
Abstract
Recent breakthrough results from immune checkpoint inhibitors (ICI) have paved the way to a new era of cancer immunotherapy. In particular, inhibition of programmed death-1 (PD-1)/programmed death-ligand 1 (PD-L1) axis with ICI including nivolumab and pembrolizumab has been emerging as a novel treatment strategy for advanced gastric cancers (GC). In a meta-analysis for anti-PD-1/PD-L1 therapy in GC, the objective response rate was 12.0% and the disease control ratio was 34.7%. The ICI treatment in GC provided modest survival benefit and especially, anti-PD-1 treatment could improve the 12-month and 18-month overall survival rate and prolonged the duration of the response. Moreover, it is likely that anti-PD-1/PD-L1 therapy is more effective in subgroups with microsatellite instability-high, Epstein-Barr virus-positive or high mutation burden in advanced GC. The next steps for developing ICI in GC are mainly two challenges as follows. First is the identification of accurate biomarkers that can predict the response to ICI. The second challenge is the clinical development of combinatorial approaches to maximize the efficacy of ICI. In this review, recent advances in ICI for GC are discussed from a viewpoint of translational aspect including biomarkers and tumor microenvironment, and from a viewpoint of clinical aspects including combination therapies.
Similar content being viewed by others

Introduction
Gastric cancer (GC) is the second leading cause of cancer-related deaths and the sixth most frequent cancers worldwide, and the prevalence is relatively higher in East Asia region in comparison to Western counties (GLOBOCAN 2018 [1]). Currently, curative resection with or without perioperative chemotherapy is a standard treatment for GC and, for unresectable or metastatic advanced GC, chemotherapy using drugs such as platinum compounds, fluoropyrimidines, docetaxel, paclitaxel and irinotecan is a standard therapy [2,3,4,5]. In addition, trastuzumab, a monoclonal antibody (mAb) for human epidermal growth factor receptor 2 (HER2), has been established as a first-line treatment for HER2-positive advanced GC patients [6]. Moreover, ramucirumab, an anti-vascular endothelial growth factor receptor 2 (VEGFR2) mAb, has also been proven efficient for a second-line treatment for advanced GC [7, 8].
Recent breakthrough results from immune checkpoint inhibitors (ICI) such as an anti-cytotoxic T-lymphocyte antigen 4 (CTLA4) mAb (ipilimimab) and anti-programmed death-1 (PD-1) mAbs (nivolumab and pembrolizumab) have paved the way to a new era of cancer immunotherapy, leading to the paradigm shift of cancer treatment [9,10,11,12]. In particular, inhibition of PD-1/programmed death-ligand 1 (PD-L1) axis with ICI including nivolumab and pembrolizumab has been emerging as a novel treatment strategy for advanced GC [13, 14]. For example, the ATTRACTION-2 study treated with nivolumab in patients with unresectable advanced or recurrent GC showed an objective response rate (ORR) of 11.2% [13] and proved prolonged overall survival (OS), leading to the establishment of standard options for a third-line treatment for advanced GC in the Japanese guideline. However, although anti-PD-1 mAb is a promising approach for advanced GC patients, the response rate still limited and developing novel strategies is necessary to maximize the efficacy of ICI.
In this review, recent advances in ICI for GC are discussed from a viewpoint of translational aspect including biomarkers and tumor microenvironment, and from a viewpoint of clinical aspects including combination therapies.
ICI for GC from clinical points of view
Clinical trials with ICI for GC
We have searched clinical trials as keywords with phase III, gastric cancer and ICI, through the following public database as of 1st March 2020: ClinicalTrials.gov (https://clinicaltrials.gov/), World Health Organization(https://www.who.int/), EU Clinical Trials Register (https://www.clinicaltrialsregister.eu/ctr-search/search), Cancer Council SA (https://www.cancersa.org.au/), Japan Registry of Clinical Trials (https://jrct.niph.go.jp/), UMIN-CTR (https://www.umin.ac.jp/) and Japic Clinical Trials Information (https://www.clinicaltrials.jp/cti-user/common/Top.jsp).
As a result, 16 clinical trials were listed up and the trial information is summarized in Table 1. The results from several clinical trials are already reported and others are still under recruiting status. The listed ICI in Table 1 is briefly presented as follows.
Nivolumab
Nivolumab is a human IgG4 mAb that can act against PD-1 and block a co-inhibitory signal on PD-1/PD-L1 and PD-1/PD-L2 axis. In a pivotal phase III trial (ATTRACTION-2), nivolumab significantly prolonged OS in patients with advanced GC in a third-line setting (hazard ratio = 0.63, 95% CI 0.51–0.78; p < 0.0001) [13], and the MST was reported to be 5.3 months with nivolumab in comparison to 4.1 months in the placebo group, and 12 month-OS in nivolumab and placebo groups were 26.6% and 10.9%, respectively.
Pembrolizumab
Pembrolizumab is a humanized high-affinity IgG4 mAb that can act against PD-1 and can block interactions on PD-1/PD-L1 and PD-1/PD-L2 axis. In the KEYNOTE-059/Cohort 1, advanced GC patients treated with pembrolizumab achieved the ORR of 11.6% [15]. However, the phase III trial, pembrolizumab compared with paclitaxel as second-line treatment for advanced GC (KEYNOTE-061, NCT02370498) has failed to meet the primary endpoint to show the clinical efficacy of pembrolizumab monotherapy (OS: hazard ratio = 0.82, 95% CI 0.66–1.03; one-sided p = 0.0421) (PFS: hazard ratio = 1.27, 95% CI 1.03–1.57) [16].
Ipilimumab
Ipilimumab is a human IgG1 mAb that can react against CTLA-4/B7 interaction on mainly antigen-presenting cells/T cells to restore T cell function. In a phase II trial, nivolumab + ipilimumab group showed a higher ORR than the nivolumab alone group (26% vs 14%) (Checkmate-032 [17]). A phase III trial of nivolumab + ipilimumab vs. nivolumab + chemotherapy in patients with advanced GC is ongoing (NCT02872116).
