Validation of Pharmacological Protocols for Targeted Inhibition of Canalicular MRP2 Activity in Hepatocytes Using [99mTc]mebrofenin Imaging in Rats

 , ,
, ,
Abstract
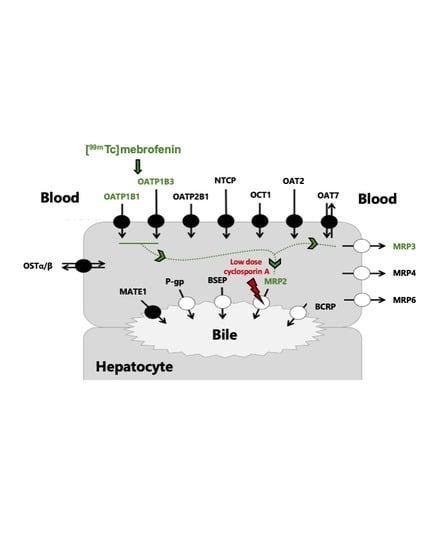
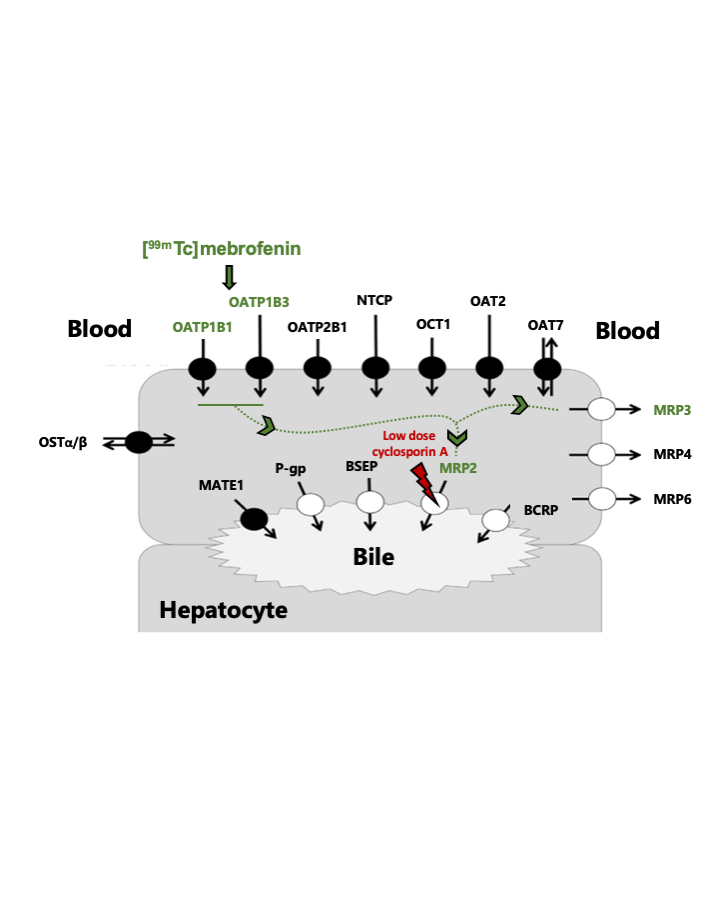
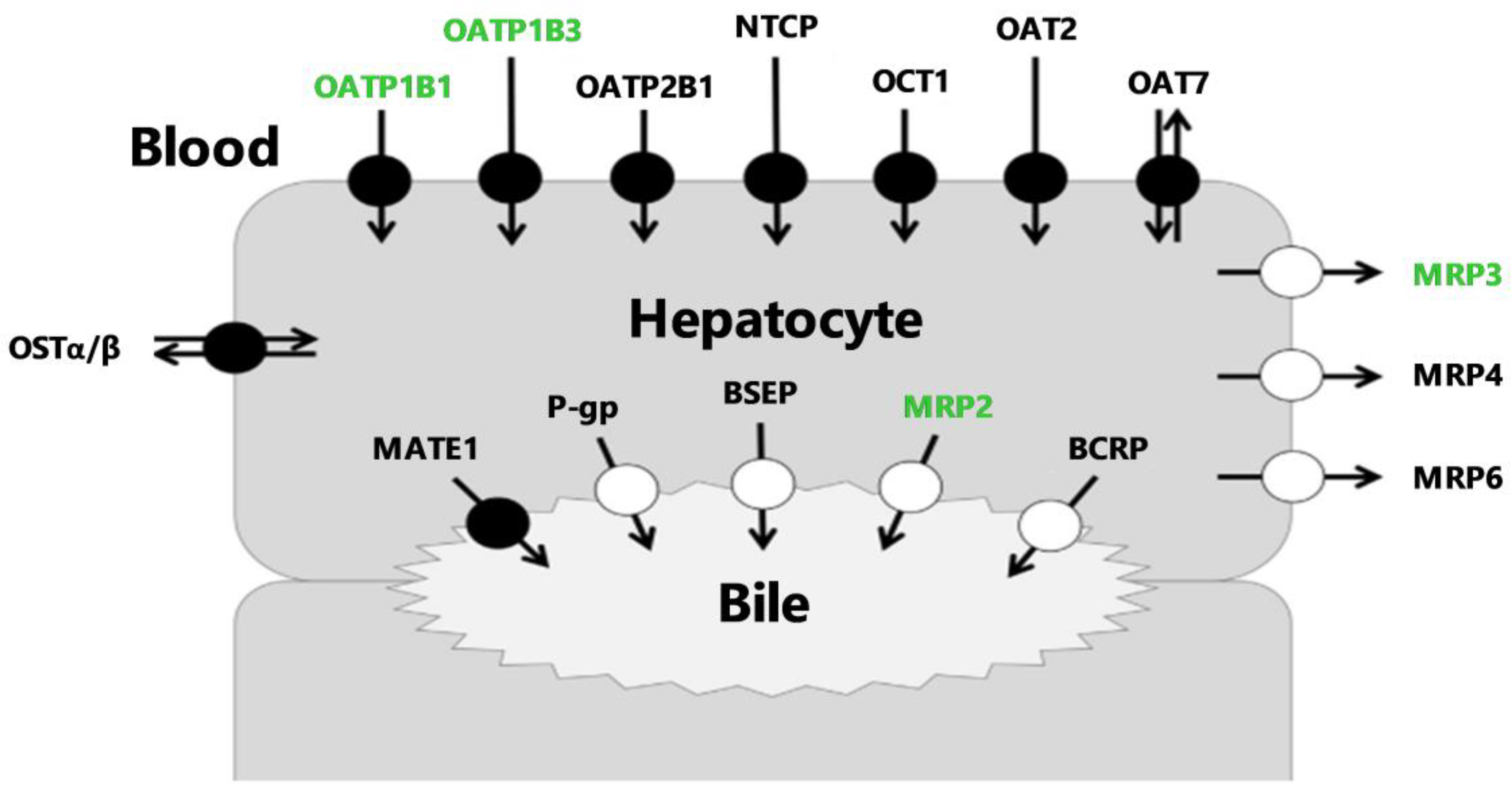
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Radiochemicals
2.2. Animals
2.3. In Vitro Inhibition Assays
2.4. [99mTc]mebrofenin Scintigraphy
2.5. Analysis of [99mTc]mebrofenin Scintigraphy Data
2.6. Pharmacokinetic Modeling of [99mTc]mebrofenin Hepatobiliary Distribution and Elimination
2.7. Assessment of Liver Perfusion with Doppler Ultrasound
2.8. Statistical Analysis
3. Results
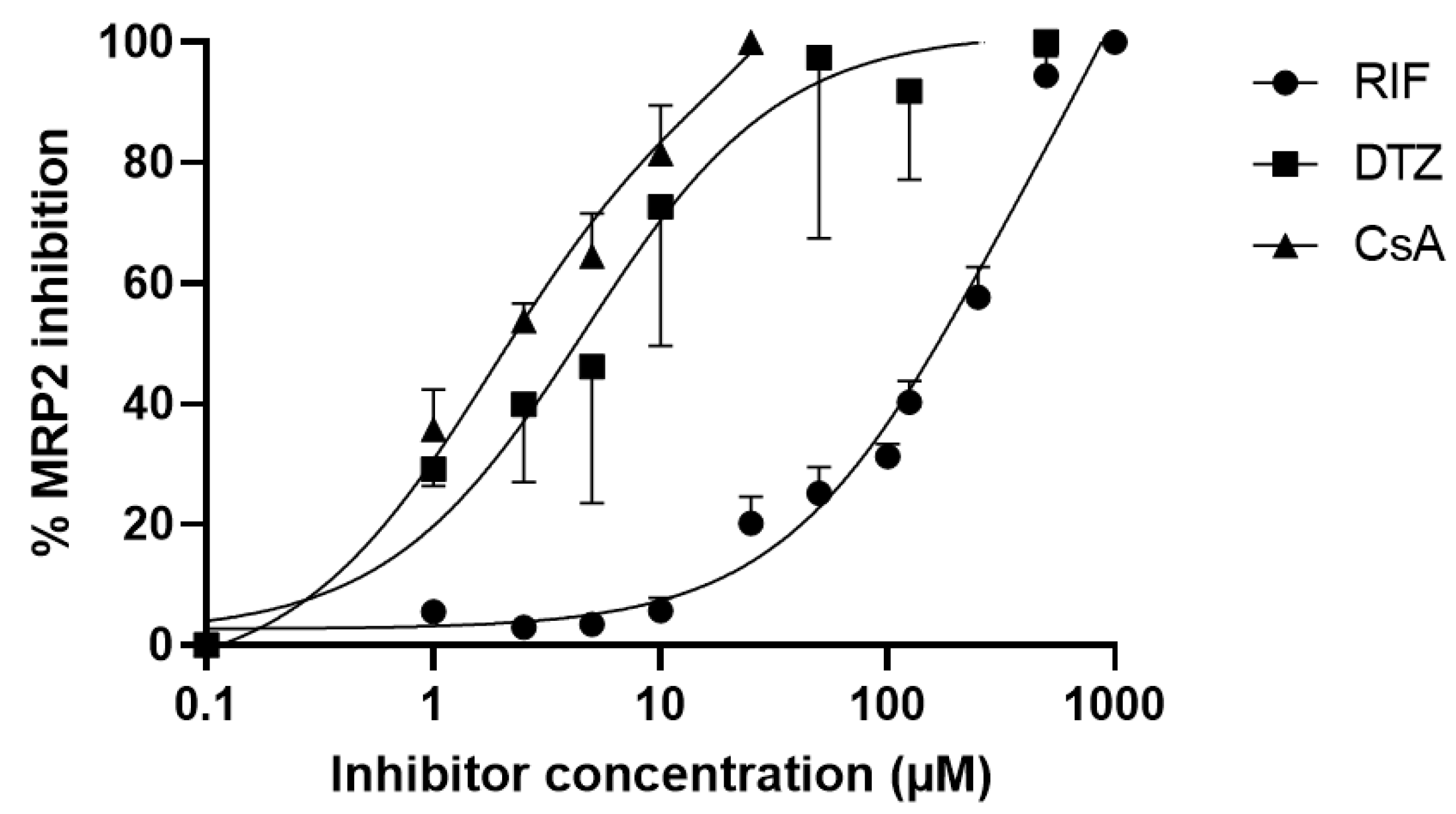
3.1. In Vitro Inhibition Assays
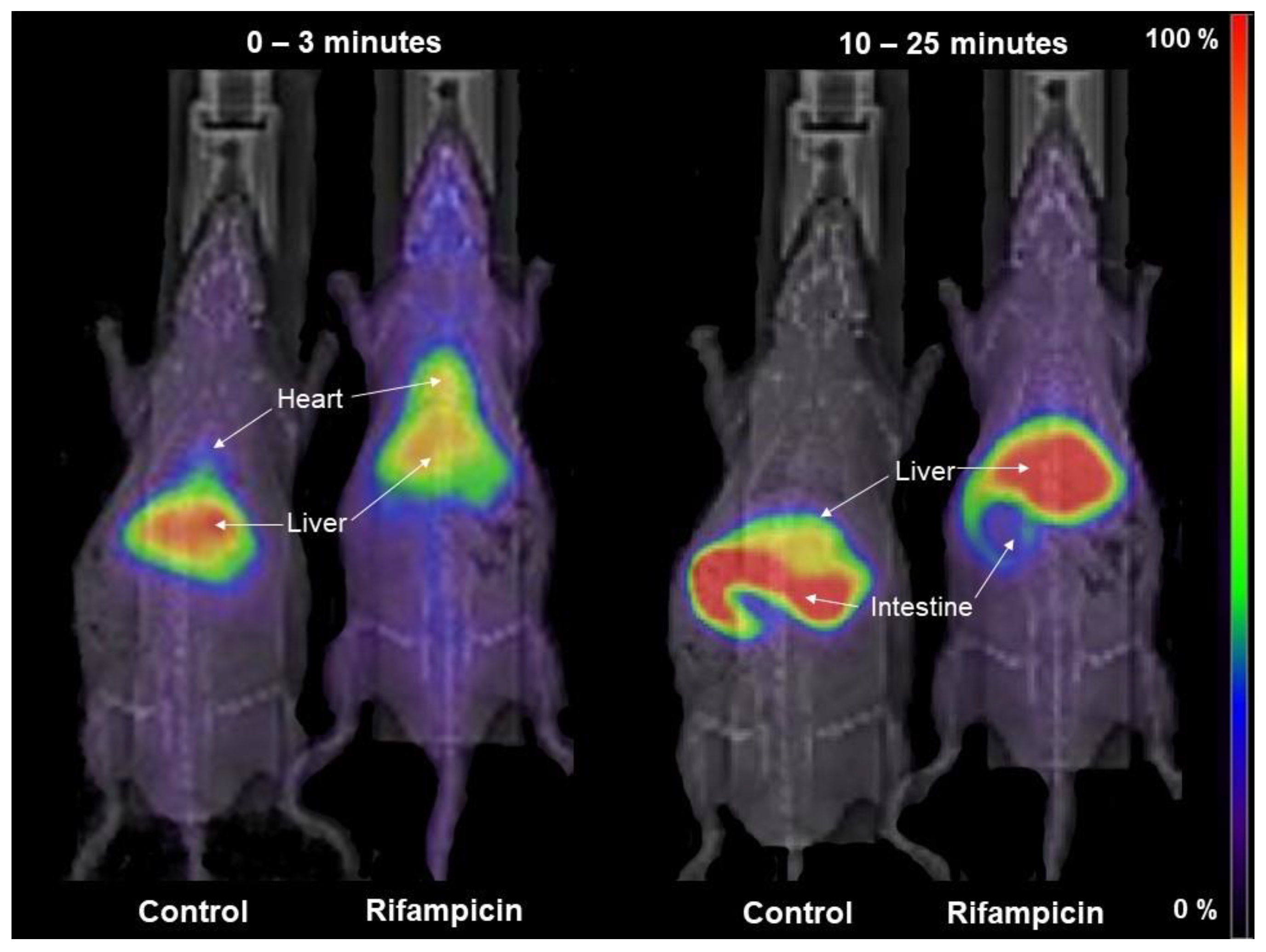
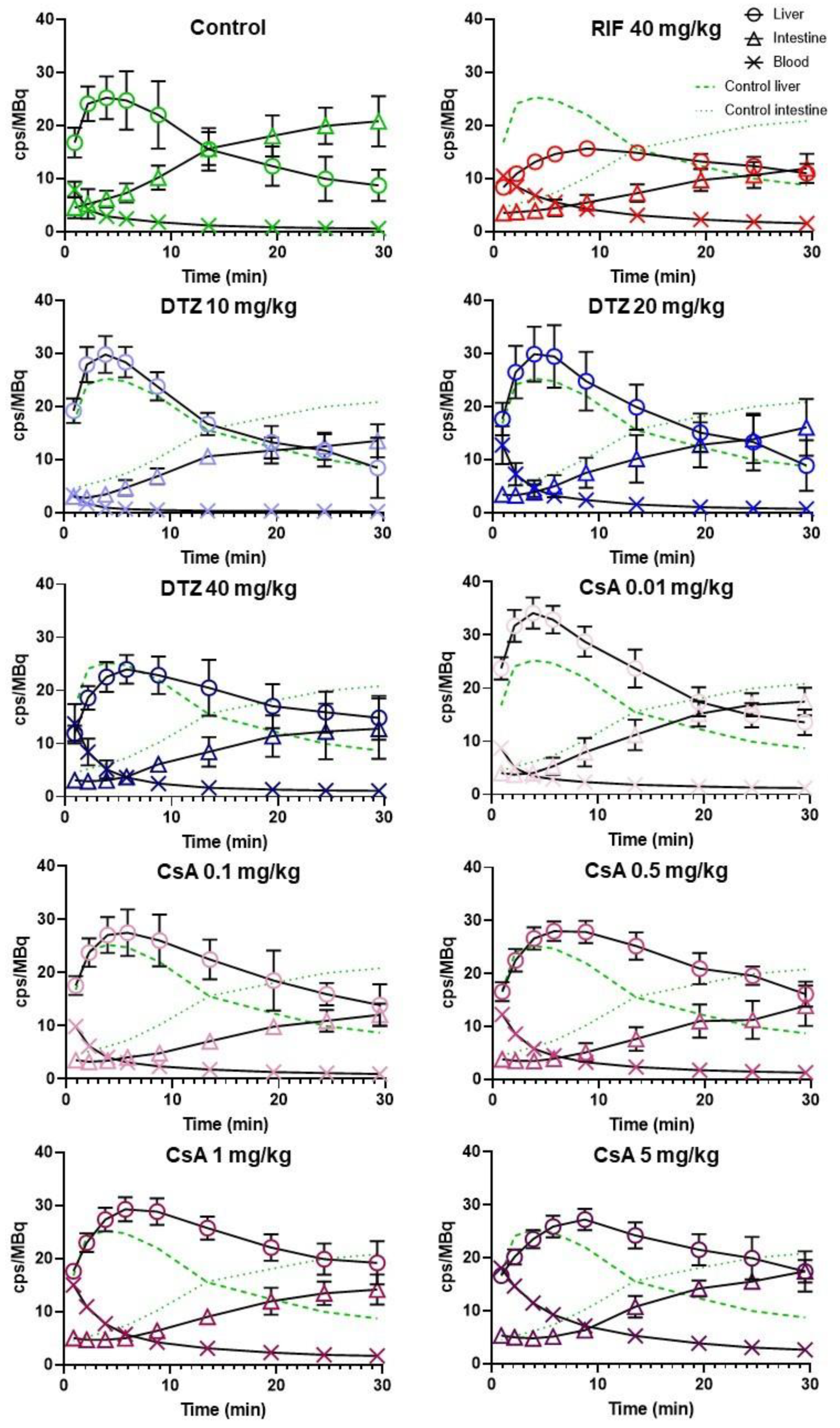
3.2. Impact of Inhibitors on the Kinetics of [99mTc]mebrofenin In Vivo
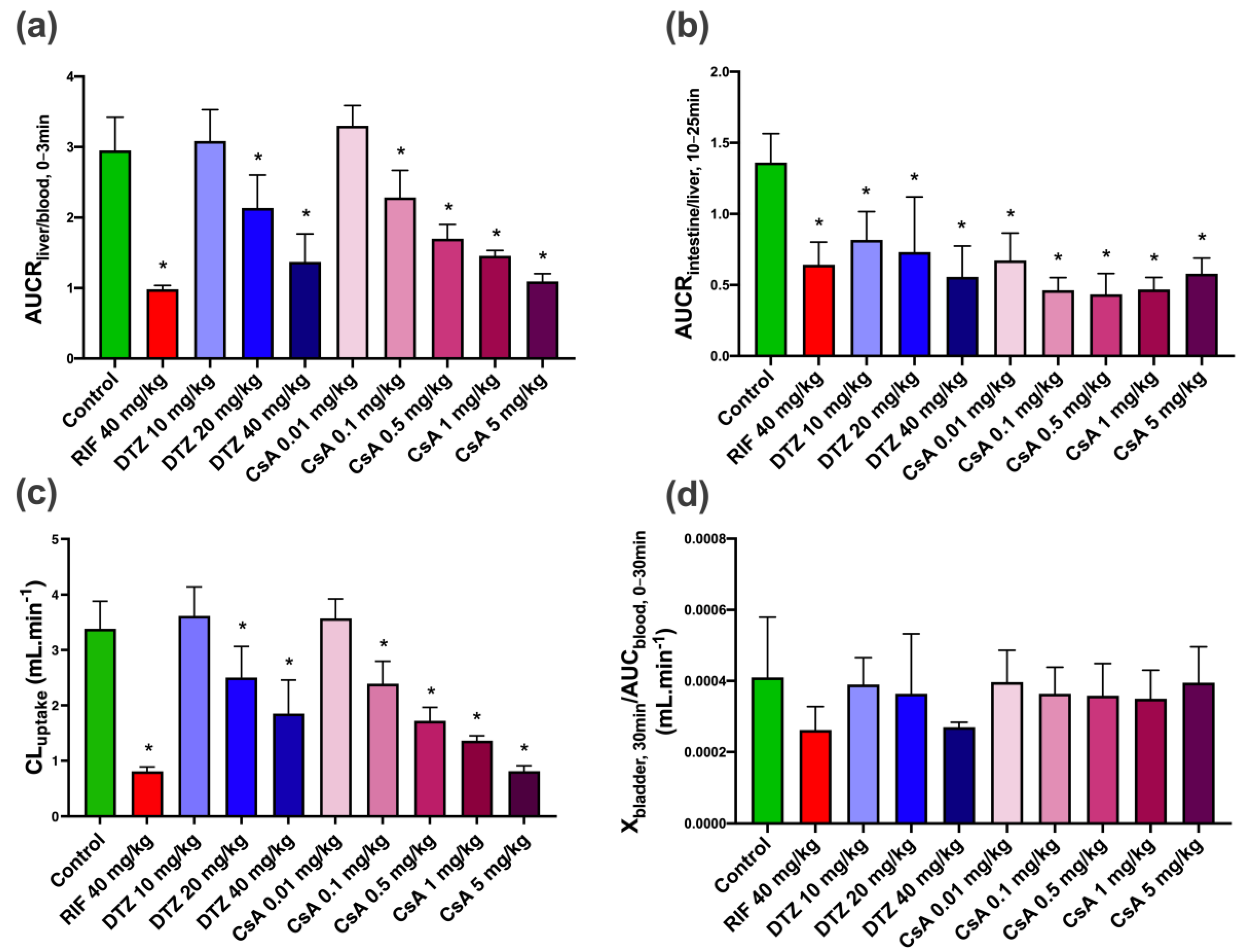
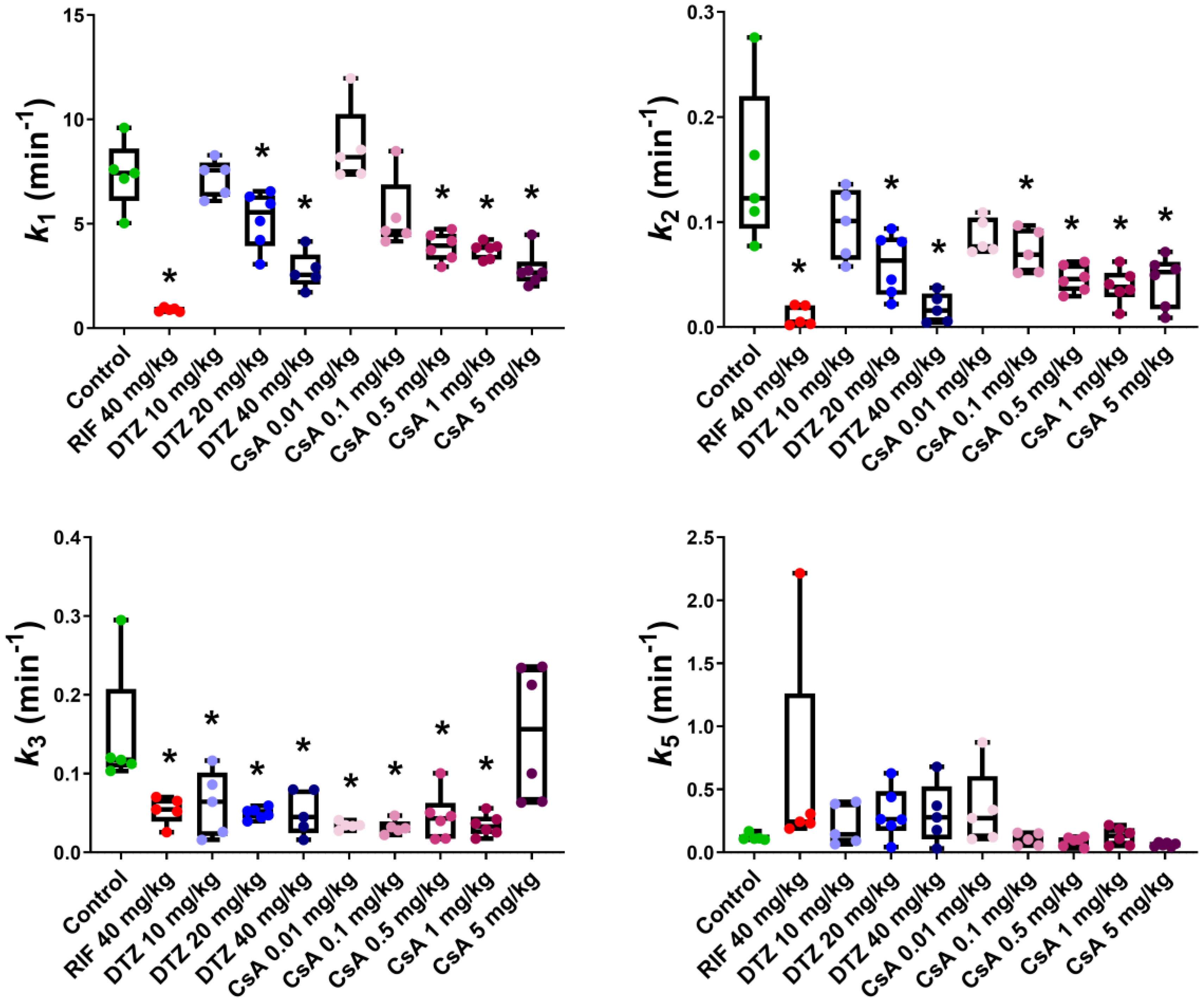
3.3. Pharmacokinetic Modeling
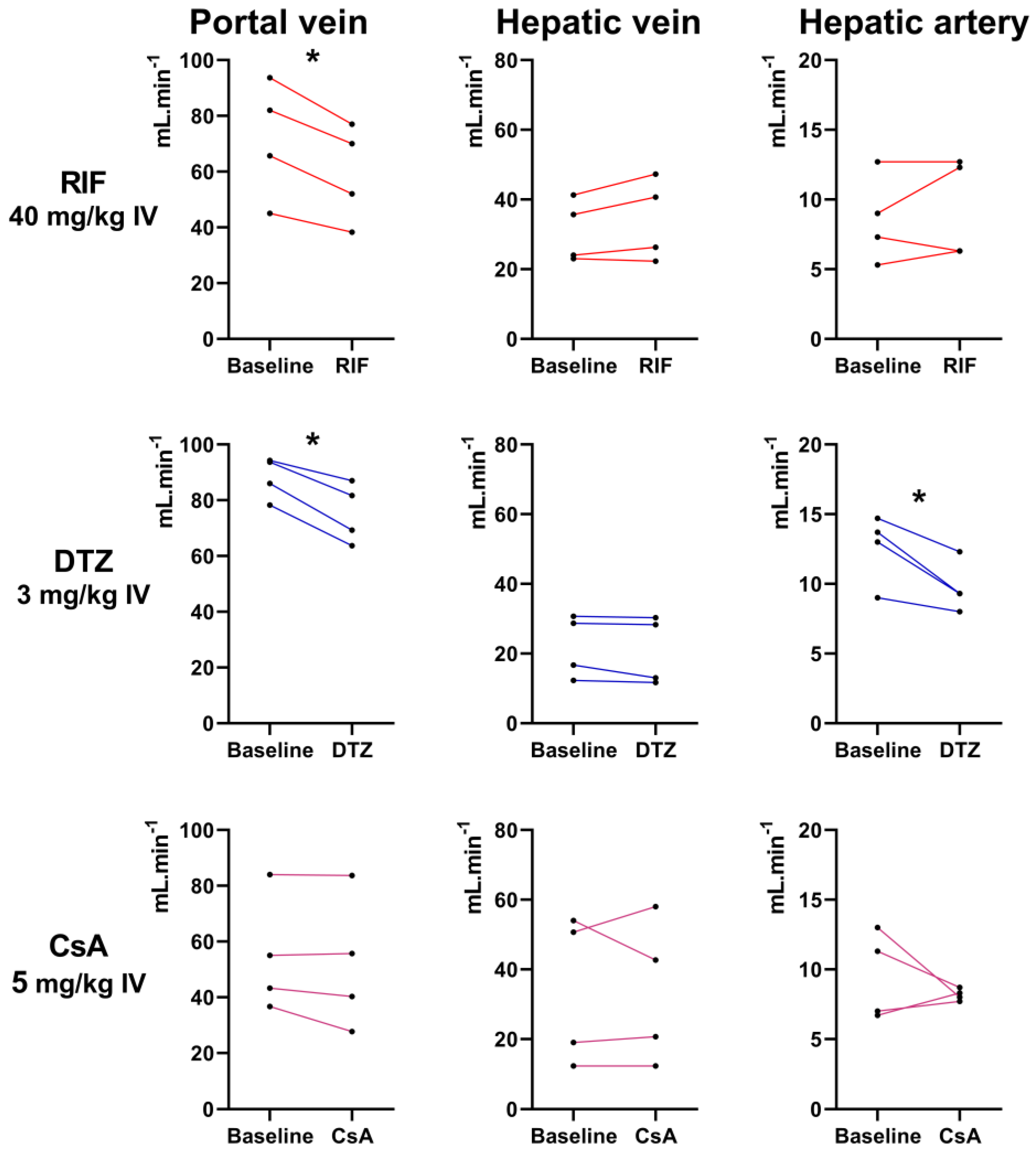
3.4. Assessment of Liver Perfusion with Doppler Ultrasound
