
Abstract
We present a new solid-state material phase which is a disordered solid solution but offers many ordered line-compound features. The emergent physical phenomena are rooted in the perfect short-range order which conserves the local octet rule. We model the dual-sublattice-mixed semiconductor alloy \({\mathrm{(ZnSnN}}_{\mathrm{2}}{\mathrm{)}}_{1 - x}{\mathrm{(ZnO)}}_{2x}\) using first-principles calculations, Monte-Carlo simulations with a model Hamiltonian, and an extension of the regular solution model by incorporating short-range order. We demonstrate that this unique solid solution, occurring at a “magic” composition, can provide an electronically pristine character without disorder-induced charge localization and, therefore, a superior carrier transport similar to ordered phases. Interestingly, this phase shows singularities that are absent in the conventional solid-solution models, such as the regular solution and band-gap bowing model. Thermodynamically, this alloy phase has a sharply reduced enthalpy at its composition (like a line compound), but it still requires the entropy from long-range disorder to be stabilized at experimentally accessible temperatures.
Similar content being viewed by others

Introduction
Materials properties depend on composition1,2 and atomic arrangement3,4. Since the Bronze Age, solid solutions, formed by alloying elements to occupy interstitial or substitutional lattice sites of metal crystals, have been used to increase the mechanical strength. Compared to the pure metal phase, this type of alloying creates local lattice strains that impede dislocation movement5. More recently, solid solutions have become a common protocol for functional materials design, such as achieving band convergence for thermoelectric materials6,7,8, engineering electronic band-gap for photocatalysis9,10, suppressing deep level defects in semiconductors11, and realizing different topological semimetal phases12. In solid solutions, properties of interest can be tuned by composition without changing the underlying lattice structure. This flexibility contrasts with “line compounds” which usually occur within a narrow composition range by forming a distinct crystal structure with fixed properties13. Line compounds are stabilized by an enthalpy reduction relative to the constituents, but solid solutions can be either thermodynamically stabilized by the configurational entropy, or constrained in metastable states14. However, the benefits of property tunability in solid solutions come along with side effects. For example, unlike ordered line-compounds15, the atoms in solid solutions are disordered. The absence of translational symmetry in solid solutions causes charge localization which is adverse to carrier transport16. To solve this dilemma, in this paper, we report a discovery of a solid-state material phase that combines solid-solution and line-compound features. In this unique alloy phase, perfect short-range order restores properties that are otherwise typical of ordered phases, such as the absence of charge localization.
Many simple alloy systems can be modeled by a random alloy approximation17. However, short-range order (SRO) often develops in more complex systems with multiple constituents. The importance of SRO has been recognized in different fields, such as semiconductor physics18,19, high-temperature superconductor physics20,21, and mineralogy, where, e.g., amphiboles22 crystallize from a solution of a large number of chemical species differing in size, electronegativity, or valence state. With increased chemical complexity, a certain degree of SRO will generally develop as a compromise between strain, chemical bond, and electrostatic energy4,18,19,23. Particularly, strong SRO effects must be expected when there are significant attractive forces between the constituents, thereby precluding the use of random alloy models. This is the case, e.g., for the mutually compensating strain fields of III–V alloys with both large- and small-atom mismatch24, and for the electrostatic interactions in non-isovalent alloys23,25,26.