Avelumab
Avelumab is a human IgG1 mAb that can react against PD-L1 and block the interaction of PD-L1 with PD-1 and CD80 molecules. JAVELIN Gastric 300 with avelumab in comparison to chemotherapy in a third-line setting for advanced GC has been reported with a negative result (NCT02625623) (hazard ratio = 1.1, 95% CI 0.9–1.4; p = 0.81) [18].
Durvalumab
Durvalumab is a human IgG1 mAb that can react against PD-L1 and block the interaction of PD-L1 with PD-1 and CD80 molecules. A phase Ib/II trial to evaluate the efficacy of durvalumab in patients with metastatic or recurrent GC is ongoing.
Meta-analysis for clinical trials with ICI in GC
Chen et al. performed a meta-analysis for clinical trials with ICI (anti-PD1, anti-PD-L1 and anti-CTLA4) for advanced GC or esophago-gastric junction tumors in a total of 2003 patients from nine clinical trials [19]. As a result, the ORR of all ICI patients, anti-PD-1/PD-L1 subgroup and anti-CTLA-4 subgroup were 9.9%, 12.0% and 2.1%, respectively, and the disease control ratio were 33.3%, 34.7% and 30.1%, respectively. The median progression-free survival of all ICI, anti-PD-1/PD-L1 subgroup and anti-CTLA-4 subgroup were 1.6, 1.6 and 2.9 months, respectively. The median OS of the three groups was 6.0, 5.4 and 7.7 months, respectively.
As for PD-L1 positive tumors in the meta-analysis, the ORR and disease control ratio of anti-PD-1 treatment in PD-L1 positive patients were significantly higher than that of PD-L1 negative patients. As for microsatellite instability (MSI) in the meta-analysis, the ORR and disease control ratio in MSI-high GC was 55.0% and 74.8%, while the ORR and disease control ratio in microsatellite stable patients were 16.6% and 34.4%, respectively. Therefore, the clinical effect of anti-PD-1 therapy in MSI-high GC was markedly better than microsatellite stable, and ORR could be increased at 3.4 times and disease control ratio could be increased at 2.2 times.
Overall, Chen et al. reported that ICI treatment could provide modest survival benefit and especially, anti-PD-1 treatment could improve the 12-month and 18-month OS and prolonged the duration of the response. Moreover, they suggested that anti-PD-1/PD-L1 therapy is more effective in subgroups with PD-L1 positive, MSI-high, Epstein-Barr virus (EBV) positive or high mutation burden in advanced GC. ICI-related biomarkers are discussed in the later parts of this review.
Treatment-related adverse events of ICI in GC
In the meta-analysis by Chen et al. [19] for advanced GC or esophago-gastric junction tumors, the overall incidence of treatment-related adverse events in ICI was 56.8%, and the incidence of ≥ grade 3 treatment-related adverse events was 14.6%. The common treatment-related adverse events in the ICI treatment were fatigue (14.1%), pruritus (10.3%), rash (9.8%), diarrhea (8.2%), hypothyroidism (7.0%), decreased appetite (6.1%), nausea (5.7%), and anemia (4.4%). Overall, it is confirmed that treatment-related adverse events with ICI were manageable and less frequent than chemotherapy.
Translational aspect of tumor microenvironment related to ICI in GC
PD-L1 expression by intrinsic factors including genomic alterations, epigenetic regulations and oncogenic signaling activation
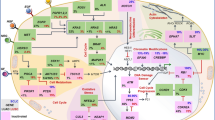
Increased expression of PD-L1 in various types of solid tumors has been reported [20], and positive PD-L1 expression has been shown in the range of 25–65% in GC [21]. Meta-analysis including a total of ten studies involving 1901 patients with GC revealed that PD-L1 expression was associated with a shorter OS [22]. PD-L1 expression is recognized to be regulated by various mechanisms including genetic, epigenetic and extrinsic regulations in GC (Fig. 1).

The regulation of PD-L1 expression by multiple factors in GC cell. The intrinsic factors (genomic alterations, epigenetic regulations and oncogenic signaling activations) and extrinsic factors (cytokines, chemokines, EBV infection and H. pylori infection) induce PD-L1 expression in GC cell. 3′-UTR 3′-untranslated region, CagA cytotoxin-associated gene A, CXCL CXC-chemokine ligand, CXCR CXC-chemokine receptor, EBNA1 EBV nuclear antigen 1, EBV Epstein-Barr virus, GC gastric cancer, HIF-1α hypoxia-inducible factor-1α, H. pyloriHelicobacter pylori, IFNAR type I interferon receptor, IFN-γ interferon-γ, IL-8 interleukin-8, IRF interferon regulatory factor, JAK Janus kinase, MAPK mitogen-activated protein kinase, mTOR mammalian target of rapamycin, NOD-1 nucleotide-binding oligomerization domain-1, PD-L1 programmed cell death ligand-1, PI3K phosphoinositide 3-kinase, PIK3CA PI3K catalytic alpha, PTEN phosphatase and tensin homologue, STAT signal transducer and activator of transcription, T4SS type IV secretion system
Genomic alterations and epigenetic regulations of PD-L1 are observed in GC, and these events are closely associated with overexpression of PD-L1. The Cancer Genome Atlas revealed that amplification of the PD-L1 gene occurs in GC. PD-L1 resides on chromosome 9p24.1 locus, and GC exhibits copy number gains of 9p24, leading to overexpression of PD-L1 [23]. Especially, 9p amplifications were enriched in EBV-positive GC. In addition to the gene amplification, the structural variation of 3′-UTR in PD-L1 gene has also been involved in up-regulation of PD-L1 expression in GC. Kataoka et al. found that the disruption of 3′-UTR of PD-L1 gene caused aberrant PD-L1 expression in multiple cancers including GC [24]. 3′-UTR of PD-L1-disrupted tumor cells attenuated CD8+ T-cell reaction, suggesting that PD-L1 overexpression by 3′-UTR truncation is involved in immune evasion of tumor cells [24]. Moreover, genetic polymorphisms of PD-L1 gene are also associated with increased PD-L1 expression in GC. A polymorphism rs10815225 in the PD-L1 promoter region locates in the binding-site of Sp1 transcription factor, and thereby increased binding of Sp1 to the region significantly up-regulated expression of PD-L1 mRNA in GC [25]. A new polymorphisms rs2297136 (AA + AG genotype) in 3′-UTR of the PD-L1 was also positively correlated with PD-L1 expression in GC [26].