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patel, M.; Taskar, K.; Zamek-Gliszczynski, M. Importance of hepatic transporters in clinical disposition of drugs and their metabolites. J. Clin. Pharmacol. 2016, 56, S23–S39. [Google Scholar] [CrossRef] [Green Version]
- Marin, J. Plasma membrane transporters in modern liver pharmacology. Scientifica 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Giacomini, K.; Galetin, A.; Huang, S. The international transporter consortium: Summarizing advances in the role of transporters in drug development. Clin. Pharmacol. Ther. 2018, 104, 766–771. [Google Scholar] [CrossRef]
- Evers, R.; Piquette-Miller, M.; Polli, J.; Russel, F.; Sprowl, J.A.; Tohyama, K.; Ware, J.; de Wildt, S.; Xie, W.; Brouwer, K. Disease-associated changes in drug transporters may impact the pharmacokinetics and/or toxicity of drugs: A white paper from the international transporter consortium. Clin. Pharmacol. Ther. 2018, 104, 900–915. [Google Scholar] [CrossRef]
- Kalliokoski, A.; Niemi, M. Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 2009, 158, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Zamek-Gliszczynski, M.; Taub, M.; Chothe, P.; Chu, X.; Giacomini, K.M.; Kim, R.; Ray, A.; Stocker, S.; Unadkat, J.; Wittwer, M.; et