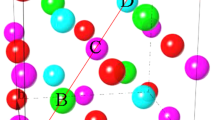
SRO in long-range-disordered systems often has a profound effect on the electronic and optical properties4,27,28,29. Thus, we require an order parameter as a measure of the degree of SRO. We note that previously defined SRO parameters based on bond statistics, such as ξ in ref. 18 and s in ref. 30, provide only partial SRO information. For example, in (GaN)1−x(ZnO)x, a pair of N–Ga3Zn1 and N–Ga1Zn3 tetrahedral motifs gives the same number of N–Ga/N–Zn bonds as two N–Ga2Zn2 motifs. But they are different from the view of local coordination and, more importantly, energetics29. Figure 1 shows a cluster of the wurtzite (ZnSnN2)1−x(ZnO)2x system (ZTNO), illustrating the anion-centered N–ZniSnj and O–ZniSnj motifs, where i + j = 4. The (i,j) distribution of these motifs serves as a measure of SRO and allows for an expansion of the local (SRO) contribution to alloy mixing enthalpy
where \(n_{i,j}^{\mathrm{N}}\) and \(n_{i,j}^{\mathrm{O}}\) are the number of respective N and O centered motifs, NM is the total number of motifs, and ei,j is the energy-expansion parameters, as previously determined in ref. 31. This model Hamiltonian reflects the local octet rule32,33,34, which can be expressed as \(Q_{\mathrm{M}} = q^{{\mathrm{N/O}}} + 1/4\mathop {\sum}\nolimits_{i,j} (iq^{{\mathrm{Zn}}} + jq^{{\mathrm{Sn}}}) = 0\), where QM is the local charge of a N or O centered motif, and q = +2, +4, −3, −2 are the formal oxidation states of Zn, Sn, N, and O, respectively. The octet-rule-conserving N–Zn2Sn2 and O–Zn4Sn0 motifs do not contribute to the mixing enthalpy, i.e., \(e_{2,2}^{\mathrm{N}} = e_{4,0}^{\mathrm{O}} = 0\). The energy contributions ei,j increase with the deviation from QM = 031. Hence, we have ΔHSRO = 0 for perfect SRO (PSRO) structures within this model Hamiltonian. This definition of SRO in terms of local motif coordination allows us to explore whether PSRO can be achieved in A1−xBx. In general, this question depends on the topology of the crystal structure connecting the motifs. In most compounds, the local motif structures connect to each other through corner sharing, as in the wurtzite structure33; edge sharing, e.g., in the rock-salt structure35; or face sharing, e.g., in some perovskites36. Thus, PSRO cannot be expected for arbitrary compositions x in A1−xBx. For example, in the corner-sharing wurtzite (GaN)1−x(ZnO)x alloy, octet-rule-breaking motifs, such as O–Zn3Ga1, are required to connect the octet-rule-conserving motifs4. However, we here address the question whether, depending on the topology of the lattice, PSRO can occur at certain “magic” compositions. It is important to mention that we are interested in short-range-ordered and three-dimensionally mixed phases, but not phase separated or layered ZnSnN2/ZnO heterostructures.

A cluster of the ZTNO wurtzite crystal structure, highlighting the anion-centered tetrahedral motifs and the motif–motif coordination with associated interaction strength parameters: εNN, εNO, and εOO.
The dual-sublattice mixed alloy ZTNO serves as a good proof-of-concept example for the problem. ZnSnN2 and related ternary nitrides are of potential technological relevance, receiving increasing interest, e.g., for photovoltaics37,38, solar water splitting39, and solid-state lighting40,41. Considerable efforts have recently been devoted to the effects of defects31,42,43 and disorder44,45,46. However, most samples are not pure nitrides and contain considerable amounts of unintentional oxygen. We have shown that intentional tuning of the Zn/Sn and the correlated O/N stoichiometry can be used to control the doping behavior31,47. In this paper, we demonstrate that perfectly short-range ordered but long-range disordered ZTNO solid solutions can occur at a “magic” composition x = 0.25. We found that short-range order creates a singularity in enthalpy and materials properties that lies outside of smooth interpolations, such as the regular solid solution and band-gap bowing models. Even more importantly, the perfect SRO completely removes electronic localization effects that can otherwise be prominent in disordered semiconductor alloys28,48. Thus, (ZnSnN2)0.75(ZnO)0.5 is a compound that combines the features of line compounds and solid solutions, and it illustrates a novel way of how atomic order can be utilized for materials design.
Results
Thermodynamics
We extended the quasi-chemical approach in the regular solid-solution model30 to study the thermodynamics of ZTNO solid solutions. Rather than atoms in binary alloys, the building blocks are now formed by the local motif structures. Statistically, this treatment separates the degrees of freedom into the SRO (first shell) and long-range order (LRO, beyond the first shell) parts in the partition function as Z = ZSROZLRO. Here, ZSRO depends on the internal degrees of freedom in the local structure and ZLRO relates to the arrangement of motifs. The mixing enthalpy (ΔHmix) is then expressed by this motif-based regular solid-solution model as
with ΔHSRO as defined in Eq. (1). The term ΔHLRO = Ωx(1 − x) describes the LRO contribution to the mixing enthalpy assuming a random motif distribution. As illustrated in Fig. 1, ΔHLRO is controlled by the arrangement of the anion-centered motifs, i.e., ZLRO. The motif-interaction parameter can be expressed as Ω = zΔε, where z is the anion–anion coordination number (z = 12 in the wurtzite lattice). \(\Delta \varepsilon = \varepsilon _{{\mathrm{NO}}} - \frac{1}{2}(\varepsilon _{{\mathrm{NN}}} + \varepsilon _{{\mathrm{OO}}})\) denotes the differences in motif–motif interactions, in analogy to the bond energies in binary alloys30.