MicroRNAs also contribute to the up-regulation of PD-L1 expression in GC. mRNA expression of PD-L1 is down-regulated by miR-570, and somatic mutation (guanine-to-cytosine mutation) at the 3′-UTR of PD-L1 led to overexpression of the protein by disrupting miR-570 binding in GC [27, 28]. miR-152 and miR-200b have been reported to be involved in carcinogenesis in GC. Although miR-152 and miR-200b usually function as down-regulators of PD-L1 expression through the binding to the 3′-UTR of PD-L1, the expression of these miRNAs was decreased in human GC tissues compared with normal tissues, leading to up-regulation of PD-L1 expression in GC [29, 30]. Similarly, miR-186 was down-regulated in human GC tissues and cell lines [31]. Overexpression of miR-186 inhibited cell proliferation and the expression of PD-L1 by suppressing hypoxia-inducible factor 1α in human GC cell lines [31].
The activation of oncogenic signaling pathways is closely associated with PD-L1 expression in many cancers [32]. The PI3K/Akt pathway is frequently activated in cancer cells, and its activation triggered by the loss of PTEN expression or mutation of PIK3CA gene up-regulates PD-L1 expression in human glioma and triple-negative breast cancer [33, 34]. PIK3CA is known as frequently mutated gene in the range of 4 to 25% of GC [35], and Kim et al. reported that PIK3CA mutations were associated with increased tumor aggressiveness and activation of Akt pathway in GC [36]. Indeed, GC patients with PTEN loss of heterozygosity showed higher PD-L1 expression [37]. Moreover, PD-L1 expression was significantly increased in GC with PIK3CA mutation compared GC with PIK3CA wild-type [38]. Furthermore, PIK3CA mutation was found in 80% of EBV-positive GC, which have a robust expression of PD-L1 [23]. Therefore, activation of the oncogenic PI3K/Akt pathway is involved in PD-L1 expression in GC.
PD-L1 expression by extrinsic factors
Interferon (IFN)-γ produced by mainly tumor-infiltrating lymphocytes is a main extrinsic factor that up-regulates PD-L1 expression in cancer cells [39]. We found that IFN-γ up-regulated PD-L1 expression through the JAK/STAT signaling pathway in GC. PD-L1 expression on tumor cells was positively correlated with IFN-γ level in the GC tissues [40]. Similarly, Moon et al. reported that IFN-γ triggered PD-L1 expression through the JAK2/STAT1/IRF-1 axis in EBV-associated GC [41]. EBV-positive GC expressed EBV nuclear antigen 1, a transcription factor that maintains the copy number of EBV genomes during cell division, which might be involved in IFN-γ-inducible PD-L1 expression [41].
Other cytokines and chemokines have also been reported to up-regulate PD-L1 expression in GC cells. IL-8 is a pro-inflammatory cytokine and functions as a chemokine to act on CXCR1 and CXCR2 receptors [42,43,44]. Sun et al. reported that GC mesenchymal stem cells produced IL-8 and GC mesenchymal stem cells-derived IL-8 triggered PD-L1 expression through activation of the STAT3/mTOR/c-myc signaling pathway in GC cells [45]. IL-8-induced PD-L1 expression caused the resistance of GC cells to the cytotoxic effect of CD8+ T cells [46].
Based on The Cancer Genome Atlas dataset, Zhang et al. revealed that chemokine signaling pathway especially the CXCL9/10/11-CXCR3 axis was most likely associated with PD-L1 expression in GC [47]. CXCL9/10/11 belong to the Glu-Leu-Arg-negative CXC chemokine subfamily [48], and all of these chemokines bind the CXCR3 [49]. The expression of CXCR3 was positively associated with PD-L1 expression in GC cell lines and tissues, and treatment of GC cells with CXCL9/10/11 upregulated PD-L1 expression through activation of the STAT and PI3K-Akt pathways [47].
Overexpression of PD-L1 has been observed in Helicobacter pylori (H. pylori) infected GC. Reyes et al. found that PD-L1 expression was up-regulated on the gastric epithelium during H. pylori infection, which inhibited T cell activation and rather enhanced frequency of regulatory T cell phenotype in gastric mucosa [50, 51]. The expression of PD-L1 by H. pylori depends on its type 4 secretion system components including the cytotoxin-associated gene A protein and peptidoglycan fragments. Type 4 secretion system components activated the oligomerization domain containing 1-p38 MAPK pathway to increase PD-L1 expression in H. pylori-infected gastric epithelial cells [52]. A further investigation by Xie et al. revealed that H. pylori also affected miRNA expression which is related to PD-L1 expression in GC cells. Infection of GC cells with H. pylori suppressed expression of miR-152 and miR-200b, suppressors of PD-L1 expression in GC as mentioned above, leading to overexpression of PD-L1 [30].
