharomyces cerevisiae | [62] |
| egKnock | 2019 | Find gene deletion targets for maximization of minimum target flux of industrial objective in flux variability analysis This tool takes gene-protein-reaction relationships into consideration | E. coli | [63] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hamdi, A.; Széliová, D.; Ruckerbauer, D.E.; Rocha, I.; Borth, N.; Zanghellini, J. Key Challenges in Designing CHO Chassis Platforms. Processes 2020, 8, 643. https://doi.org/10.3390/pr8060643
AMA Style
Hamdi A, Széliová D, Ruckerbauer DE, Rocha I, Borth N, Zanghellini J. Key Challenges in Designing CHO Chassis Platforms. Processes. 2020; 8(6):643. https://doi.org/10.3390/pr8060643
Chicago/Turabian StyleHamdi, Anis, Diana Széliová, David E. Ruckerbauer, Isabel Rocha, Nicole Borth, and Jürgen Zanghellini. 2020. "Key Challenges in Designing CHO Chassis Platforms" Processes 8, no. 6: 643. https://doi.org/10.3390/pr8060643
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.