The formulation of Eq. (2) allows us to describe the non-random short-range ordered but long-range disordered ZTNO solid solution. To obtain atomic structure models with realistic degrees of SRO, we performed Monte-Carlo (MC) simulations at fixed compositions using the motif Hamiltonian, i.e., Eq. (1), in 128-atom ZTNO supercells. Random seeds were equilibrated at 5000 K and then cooled at a rate of 6000 MC steps per K. After equilibration at 700 K, a typical growth temperature for thin-film deposition47, the structures are relaxed by statistically minimizing the number of octet-rule-breaking motifs, resulting in a minimized ΔHSRO31. The remaining octet-rule-breaking motifs are due to geometric frustration in non-magic compositions and cannot be removed by further lowering the MC temperature (It should be noted that thin-film growth can result in non-equilibrium disorder described by a higher “effective temperature,” reflecting disorder due to limited kinetics49.). For MC equilibrated structures at 700 K, the energies and corresponding mixing enthalpies ΔHmix were calculated from density functional theory (DFT) calculations50,51,52.
Figure 2a shows the DFT-calculated mixing enthalpy of the MC equilibrated structures as a function of composition. Most data points follow the expected quadratic dependence of x, except for the singular points at x = 0, 0.25, and 0.5. For these compositions, we observe PSRO, i.e., only the octet-rule-conserving motifs occur. At x = 0, the PSRO is trivially fulfilled for ZnSnN2 with its ground-state crystal structure (space group 33, Pna21); but for x = 0.25 and 0.5, these are non-trivial results from the MC simulation. We then fit ΔHmix of the non-PSRO structures to Eq. (2) and obtain ΔHSRO = 12.9 ± 0.9 meV/motif and Ω = 113.8 ± 6.3 meV/motif. Notably, we find for the non-PSRO structures that the contribution from ΔHSRO is in good approximation a constant across the composition range 0 < x < 0.5 (see Supplementary Fig. 1), indicating a more or less constant fraction of residual non-ideal motifs. At the “magic” compositions, the MC optimization removes all octet-rule-breaking motifs and ΔHSRO = 0. We further performed an exhaustive search over the full composition range in a 64-atom (2 × 2 × 1) cell and confirmed that perfect SRO structures occur only at x = 0.25 and 0.5, besides the trivial cases of x = 0 and 1. We note that unlike in other cases of solid solutions and line compounds that show symmetry with respect to composition, e.g., the Ni3Al and NiAl3 phases in the Ni–Al system53, this symmetry is here broken due to alloying of a ternary (ZnSnN2) and a binary (ZnO) phase. Indeed, the exhaustive search did not reveal a PSRO phase at x = 0.75 on the oxygen-rich side of the composition range. As seen in Fig. 2b, the case of x = 0.5 is a layered superlattice formed by N–Zn2N2 and O–Zn4Sn0 motifs. While such superlattices could accommodate in principle all compositions via variation of the layer thickness and periodicity, we are here interested in the case where the two motif types are three-dimensionally mixed, which was found only for the PSRO phase at x = 0.25. This phase also showed a larger number of configurational representations (see Supplementary Table 1), suggesting a considerable residual entropy from long-range disorder (Exemplary PSRO structure files are available in the Supplementary data 1 to 4).

a DFT-calculated mixing enthalpy (ΔHmix) as a function of composition x for MC structures equilibrated at T = 700 K. Non-PSRO structures are shown as purple squares and PSRO structures as green squares. The black dotted line shows the fitted extended regular solid-solution model, i.e., Eq. (2), and the shaded area denotes the 3σ prediction band resulting from the fit. b Examples of PSRO ZTNO structures at x = 0.25 and 0.5. c ΔG as a function of temperature at x = 0.25 shows three regimes: phase separation (SEP), PSRO phases, and random phases.