PD-L2 expression on GC
Since PD-1 binds to PD-L2 as well as PD-L1, PD-1/PD-L2 interaction should be involved in the anti-tumor effect of cytotoxic T lymphocyte (CTL) and the clinical response to anti-PD-1 mAb. In fact, several reports showed that PD-L2 expression was independently associated with clinical response to ICI in patients with head and neck squamous cell carcinoma [53], renal cell carcinoma and lung squamous cell carcinoma [54]. However, regulation of PD-L2 expression has not yet been elucidated [53,54,55], although the mechanism behind PD-L1 regulation has been well demonstrated such as intrinsic and extrinsic factors (Fig. 1). We and Garcia-Diaz et al. reported that PD-L2 expression is induced on tumor cells in the presence of IFN-β and IFN-γ [40, 56, 57]. Furthermore, we have recently reported that 28.4% of GC patients expressed PD-L2 on tumor cells, and IFN-γ signature significantly correlated with PD-L2 expression, and anti-PD-L2 mAb enhanced the anti-tumor activity of CTL against GC cells expressing PD-L2 [58]. Considering the previous and our results, it is likely that PD-L2 is expressed on GC cells and PD-1/PD-L2 interactions are functionally involved in anti-tumor CTL activities.
The role of tumor-associated macrophage in GC
Tumor-associated macrophages (TAM) are the most abundant immune components in the tumor microenvironment, and they predominantly present as an M2-phenotype (anti-inflammatory phenotype), which associate with tumor progression, invasion and angiogenesis [59]. Indeed, previous reports suggest that high density of TAM predicted a poor prognosis in many types of solid tumors including GC [60]. TAM produce immunosuppressive mediators such as IL-10, tumor growth factor-β, prostaglandins and indoleamine 2, 3-dioxygenase, that suppress functions of CTL and promote the immunosuppressive activity of regulatory T cells [60]. It has been also reported that TAM could generate PD-L1-positive tumor cells via IFN-γ-independent pathway, leading to aggressive cancer [61, 62]. In fact, a significant association between PD-L1 expression in tumor cells and TAM infiltration was observed in GC [63], suggesting that TAM is also likely one of the extrinsic factors inducing PD-L1 expression in tumor cells.
Recent accumulating evidence suggests that TAM often express immune checkpoint molecules including PD-L1, PD-L2, VISTA (V-domain Ig-containing suppressor of T-cell activation) and B7-H4, which tames protective adaptive immunity for tumor progression. The expression of PD-L1 on TAM might be triggered by cytokine stimulation and hypoxia [64, 65] and PD-L1 expression in stroma was significantly associated with dense infiltration of TAM in GC [63]. Lin et al. reported that GC cell-derived colony-stimulating factor (CSF)-2 facilitated secretion of IL-8/CXCL8 by macrophages, and the secreted IL-8/CXCL8 inhibited CD8+ T cells function by inducing autonomous PD-L1 expression on macrophages. The CXCL-8-PD-L1 axis in TAM was associated with poor prognosis in human GC [66]. In addition, it has been reported that TAM expressed not only PD-L1 but also its ligand PD-1 in human solid tumors [67]. Wang et al. identified a pro-tumorigenic subset of macrophages that constitutively expressed PD-1 in GC tissues and activation of PD-1 signaling on TAM augmented their immunosuppressive activity [68].
Inhibition of TAM function by chemicals has been shown to reduce its infiltration into tumor [69, 70]. CSF-1 is a crucial mediator of macrophage differentiation and survival, and blockade of CSF-1-signaling by antagonists of CSF-1 receptor could suppress the activity of TAM in the tumor microenvironment [69, 70]. Interestingly, the preclinical study demonstrated that combination of PLX-3397 (Pexidartinib), a small molecule designed to target CSF-1 receptor, and antagonists of PD-1 and CTLA-4 enhanced productive anti-tumor T-cell responses in pancreatic cancer models [71]. In addition, clinical trials with ICI in combination with CSF-1 receptor inhibitors such as PLX-3397 have been initiated in multiple solid tumors including GC [59]. Inhibition of TAM by CSF-1 receptor inhibitor might synergistically enhance the effects of ICI.
Predictive biomarkers for checkpoint-based immunotherapy in GC
The Cancer Genome Atlas classified GC into four molecular subtypes: EBV positive (9%), MSI-high (22%), genomically stable (20%) and chromosomal instability (50%). Within these subtypes, EBV-positive tumor and MSI-high tumor have shown better response to ICI [23, 72]. In addition, high tumor mutational burden (TMB) [73], T-cell inflamed gene profiling and interferon-γ gene signature [74], and circulating tumor DNA (ctDNA) [75] have been also reported as possible biomarkers predicting clinical outcomes of immunotherapy by ICI (Table 2). The USA Food and Drug Administration has approved the use of pembrolizumab for patients with refractory advanced GC expressing PD-L1, or patients with unresectable or metastatic MSI-high/mismatch repair deficiency (dMMR) of solid tumors including GC [76, 77]. Thus, PD-L1 expression and MSI-high/dMMR are most likely promising predictive biomarkers of checkpoint-based immunotherapy in GC.
PD-L1 as a biomarker
PD-L1 expression in tumor tissues is measured by immunohistochemistry using FFPA sections, and evaluated by the ratio of PD-L1+ tumor cells so-called tumor proportion score (TPS) or by the ratio of PD-L1-stained tumor and immune cells so-called combined positive score (CPS). In the ATTRACTION-2 trial, although approximately 40% of patients were assessed for PD-L1 tumor expression using TPS, survival benefits of nivolumab was shown to be irrespective of PD-L1 status [13]. Moreover, in the CheckMate 032 trial, the responses were shown regardless of tumor PD-L1 expression [17]. On the other hand, in the phase II KEYNOTE-059 trial [76], patients who had PD-L1-positive tumors (CPS ≧ 1) showed an ORR of 22.7% including 2.7% complete response, while patients who had PD-L1-negative tumors (CPS < 1) demonstrated an ORR of 8.6% including 3.4% complete response. In addition, in phase III KEYNOTE-061 trial [16], patients with a PD-L1 CPS of 10 or higher (CPS ≧ 10), but not CPS ≧ 1, had an OS of 10.4 months with pembrolizumab in comparison to 8.0 months with paclitaxel, suggesting that pembrolizumab had a greater effect for patients with higher PD-L1 expression (CPS ≧ 10). Taken together, the significance of PD-L1 as a predictive biomarker still remains debatable.