al. Transporters in drug development: 2018 ITC recommendations for transporters of emerging clinical importance. Clin. Pharmacol. Ther. 2018, 104, 890–899. [Google Scholar] [CrossRef]
- Nishino, A.; Kato, Y.; Igarashi, T.; Sugiyama, Y. Both cMOAT/MRP2 and another unknown transporter(s) are responsible for the biliary excretion of glucuronide conjugate of the nonpeptide angiotensin II antagonist, telmisaltan. Drug Metab. Dispos. 2000, 28, 1146–1148. [Google Scholar]
- Matsson, P.; Pedersen, J.; Norinder, U.; Bergström, C.; Artursson, P. Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharm. Res. 2009, 26, 1816–1831. [Google Scholar] [CrossRef]
- Pauli-Magnus, C.; Meier, P. Hepatobiliary transporters and drug-induced cholestasis. Hepatology 2006, 44, 778–787. [Google Scholar] [CrossRef]
- Thakkar, N.; Slizgi, J.; Brouwer, K. Effect of Liver disease on hepatic transporter expression and function. J. Pharm. Sci. 2017, 106, 2282–2294. [Google Scholar] [CrossRef] [Green Version]
- Bonnaventure, P.; Pastor, C. Quantification of drug transport function across the multiple resistance-associated protein 2 (Mrp2) in rat livers. Int. J. Mol. Sci. 2015, 16, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghibellini, G.; Leslie, E.; Brouwer, K. Methods to evaluate biliary excretion of drugs in humans: An updated review. Mol. Pharm. 