We can attribute the emergence of this phase at a specific composition to crystallographic “frustration” due to the connectivity of the motif structure within the topology of the underlying wurtzite lattice. For example, in any given octet-rule conserving structure (e.g., x = 0 or 1), adding a single charge-conserving (2ON + ZnSn) or (2NO + SnZn) unit immediately creates numerous octet-rule violating motifs in the neighborhood. In order to maintain the octet-rule conserving structure, a minimum concentration of such additions is required in conjunction with a global rearrangement of the ionic distribution on the wurtzite lattice. A comparable phenomenon has led to the identification of extended anti-site defects in chalcogenides54. The interest in the PSRO phase at x = 0.25 is further motivated from an electronic structure perspective, since either superlattices or O rich compositions are expected to cause charge localization effects that deteriorate electronic properties. Indeed, it is now well established that more dilute N concentrations in ZnO cause defect-like mid-gap states55,56.
The singularity in ΔHmix(x) at x = 0.25 (Fig. 2a) resembles the situation in a line compound, where a large enthalpy reduction results from crystallization into a crystal structure that accommodates the respective stoichiometry57. Despite the large reduction, ΔHmix remains positive, implying the phase separation into ZnSnN2 and ZnO at low temperatures. However, at finite temperatures, the minimization of Gibbs free energy (ΔG = ΔH − TΔS) defines the thermodynamic equilibrium of the NpT ensemble. For the PSRO structure to form, there are two critical temperatures: TSS, below which the material tends to be phase-separated, and TSR, above which random structures are thermodynamically preferred. To determine these transitions, we determined ΔG for the fully random alloy (no SRO) using ΔHmix(0.25) = 149 meV/motif calculated from DFT and the entropy for random mixing on both sublattices, ΔS ≈ 1.16 kB/motif. For estimating the entropy of the PSRO structure, the exact enumeration of configurations becomes impractical at the 128-atom supercell level. We also noticed some well-known methods such as Pauling’s technique58,59 are not practical due to the additional complication from two types of tetrahedra, i.e., N–Zn2Sn2 and O–Zn4. Instead, we use the N–O two-center 20-motif cluster (Fig. 1) as a model to estimate the entropy from long-range disorder in the presence of PSRO. We enumerate all configurations observing the octet rule while varying nO and nN (see Supplementary Fig. 2). A maximum entropy of ΔSSRO ≈ 8.44 kB is obtained for the cluster at nO/nN = 3:9, which locally satisfies the composition x = 0.25. The corresponding entropy ΔSSRO ≈ 0.42 kB/motif is considerably smaller than the random entropy. In addition, it is worth noting that the enumeration of configurations from small clusters underestimates the total configurational entropy60. However, it is sufficient to stabilize the PSRO (ZnSnN2)0.75(ZnO)0.5 alloy at finite temperatures, given the substantial enthalpy reduction relative to non-PSRO structures. Figure 2c shows the stability of the PSRO phase relative to the phase-separated (T < Tss) and fully random (T > TSR) structures, with a stability range between about 500 and 2100 K. The stabilization temperature of the fully random structure is close to the “effective temperature” that has been inferred in thin-film growth28,47,49. Therefore, suitable post-deposition treatments may be necessary in practice to stabilize the PSRO structure at the magic composition.
Electronic structure
The band gap is often sensitive to atomic ordering and distribution3. Because the MC relaxed supercells have 128 atoms, direct GW calculations61 as performed for the primitive cell of ordered ZnSnN2 become impractical28. Thus, we used a simplified single-shot hybrid+U (SSH+U) functional to calculate the band structures. The parameters in the SSH+U functional were tuned to reproduce the GW results at a small cell level28,31. The calculated band gaps as a function of composition x are shown in Fig. 3. We observe that the PSRO structures at x = 0.25 behave very differently from non-PSRO structures, with the band gap falling outside the 3σ band of the band gap bowing model fitted to non-PSRO structures
where we obtained a = 1.41 ± 0.07 eV and the bowing parameter b = 1.40 ± 0.14 eV. \(E_{\mathrm{g}}^{{\mathrm{ZnO}}}\) is the wurtzite ZnO band gap (3.4 eV). Because we are fitting the band gaps of the non-PSRO, we include the x = 0 limit as a free parameter a, instead of using the gap of the ordered ZnSnN2 phase. Similar to the behavior in the mixing enthalpy, the band gap shows a singularity at x = 0.25, peaking at Eg = 1.87 eV, outside of the 3σ band. By eliminating high Zn-coordinated N motifs, such as N–Zn3Sn1, the PSRO structures experience less hybridization between N-p and Zn-d orbitals. This causes a lower valence band maximum (VBM) and an increased band gap. The band gap extrapolated to x = 0 is close to the gap of pure ZnSnN2. This observation reflects the fact that even though the Zn-rich motifs cause an upward bowing of the VBM, they do not cause a defect state inside the gap at low concentrations28. At x = 0.5, ZTNO forms a ZnSnN2/ZnO superlattice structure, i.e., Fig. 2b. In superlattices, the band gap is somewhat ill-defined because it is formed by two subsystems (see Supplementary Fig. 3).