MSI-high as a biomarker
Microsatellites are repeated DNA sequences scattered throughout the human genome, and DNA polymerases more likely to make errors in microsatellites. Because mismatch DNA is usually repaired by the MMR system, dMMR is frequently associated with MSI-high. MSI-high/dMMR are occurred in tumor cells and generate hypermutation and formation of immunogenic neoantigens that could be recognized by immune cells. Thus, tumors with MSI-high/dMMR exhibit dense infiltration of immune cells especially CD8+ tumor-infiltrating lymphocytes and are thought to be sensitive to ICI. In fact, in the KEYNOTE-059 trial, ORR of pembrolizumab was 57.1% in patients with MSI-high GC, while that was 9.0% in patients with non-MSI-high GC [76]. Moreover, the KEYNOTE-061 trial demonstrated that pembrolizumab monotherapy had better response compared with paclitaxel monotherapy in patients with MSI-high GC [16]. Furthermore, dramatic responses to pembrolizumab were obtained in patients with MSI-high metastatic GC (ORR of 85.7%) [75].
Tumor mutation burden (TMB) as a biomarker
TMB is the total number of mutations per coding area of a tumor genome, and a significant correlation between high TMB and enhanced ORR and progression-free survival for ICI treatment has been observed in certain cancers [73]. A recent phase Ib/II clinical trial (NCT02915432) evaluated the value of TMB as a biomarker for chemo-refractory GC treated with toripalimab, a humanized IgG4 mAb against PD-1 [78]. Patients with high TMB tumors showed significant better responses and longer survival advantage than patients with low TMB tumors (ORR: 33.3% vs 7.1%, OS 14.6 months vs 4.0 months). Samstein et al. broadly analyzed the association between TMB and clinical response to ICI using the clinical and genomic data of 1662 advanced cancer patients including GC (n = 126) treated with ICI and 5371 non-ICI-treated patients [79]. A clear trend was observed between high TMB and increasing benefit for ICI treatment across cancer types, and better OS was also observed in most of the cancer types including GC (but not significant, p = 0.221). However, further studies are needed for better understating of TMB as a predictive biomarker of immunotherapy with ICI in GC.
EBV-positive as a biomarker
Given that EBV-positive GC exhibits, (1) increased PD-L1 expression in tumor and immune cells (PD-L1 staining in tumor cells in 50% and immune cells in 94%), (2) enrichment of IFN-γ gene signature, and (3) dense infiltration of CD8+ tumor-infiltrating lymphocytes [72], EBV-positive GC most likely responds to ICI. Surprisingly, Kim et al. reported that all patients who had EBV-positive tumors (n = 6) achieved a complete or partial response by pembrolizumab [76], suggesting that EBV-positive tumors might have a strong response to ICI treatment. However, Want et al. recently reported that one partial response, two stable disease, and one progressive disease responses for toripalimab were observed among the four GC patients with EBV-positive (n = 4; 72.7%) [78]. Thus, further investigations in larger clinical trials should be needed.
Other factors as a biomarker
Immune-related gene signatures involved in IFN-γ signaling and activated T cell biology have also been proposed to serve as predictive biomarkers for ICI treatment. IFN-γ is produced by natural killer cells and T cells, and it augments cytotoxic function of tumor-infiltrating lymphocytes, recruitment of macrophages, and expression of tumor antigen-presenting MHC molecules in tumor cells, leading to anti-tumor immune response. Six IFN-γ gene signatures including IDO1, CXCL10, CXCL9, HLA-DRA, STAT1 and IFNG were significantly associated with improved progression-free survival in pembrolizumab-treated patients with GC [14, 74]. In the KEYNOTE-059 trial, T-cell-inflamed gene expression profiles using 18 genes (CCL5, CD27, CD274, CD276, CD8A, CMKLR1, CXCL9, CXCR6, HLA-DQA1, HLA-DRB1, HLA-E, IDO1, LAG3, NKG7, PDCD1KLG2, PSMB10, STAT1, TGIT) also demonstrated that the profiling score was significantly higher in responders than in non-responder in patients with GC [76]. Moreover, IFN-γ gene signatures were strongly enriched in MSI-high GC and EBV-positive GC, which subgroups showing better response to ICI. However, clinical information related to the predictive value of immune-gene related signatures for ICI treatment in GC is still limited.
Plasma-derived cell-free ctDNA could predict patients likely to respond to ICI in non-small-cell lung carcinoma and urothelial bladder cancer [80]. ctDNA is released from necrotic or apoptotic tumor cells into bloodstream and presents a better correlation of TMB in tumors. Interestingly, Kim et al. demonstrated that ctDNA mutational load score correlated with clinical response to pembrolizumab in GC patients, suggesting that ctDNA might be able to select patients who are most likely sensitive to ICI treatment in GC [36].
Strategies for combined therapy with ICI in GC
To improve the efficacy of ICI, especially anti-PD-1 mAb, in GC, the next promising option is to develop strategies combining anti-PD-1 mAb with radiotherapy, chemotherapy, molecular target agents or other ICI.
Combined with other ICI
T cell exhaustion remains as a critical issue in the immunotherapy and co-expression of multiple immune checkpoints such as T cell immunoglobulin-3 and lymphocyte activation gene-3 is a typical feature of exhausted CD8 T cells in various types of cancers including GC [81, 82]. Therefore, the efficacy of anti-PD-1 mAb is expected to be enhanced by inhibition targeting other co-inhibitory immune checkpoints. Currently, anti-PD-1 mAb combined with ICI targeting lymphocyte activation gene-3 and T cell immunoglobulin-3 are under investigation in several clinical trials (NCT01968109, NCT03662659 and NCT02817633).