2006, 3, 198–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández Lozano, I.; Langer, O. Use of imaging to assess the activity of hepatic transporters. Expert Opin. Drug Metab. Toxicol. 2020, 16, 149–164. [Google Scholar] [CrossRef] [Green Version]
- Tournier, N.; Stieger, B.; Langer, O. Imaging techniques to study drug transporter function in vivo. Pharmacol. Ther. 2018, 189, 104–122. [Google Scholar] [CrossRef] [Green Version]
- Loberg, M.; Cooper, M.; Harvey, E.; Callery, P.; Faith, W. Development of new radiopharmaceuticals based on n-substitution of iminodiacetic acid. J. Nucl. Med. 1976, 17, 633–638. [Google Scholar]
- Krishnamurthy, S.; Krishnamurthy, G. Technetium-99m-iminodiacetic acid organic anions: Review of biokinetics and clinical application in hepatology. Hepatology 1989, 9, 139–153. [Google Scholar] [CrossRef]
- Ghibellini, G.; Leslie, E.; Pollack, G.; Brouwer, K. Use of Tc-99m mebrofenin as a clinical probe to assess altered hepatobiliary transport: Integration of in vitro, pharmacokinetic modeling, and simulation studies. Pharm. Res. 2008, 25, 1851. [Google Scholar] [CrossRef] [Green Version]