Electronic band gap (Eg) as a function of composition (x): the black dotted line represents fitting according to the band gap bowing model between disordered ZnSnN2 and ZnO. The shaded area denotes the 3σ prediction band from the fitting. The green rectangles are the band gaps of PSRO structures.
Charge localization
Electronic charge localization effects are often prominent in disordered semiconductors and are technologically relevant because they can impede carrier transport18,28. To evaluate the effect of SRO on carrier localization, we calculated the inverse participation ratio (IPR) from the electronic density of states (DOS)28,
where pi(E) is the local density of states (LDOS) projected on each atom i as a function of energy E, and NA is the total number of atoms in the cell. The IPR describes the energy-resolved atomic localization ratio of the DOS, with IPR = 1 for perfect delocalization (all LDOS are equal). IPR increases with localization. Figure 4 shows the IPR results for pure ZnSnN2, the PSRO structures at x = 0.25, and non-PSRO structures at x = 0.25 with different degrees of SRO (ΔHSRO). We observe that the IPR is close to 1 for ordered ZnSnN2, but shows stronger localization effects near the VBM with increasing fraction of Zn-rich motifs. Remarkably, the PSRO structures exhibit no localization effects, with an IPR spectrum closely resembling the ordered ZnSnN2 (one representative PSRO structure is shown in Fig. 4, but others are very similar). This finding highlights the fact that the perfectly short-range-ordered ZTNO phase is electronically pristine, despite the presence of long-range disorder.

The calculated inverse participation ratio (IPR) for PSRO and non-PSRO ZTNO structures at x = 0.25 with different degree of SRO (ΔHSRO, meV/motif). Note that IPR = 1 stands for an ideal charge-delocalized situation and the offsets of each spectrum are for graphical clarity. ni,j denotes the fractions of octet-rule-breaking N–ZniSnj motifs. The IPR of the ordered ZnSnN2 is included for comparison.
Discussion
We described a hitherto unrecognized solid-state material phase borne out of a disordered solid solution but having ordered line-compound features. The development of perfect short-range order in the solid solution is responsible for this interesting physical phenomenon. We performed first-principles and Monte-Carlo calculations for the dual-sublattice mixed semiconductor alloy (ZnSnN2)1−x(ZnO)2x. At a magic composition x = 0.25, we obtained this special solid-solution phase, which is predicted to be stable in an experimentally accessible temperature window. Singularities in the mixing enthalpy and the band gap highlight the special character of this phase from both thermodynamic and electronic-structure perspectives. More importantly, charge localization disappears in this unique solid-solution phase, which signals a superior carrier transport. We envision that this work will enable the search for new materials, such as superconductors, thermoelectrics, where the properties are not negatively affected by disorder. This work about SRO can also be connected to many emergent areas in the materials research community, such as poly-cation/-anion materials and high-entropy oxides.
Methods
First-principles total-energy calculation
The supercells of ZTNO structures were relaxed using the Vienna ab initio simulation package (VASP) with the projector-augmented-wave implementations for DFT50. The ground-state energies of a 2 × 2 × 2 supercell were calculated with a cutoff energy of 380 eV for the plane-wave basis set and in a 2 × 2 × 2 Gamma k-point mesh (or similar k-point density for smaller cell size). The reference energies of ZnSnN2 and ZnO were calculated from ordered orthorhombic ZnSnN2 (SG 33, Pna21) and wurtzite ZnO (SG 186, P63mc) The convergence criteria is that the total energy is smaller than 10−5 eV/supercell for the electronic steps and the total force on each atom is smaller than 0.02 eV Å−1. The generalized-gradient-approximation in the Perdew–Burke–Ernzerhof flavor was used for the electron exchange and correlation52. Due to the fact that the valence band top is affected by the Zn-d shell, a Coulomb potential (U − J = 6 eV) was applied to Zn-d orbit with the Dudarev approach51. The choice of the U − J value is consistent with the literature31,55.