Combined with anti-VEGF therapy
In general, it is recognized that tumor-induced angiogenic factors including VEGF can promote tumor neovascularization, and tumor neovascularization is a condition that anti-tumor CTL fail to infiltrate into tumor microenvironment including GC [83]. Therefore, vascular normalization with anti-VEGF therapy have a potential to restore the infiltration of anti-CTL [84].
In addition, within tumor microenvironment in GC, there are several types of immune suppressive cells including regulatory T cells and TAM. It is reported that activity of regulatory T cells and TAM is partially dependent on VEGF/VEGF receptor (VEGFR) axis, and tumor-induced angiogenic factors or hypoxic condition can further promote regulatory T cell and TAM function via VEGF/VEGFR axis, leading to downregulation of anti-tumor CTL activity [85]. Thus, it is likely that inhibition of VEGF/VEGFR axis with chemicals can block the regulatory T cell and TAM function, resulting in enhancement of anti-tumor immunity and synergistic effect is expected in combination of ICI with anti-VEGF therapy (Fig. 2). In fact, Kato el al. reported that anti-VEGFR2 mAb, ramucirumab had a potential to induce clinical synergism in combination with nivolumab, based on subgroup analysis for ATTRACTION-2 trial [86].

Figure 2 was modified from Nakajima S and Kono K [90] with permission from Hokuryokan Co., Ltd.
Combined therapy with ICI. Combined cancer immunotherapy with anti-VEGF therapy (upper) and radiotherapy (lower). CTL cytotoxic T lymphocyte, HLA human leukocyte antigen, HMGB1 high mobility group box 1, ICI immune checkpoint inhibitors, TAM tumor-associated macrophage, Treg regulatory T cell, VEGF vascular endothelial growth factor, VEGFR VEGF receptor
Combined with radiotherapy
In combination of ICI with radiotherapy, radiotherapy is expected to induce immunogenic cell death [87] and the combination can lead to synergistic effect of anti-tumor CTL (Fig. 2) [87]. Irradiation can induce several degree of tumor death and thereafter, some dying components in certain condition could become immunogenic antigen. Then, immunogenic cell death could trigger the uptake of antigenic components by dendritic cells and transfer antigenic signals to T-cell–mediated immunity, resulting in the expansion of anti-tumor CTL. We and others have shown that high mobility group box 1 and calreticulin induced by cytotoxic stresses such as chemoradiation are important mediators to induce immunogenic cell death [87, 88].
Although a promising result from a clinical trial with irradiation followed by anti-CTLA-4 mAbs in lung cancer was reported [89], there is currently no report to prove the synergistic effect of the combination of irradiation with ICI for GC. We are currently performing a phase II clinical trial of irradiation followed by nivolumab for advanced GC (NCT03453164).
Conclusion
The next steps for developing ICI in GC are mainly two challenges as follows. First is the identification of accurate biomarkers that can predict the response to ICI. The second challenge is the clinical development of combinatorial approaches to maximize the efficacy of ICI.
References
Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Pineros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144:1941–53.
Japanese gastric Association. Japanese gastric cancer treatment guidelines 2014 (ver. 4). Gastric Cancer. 2017;20:1–19.
Sasako M, Sakuramoto S, Katai H, Kinoshita T, Furukawa H, Yamaguchi T, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol. 2011;29:4387–93.
Ajani JA, Lee J, Sano T, Janjigian YY, Fan D, Song S. Gastric adenocarcinoma. Nat Rev Dis Primers. 2017;3:17036.
Koizumi W, Narahara H, Hara T, Takagane A, Akiya T, Takagi M, et al. S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol. 2008;9:215–21.
Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–97.
Fuchs CS, Tomasek J, Yong CJ, Dumitru F, Passalacqua R, Goswami C, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2014;383:31–9.
Wilke H, Muro K, Van Cutsem E, Oh SC, Bodoky G, Shimada Y, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15:1224–355.
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–9.
Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–14.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61.
Kono K. Advances in cancer immunotherapy for gastroenterological malignancy. Ann Gastroenterol Surg. 2018;2:244–5.
Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:2461–71.
Muro K, Chung HC, Shankaran V, Geva R, Catenacci D, Gupta S, et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol. 2016;17:717–26.
Fashoyin-Aje L, Donoghue M, Chen H, He K, Veeraraghavan J, Goldberg KB, et al. FDA approval summary: pembrolizumab for recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma expressing PD-L1. Oncologist. 2019;24:103–9.
Shitara K, Ozguroglu M, Bang YJ, Di Bartolomeo M, Mandala M, Ryu MH, et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet. 2018;392:123–33.
Janjigian YY, Bendell J, Calvo E, Kim JW, Ascierto PA, Sharma P, et al. CheckMate-032 study: efficacy and safety of nivolumab and nivolumab plus ipilimumab in patients with metastatic esophagogastric cancer. J Clin Oncol. 2018;36:2836–44.
Bang YJ, Ruiz EY, Van Cutsem E, Lee KW, Wyrwicz L, Schenker M, et al. Phase III, randomised trial of avelumab versus physician's choice of chemotherapy as third-line treatment of patients with advanced gastric or gastro-oesophageal junction cancer: primary analysis of JAVELIN Gastric 300. Ann Oncol. 2018;29:2052–60.
Chen C, Zhang F, Zhou N, Gu YM, Zhang YT, He YD, et al. Efficacy and safety of immune checkpoint inhibitors in advanced gastric or gastroesophageal junction cancer: a systematic review and meta-analysis. Oncoimmunology. 2019;8:e1581547.
Dong P, Xiong Y, Yue J, Hanley SJB, Watari H. Tumor-Intrinsic PD-L1 signaling in cancer initiation, development and treatment: beyond immune evasion. Front Oncol. 2018;8:386.
Togasaki K, Sukawa Y, Kanai T, Takaishi H. Clinical efficacy of immune checkpoint inhibitors in the treatment of unresectable advanced or recurrent gastric cancer: an evidence-based review of therapies. OncoTargets Ther. 2018;11:8239–50.