- Ryan, J.; Cooper, M.; Loberg, M.; Harvey, E.; Sikorski, S. Technetium-99m-Labeled N-(2,6-Dimethylphenylcarbamoylmethyl) Iminodiacetic Acid (Tc-99m HIDA): A new radiopharmaceutical for hepatobiliary imaging studies. J. Nucl. Med. 1977, 18, 997–1004. [Google Scholar]
- Marie, S.; Cisternino, S.; Buvat, I.; Declèves, X.; Tournier, N. Imaging probes and modalities for the study of solute carrier O (SLCO)-transport function in vivo. J. Pharm. Sci. 2017, 106, 2335–2344. [Google Scholar] [CrossRef] [Green Version]
- de Graaf, W.; Häusler, S.; Heger, M.; van Ginhoven, T.; van Cappellen, G.; Bennink, R.; Kullak-Ublick, G.; Hesselmann, R.; van Gulik, T.; Stieger, B. Transporters involved in the hepatic uptake of 99mTc-mebrofenin and indocyanine green. J. Hepatol. 2011, 54, 738–745. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, K.; Joseph, B.; Ananthanarayanan, M.; Balasubramaniyan, N.; Tronco, G.; Palestro, C.; Gupta, S. Adenosine triphosphate–binding cassette subfamily c member 2 is the major transporter of the hepatobiliary imaging agent 99mTc-mebrofenin. J. Nucl. Med. 2009, 50, 