Electronic structure calculation
In the SSH+U approach, we calculate the diagonal matrix elements for the DFT+U wavefunctions without diagonalization. Formally, this is analogous to the common single-shot GW approach, when replacing the self-energy by the scaled nonlocal Fock potential plus the on-site potential U. Expressing the composition as Zn1+xSn1−xN2−2xO2x, fitting to GW reference calculations yields the hybrid functional scaling factor α = 0.144 − 0.052x and the composition independent U − J = 4.2 eV31. The latter value is somewhat smaller than in DFT+U, because part of the on-site Coulomb interaction is already corrected by the Fock potential.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
References
Shevlin, S. A., Curioni, A. & Andreoni, W. Ab initio design of high-k dielectrics: LaxY1-xAlO3. Phys. Rev. Lett. 94, 146401 (2005).
Stevanović, V., Zakutayev, A. & Lany, S. Composition dependence of the band gap and doping in Cu2O-based alloys as predicted by an extension of the dilute-defect model. Phys. Rev. Appl. 2, 044005 (2014).
Mäder, K. A. & Zunger, A. Short- and long-range-order effects on the electronic properties of III–V semiconductor alloys. Phys. Rev. B 51, 10462–10476 (1995).
Li, L., Muckerman, J. T., Hybertsen, M. S. & Allen, P. B. Phase diagram, structure, and electronic properties of (Ga1−xZnx)(N1−xOx) solid solutions from DFT-based simulations. Phys. Rev. B 83, 134202 (2011).
Li, X. & Lu, K. Improving sustainability with simpler alloys. Science 364, 733–734 (2019).
Pei, Y. et al. Convergence of electronic bands for high performance bulk thermoelectrics. Nature 473, 66–69 (2011).
Ohno, S. et al. Phase boundary mapping to obtain n-type Mg3Sb2-based thermoelectrics. Joule 2, 141–154 (2018).
Liu, W. et al. Convergence of conduction bands as a means of enhancing thermoelectric performance of n-type Mg2Si1−xSnx solid solutions. Phys. Rev. Lett. 108, 166601 (2012).
Maeda, K. et al. GaN:ZnO solid solution as a photocatalyst for visible-light-driven overall water splitting. J. Am. Chem. Soc. 127, 8286–8287 (2005).
Yang, C. et al. Epitaxy of (GaN)1−x(ZnO)x solid-solution thin films with widely tunable chemical composition and strong visible absorption. Phys. Rev. Appl. 10, 044001 (2018).
Huang, B., Yoon, M., Sumpter, B. G., Wei, S.-H. & Liu, F. Alloy engineering of defect properties in semiconductors: suppression of deep levels in transition-metal dichalcogenides. Phys. Rev. Lett. 115, 126806 (2015).
Huang, H., Jin, K.-H. & Liu, F. Alloy engineering of topological semimetal phase transition in MgTa2−xNbxN3. Phys. Rev. Lett. 120, 136403 (2018).
DeHoff, R. Thermodynamics in Materials Science (CRC Press, 2006).
Stringfellow, G. B. Microstructures produced during the epitaxial growth of InGaN alloys. J. Cryst. Growth 312, 735–749 (2010).
A line compound is defined as an ordered stoichiometric phase corresponding to a “line” in the phase diagram in the low temperature limit. Notwithstanding this definition, line compounds are generally subject to the formation of point defects, causing a phase width in composition7 and disorder at elevated temperatures.
Anderson, P. W. Absence of diffusion in certain random lattices. Phys. Rev. 109, 1492–1505 (1958).
Wei, S.-H., Ferreira, L. G., Bernard, J. E. & Zunger, A. Electronic properties of random alloys: special quasirandom structures. Phys. Rev. B 42, 9622–9649 (1990).
Kim, K. & Zunger, A. Spatial correlations in GaInAsN alloys and their effects on band-gap enhancement and electron localization. Phys. Rev. Lett. 86, 2609–2612 (2001).
Albrecht, M. et al. Compositional correlation and anticorrelation in quaternary alloys: competition between bulk thermodynamics and surface kinetics. Phys. Rev. Lett. 99, 206103 (2007).
Cava, R. J. et al. Structural anomalies, oxygen ordering and superconductivity in oxygen deficient Ba2YCu3Ox. Physica C 165, 419–433 (1990).