Zhang M, Dong Y, Liu H, Wang Y, Zhao S, Xuan Q, et al. The clinicopathological and prognostic significance of PD-L1 expression in gastric cancer: a meta-analysis of 10 studies with 1,901 patients. Sci Rep. 2016;6:37933.
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9.
Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3'-UTR disruption in multiple cancers. Nature. 2016;534:402–6.
Tao LH, Zhou XR, Li FC, Chen Q, Meng FY, Mao Y, et al. A polymorphism in the promoter region of PD-L1 serves as a binding-site for SP1 and is associated with PD-L1 overexpression and increased occurrence of gastric cancer. Cancer Immunol Immunother. 2017;66:309–18.
Wu Y, Zhao T, Jia Z, Cao D, Cao X, Pan Y, et al. Polymorphism of the programmed death-ligand 1 gene is associated with its protein expression and prognosis in gastric cancer. J Gastroenterol Hepatol. 2019;34:1201–7.
Wang W, Sun J, Li F, Li R, Gu Y, Liu C, et al. A frequent somatic mutation in CD274 3'-UTR leads to protein over-expression in gastric cancer by disrupting miR-570 binding. Hum Mutat. 2012;33:480–4.
Wang W, Li F, Mao Y, Zhou H, Sun J, Li R, et al. A miR-570 binding site polymorphism in the B7–H1 gene is associated with the risk of gastric adenocarcinoma. Hum Genet. 2013;132:641–8.
Wang Y, Wang D, Xie G, Yin Y, Zhao E, Tao K, et al. MicroRNA-152 regulates immune response via targeting B7–H1 in gastric carcinoma. Oncotarget. 2017;8:28125–34.
Xie G, Li W, Li R, Wu K, Zhao E, Zhang Y, et al. Helicobacter pylori promote B7–H1 expression by suppressing miR-152 and miR-200b in gastric cancer cells. PLoS ONE. 2017;12:e0168822.
Liu L, Wang Y, Bai R, Yang K, Tian Z. MiR-186 inhibited aerobic glycolysis in gastric cancer via HIF-1alpha regulation. Oncogenesis. 2016;5:e224.
Zerdes I, Matikas A, Bergh J, Rassidakis GZ, Foukakis T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: biology and clinical correlations. Oncogene. 2018;37:4639–61.
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7–H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–8.
Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014;2:361–70.
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554.
Kim JW, Lee HS, Nam KH, Ahn S, Kim JW, Ahn SH, et al. PIK3CA mutations are associated with increased tumor aggressiveness and Akt activation in gastric cancer. Oncotarget. 2017;8:90948–58.
Oki E, Okano S, Saeki H, Umemoto Y, Teraishi K, Nakaji Y, et al. Protein expression of programmed death 1 ligand 1 and HER2 in gastric carcinoma. Oncology. 2017;93:387–94.
Menyhart O, Pongor LS, Gyorffy B. Mutations defining patient cohorts with elevated PD-L1 expression in gastric cancer. Front Pharmacol. 2018;9:1522.
Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7–h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:12737.
Mimura K, Teh JL, Okayama H, Shiraishi K, Kua LF, Koh V, et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018;109:43–53.
Moon JW, Kong SK, Kim BS, Kim HJ, Lim H, Noh K, et al. IFNgamma induces PD-L1 overexpression by JAK2/STAT1/IRF-1 signaling in EBV-positive gastric carcinoma. Sci Rep. 2017;7:17810.
David JM, Dominguez C, Hamilton DH, Palena C. The IL-8/IL-8R axis: a double agent in tumor immune resistance. Vaccines (Basel). 2016;4:22.
Xie K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 2001;12:375–91.
Macri A, Versaci A, Loddo S, Scuderi G, Travagliante M, Trimarchi G, et al. Serum levels of interleukin 1beta, interleukin 8 and tumour necrosis factor alpha as markers of gastric cancer. Biomarkers. 2006;11:184–93.
Zhu W, Huang L, Li Y, Qian H, Shan X, Yan Y, et al. Mesenchymal stem cell-secreted soluble signaling molecules potentiate tumor growth. Cell Cycle. 2011;10:3198–207.
Sun L, Wang Q, Chen B, Zhao Y, Shen B, Wang H, et al. Gastric cancer mesenchymal stem cells derived IL-8 induces PD-L1 expression in gastric cancer cells via STAT3/mTOR-c-Myc signal axis. Cell Death Dis. 2018;9:928.
Zhang C, Li Z, Xu L, Che X, Wen T, Fan Y, et al. CXCL9/10/11, a regulator of PD-L1 expression in gastric cancer. BMC Cancer. 2018;18:462.
Clark-Lewis I, Dewald B, Geiser T, Moser B, Baggiolini M. Platelet factor 4 binds to interleukin 8 receptors and activates neutrophils when its N terminus is modified with Glu-Leu-Arg. Proc Natl Acad Sci USA. 1993;90:3574–7.
Cox MA, Jenh CH, Gonsiorek W, Fine J, Narula SK, Zavodny PJ, et al. Human interferon-inducible 10-kDa protein and human interferon-inducible T cell alpha chemoattractant are allotopic ligands for human CXCR3: differential binding to receptor states. Mol Pharmacol. 2001;59:707–15.
Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. Expression of B7–H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J Immunol. 2006;176:3000–9.
Beswick EJ, Pinchuk IV, Das S, Powell DW, Reyes VE. Expression of the programmed death ligand 1, B7–H1, on gastric epithelial cells after Helicobacter pylori exposure promotes development of CD4+ CD25+ FoxP3+ regulatory T cells. Infect Immun. 2007;75:4334–411.
Lina TT, Alzahrani S, House J, Yamaoka Y, Sharpe AH, Rampy BA, et al. Helicobacter pylori cag pathogenicity island's role in B7–H1 induction and immune evasion. PLoS ONE. 2015;10:e0121841.