1140–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrikse, N.; Kuipers, F.; Meijer, C.; Havinga, R.; Bijleveld, C.; van der Graaf, W.; Vaalburg, W.; de Vries, E. In vivo imaging of hepatobiliary transport function mediated by multidrug resistance associated protein and P-glycoprotein. Cancer Chemother. Pharmacol. 2004, 54, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Hendrikse, N.H.; Franssen, E.J.F.; van der Graaf, W.T.A.; Vaalburg, W.; de Vries, E.G.E. Visualization of multidrug resistance in vivo. Eur. J. Nucl. Med. 1999, 26, 283–293. [Google Scholar] [CrossRef]
- de Graaf, W.; Bennink, R.; Veteläinen, R.; van Gulik, T. Nuclear imaging techniques for the assessment of hepatic function in liver surgery and transplantation. J. Nucl. Med. 2010, 51, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Kannan, P.; John, C.; Zoghbi, S.S.; Halldin, C.; Gottesman, M.M.; Innis, R.B.; Hall, M.D. Imaging the function of P-Glycoprotein with radiotracers: Pharmacokinetics and in vivo applications. Clin. Pharmacol. Ther. 2009, 86, 368–377. [Google Scholar] [CrossRef]
- Wanek, T.; Kuntner, C.; Bankstahl, J.; Bankstahl, M.; Stanek, J.; Sauberer, M.; Mairinger, S.; Strommer, S.; Wacheck, V.; Löscher, W.; et al. A comparative small-animal PET evaluation of [11C]tariquidar, [11C]elacridar and (R)-[11C]verapamil for detection of P-glycoprotein-expressing murine breast cancer. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, K.; Tanaka, M.; Ishii, A.; Katayama, Y.; Nakaoka, T.; Irie, S.; Kawahata, H.; Yamanaga, T.; Wada, Y.; Miyake, T.; et al. A clinical quantitative evaluation of hepatobiliary transport of [11C]Dehydropravastatin in humans using positron emission tomography. Drug Metab. Dispos. 2018, 46, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Karlgren, M.; Vildhede, A.; Norinder, U.; Wisniewski, J.; Kimoto, E.; Lai, Y.; Haglund, U.; Artursson, P. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): Influence of protein expression on drug–drug interactions. J. Med. Chem. 2012, 55, 4740–4763. [Google Scholar] [CrossRef]
- Te Brake, L.; Russel, F.; van den Heuvel, J.; de Knegt, G.; de Steenwinkel, J.; Burger, D.; Aarnoutse, R.; Koenderink, J. Inhibitory potential of tuberculosis drugs on ATP-binding cassette drug transporters. Tuberculosis 2016, 96, 150–157. [Google Scholar] [CrossRef]