Campi, G. et al. Inhomogeneity of charge-density-wave order and quenched disorder in a high-Tc superconductor. Nature 525, 359–362 (2015).
Hawthorne, F. C. Short-range atomic arrangements in minerals I, the minerals of the amphibole, tourmaline and pyroxene supergroups. Eur. J. Mineral. 28, 513–536 (2016).
Ma, J., Deng, H.-X., Luo, J.-W. & Wei, S.-H. Origin of the failed ensemble average rule for the band gaps of disordered nonisovalent semiconductor alloys. Phys. Rev. B 90, 115201 (2014).
Biswas, K. & Lany, S. Energetics of quaternary III–V alloys described by incorporation and clustering of impurities. Phys. Rev. B 80, 115206 (2009).
Liu, J., Fernández-Serra, M. V. & Allen, P. B. Special quasiordered structures: role of short-range order in the semiconductor alloy (GaN)1−x(ZnO)x. Phys. Rev. B 93, 054207 (2016).
Brown, I. D. Recent developments in the methods and applications of the bond valence model. Chem. Rev. 109, 6858–6919 (2009).
Chen, D. P., Neuefeind, J. C., Koczkur, K. M., Bish, D. L. & Skrabalak, S. E. Role of short-range chemical ordering in (GaN)1−x(ZnO)x for photodriven oxygen evolution. Chem. Mater. 29, 6525–6535 (2017).
Lany, S. et al. Monte carlo simulations of disorder in ZnSnN2 and the effects on the electronic structure. Phys. Rev. Mater. 1, 035401 (2017).
Zawadzki, P., Zakutayev, A. & Lany, S. Entropy-driven clustering in tetrahedrally bonded multinary materials. Phys. Rev. Appl. 3, 034007 (2015).
Porter, D., Easterling, K. & Sherif, M. Phase Transformations in Metals and Alloys (CRC Press, 2009).
Pan, J. et al. Interplay between composition, electronic structure, disorder, and doping due to dual sublattice mixing in nonequilibrium synthesis of ZnSnN2:O. Adv. Mater. 31, 1807406 (2019).
Bernard, J. E. & Zunger, A. Ordered-vacancy-compound semiconductors: pseudocubic CdIn2Se4. Phys. Rev. B 37, 6835–6856 (1988).
Quayle, P. C. et al. Charge-neutral disorder and polytypes in heterovalent wurtzite-based ternary semiconductors: the importance of the octet rule. Phys. Rev. B 91, 205207 (2015).
Park, J.-S. et al. Ordering-induced direct-to-indirect band gap transition in multication semiconductor compounds. Phys. Rev. B 91, 075204 (2015).
Schrön, A., Rödl, C. & Bechstedt, F. Energetic stability and magnetic properties of MnO in the rocksalt, wurtzite, and zinc-blende structures: Influence of exchange and correlation. Phys. Rev. B 82, 165109 (2010).
Søndenå, R., Stølen, S., Ravindran, P., Grande, T. & Allan, N. L. Corner- versus face-sharing octahedra in AMnO3 perovskites (A = Ca, Sr, and Ba). Phys. Rev. B 75, 184105 (2007).
Lahourcade, L. et al. Structural and optoelectronic characterization of RF sputtered ZnSnN2. Adv. Mater. 25, 2562–2566 (2013).
Qin, R. et al. Semiconducting ZnSnN2 thin films for Si/ZnSnN2 p-n junctions. Appl. Phys. Lett. 108, 142104 (2016).
Fang, D. Q., Chen, X., Gao, P. F., Zhang, Y. & Zhang, S. L. Mono- and bilayer ZnSnN2 sheets for visible-light photocatalysis: first-principles predictions. J. Phys. Chem. C 121, 26063–26068 (2017).
Karim, M. R. & Zhao, H. Design of InGaN–ZnSnN2 quantum wells for high-efficiency amber light emitting diodes. J. Appl. Phys. 124, 034303 (2018).
Han, L., Kash, K. & Zhao, H. Designs of blue and green light-emitting diodes based on type-type-II InGaN–ZnGeN2 quantum wells. J. Appl. Phys. 120, 103102 (2016).
Tsunoda, N., Kumagai, Y., Takahashi, A. & Oba, F. Electrically benign defect behavior in zinc tin nitride revealed from first principles. Phys. Rev. Appl. 10, 011001 (2018).
Adamski, N. L., Zhu, Z., Wickramaratne, D. & Van de Walle, C. G. Strategies for p-type doping of ZnGeN2. Appl. Phys. Lett. 114, 032101 (2019).