Yearley JH, Gibson C, Yu N, Moon C, Murphy E, Juco J, et al. PD-L2 expression in human tumors: relevance to anti-PD-1 therapy in cancer. Clin Cancer Res. 2017;23:3158–67.
Tanegashima T, Togashi Y, Azuma K, Kawahara A, Ideguchi K, Sugiyama D, et al. Immune suppression by PD-L2 against spontaneous and treatment-related antitumor immunity. Clin Cancer Res. 2019;25:4808–19.
Ohigashi Y, Sho M, Yamada Y, Tsurui Y, Hamada K, Ikeda N, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res. 2005;11:2947–53.
Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–201.
Nakayama Y, Mimura K, Tamaki T, Shiraishi K, Kua LF, Koh V, et al. PhosphoSTAT1 expression as a potential biomarker for antiPD1/antiPDL1 immunotherapy for breast cancer. Int J Oncol. 2019;54:2030–8.
Nakayama Y, Mimura K, Kua LF, Okayama H, Min AKT, Saito K, et al. Immune suppression caused by PD-L2 expression on tumor cells in gastric cancer. Gastric Cancer. 2020. https://doi.org/10.1007/s10120-020-01079-z.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416.
Yang L, Zhang Y. Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol. 2017;10:58.
Kasikara C, Kumar S, Kimani S, Tsou WI, Geng K, Davra V, et al. Phosphatidylserine sensing by TAM receptors regulates AKT-dependent chemoresistance and PD-L1 expression. Mol Cancer Res. 2017;15:753–64.
Wei Y, Zhao Q, Gao Z, Lao XM, Lin WM, Chen DP, et al. The local immune landscape determines tumor PD-L1 heterogeneity and sensitivity to therapy. J Clin Invest. 2019;129:3347–60.
Harada K, Dong X, Estrella JS, Correa AM, Xu Y, Hofstetter WL, et al. Tumor-associated macrophage infiltration is highly associated with PD-L1 expression in gastric adenocarcinoma. Gastric Cancer. 2018;21:31–40.
Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci USA. 2003;100:5336–411.
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–90.
Lin C, He H, Liu H, Li R, Chen Y, Qi Y, et al. Tumour-associated macrophages-derived CXCL8 determines immune evasion through autonomous PD-L1 expression in gastric cancer. Gut. 2019;68:1764–73.
Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495–9.
Wang F, Li B, Wei Y, Zhao Y, Wang L, Zhang P, et al. Tumor-derived exosomes induce PD1(+) macrophage population in human gastric cancer that promotes disease progression. Oncogenesis. 2018;7:41.
Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–59.
Gomez-Roca CA, Italiano A, Le Tourneau C, Cassier PA, Toulmonde M, D'Angelo SP, et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann Oncol. 2019;30:1381–92.
Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–69.
Derks S, Liao X, Chiaravalli AM, Xu X, Camargo MC, Solcia E, et al. Abundant PD-L1 expression in Epstein-Barr Virus-infected gastric cancers. Oncotarget. 2016;7:32925–322.
Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377:2500–1.
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–40.
Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med. 2018;24:1449–588.
Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trial. JAMA Oncol. 2018;4:e180013.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20.
Wang F, Wei XL, Wang FH, Xu N, Shen L, Dai GH, et al. Safety, efficacy and tumor mutational burden as a biomarker of overall survival benefit in chemo-refractory gastric cancer treated with toripalimab, a PD-1 antibody in phase Ib/II clinical trial NCT02915432. Ann Oncol. 2019;30:1479–86.
Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–6.
Cabel L, Proudhon C, Romano E, Girard N, Lantz O, Stern MH, et al. Clinical potential of circulating tumour DNA in patients receiving anticancer immunotherapy. Nat Rev Clin Oncol. 2018;15:639–50.
Shi JF, Xu SX, He P, Xi ZH. Expression of carcinoembryonic antigen-related cell adhesion molecule 1(CEACAM1) and its correlation with angiogenesis in gastric cancer. Pathol Res Pract. 2014;210:473–6.
Thompson ED, Zahurak M, Murphy A, Cornish T, Cuka N, Abdelfatah E, et al. Patterns of PD-L1 expression and CD8 T cell infiltration in gastric adenocarcinomas and associated immune stroma. Gut. 2017;66:794–801.
Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624.
Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol. 2018;15:310–24.
Tada Y, Togashi Y, Kotani D, Kuwata T, Sato E, Kawazoe A, et al. Targeting VEGFR2 with Ramucirumab strongly impacts effector/ activated regulatory T cells and CD8(+) T cells in the tumor microenvironment. J Immunother Cancer. 2018;6:106.
Kato K, Satoh T, Muro K, Yoshikawa T, Tamura T, Hamamoto Y, et al. A subanalysis of Japanese patients in a randomized, double-blind, placebo-controlled, phase 3 trial of nivolumab for patients with advanced gastric or gastro-esophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2). Gastric Cancer. 2019;22:344–54.
Suzuki Y, Mimura K, Yoshimoto Y, Watanabe M, Ohkubo Y, Izawa S, et al. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res. 2012;72:3967–76.
Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3:e955691.
Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med. 2018;24:1845–51.
Nakajima S, Kono K. The development of combination cancer immunotherapy. Precision medicine. 3rd ed. Tokyo: Hokuryukan Co., Ltd.; 2020. p. 1–4.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Koji Kono has received research grants from Ono pharmaceutical company, Bristol Myers Squibb and Taiho pharmaceutical company. Koji Kono has received a speaker honorarium from Ono pharmaceutical company, Bristol Myers Squibb and MSD pharmaceutical company. Kosaku Mimura has received a speaker honorarium from Ono pharmaceutical company and Bristol Myers Squibb.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kono, K., Nakajima, S. & Mimura, K. Current status of immune checkpoint inhibitors for gastric cancer. Gastric Cancer 23, 565–578 (2020). https://doi.org/10.1007/s10120-020-01090-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-020-01090-4