Jayatunga, B. H. D., Lyu, S., Radha, S. K., Kash, K. & Lambrecht, W. R. L. Ordering in the mixed ZnGeN2-GaN alloy system: crystal structures and band structures of ZnGeGa2N4 from first principles. Phys. Rev. Mater. 2, 114602 (2018).
Veal, T. D. et al. Band gap dependence on cation disorder in ZnSnN2 solar absorber. Adv. Energy Mater. 5, 1501462 (2015).
Makin, R. A. et al. Order parameter and band gap of ZnSnN2. In Proc. 2018 IEEE 7th World Conference on Photovoltaic Energy Conversion (WCPEC), 3865–3868 (2018).
Fioretti, A. N. et al. Exciton photoluminescence and benign defect complex formation in zinc tin nitride. Mater. Horiz. 5, 823–830 (2018).
Yang, T.-J., Shivaraman, R., Speck, J. S. & Wu, Y.-R. The influence of random indium alloy fluctuations in indium gallium nitride quantum wells on the device behavior. J. Appl. Phys. 116, 113104 (2014).
Ndione, P. F. et al. Control of the electrical properties in spinel oxides by manipulating the cation disorder. Adv. Funct. Mater. 24, 610–618 (2014).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Khina, B. B. Modeling nonisothermal interaction kinetics in the condensed state: a diagram of phase formation mechanisms for the Ni–Al system. J. Appl. Phys. 101, 063510 (2007).
Zawadzki, P., Zakutayev, A. & Lany, S. Extended antisite defects in tetrahedrally bonded semiconductors. Phys. Rev. B 92, 201204(R) (2015).
Lany, S. & Zunger, A. Generalized Koopmans density functional calculations reveal the deep acceptor state of NO in ZnO. Phys. Rev. B 81, 205209 (2010).
Lyons, J. L., Janotti, A. & Van de Walle, C. G. Why nitrogen cannot lead to p-type conductivity in ZnO. Appl. Phys. Lett. 95, 252105 (2009).
Swalin, R. Thermodynamics of Solids (Wiley, 1972).
Pauling, L. The structure and entropy of ice and of other crystals with some randomness of atomic arrangement. J. Am. Chem. Soc. 57, 2680–2684 (1935).
Anderson, P. W. Ordering and antiferromagnetism in ferrites. Phys. Rev. 102, 1008–1013 (1956).
Li, Y., Lousada, C. M., Soroka, I. L. & Korzhavyi, P. A. Bond network topology and antiferroelectric order in cuprice CuOH. Inorg. Chem. 54, 8969–8977 (2015).
Hedin, L. New method for calculating the one-particle green’s function with application to the electron-gas problem. Phys. Rev. 139, A796–A823 (1965).
Acknowledgements
This work was supported by the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308 with the Alliance for Sustainable Energy, LLC, the manager and operator of the National Renewable Energy Laboratory. The funding was provided by the Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division. S.L. acknowledges support from the Energy Frontier Research Center “Center for Next Generation of Materials Design.” J.C. thanks DOE Office of Science under the Science Undergraduate Laboratory Internship Program (SULI) for financial support in the summer of 2017. This work used high-performance computing resources located at NREL and sponsored by the Office of Energy Efficiency and Renewable Energy. The authors thank Dr. Eric Toberer, Dr. Yue Qi, and Dr. Yong Wan for their constructive suggestions. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. government. The U.S. government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for government purposes.
Author information
Authors and Affiliations
Contributions
J.P. and S.L. conceived the research. J.P. conducted the first-principles calculations, thermodynamics analysis, and electronic structure calculations. J.C. and J.P. prepared the atomic structures through Monte Carlo simulations. S.L. and A.C.T. supervised the work. J.P. and S.L. wrote the manuscript with contributions from all authors. All authors contributed to this work through periodic discussions. All authors have given approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pan, J., Cordell, J.J., Tucker, G.J. et al. Perfect short-range ordered alloy with line-compound-like properties in the ZnSnN2:ZnO system. npj Comput Mater 6, 63 (2020). https://doi.org/10.1038/s41524-020-0331-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-020-0331-8
This article is cited by
-
Defect modeling and control in structurally and compositionally complex materials
Nature Computational Science (2023)
-
Machine learning for alloys
Nature Reviews Materials (2021)