- Neyt, S.; Huisman, M.; Vanhove, C.; Man, H.; Vliegen, M.; Moerman, L.; Dumolyn, C.; Mannens, G.; Vos, F. In vivo visualization and quantification of (disturbed) oatp-mediated hepatic uptake and mrp2-mediated biliary excretion of 99mTc-mebrofenin in mice. J. Nucl. Med. 2013, 54, 624–630. [Google Scholar] [CrossRef] [Green Version]
- Köck, K.; Ferslew, B.; Netterberg, I.; Yang, K.; Urban, T.J.; Swaan, P.W.; Stewart, P.; Brouwer, K. Risk Factors for development of cholestatic drug-induced liver injury: Inhibition of hepatic basolateral bile acid transporters multidrug resistance-associated proteins 3 and 4. Drug Metab. Dispos. 2014, 42, 665–674. [Google Scholar] [CrossRef]
- Wen, X.; Joy, M.; Aleksunes, L.M. In vitro transport activity and trafficking of MRP2/ABCC2 Polymorphic variants. Pharm. Res. 2017, 34, 1637–1647. [Google Scholar] [CrossRef]
- Amor, D.; Goutal, S.; Marie, S.; Caillé, F.; Bauer, M.; Langer, O.; Auvity, S.; Tournier, N. Impact of rifampicin-inhibitable transport on the liver distribution and tissue kinetics of erlotinib assessed with PET imaging in rats. EJNMMI Res. 2018, 8, 81. [Google Scholar] [CrossRef]
- Yeung, P.; Alcos, A.; Tang, J.; Tsui, B. Pharmacokinetics and metabolism of diltiazem in rats: Comparing single vs. repeated subcutaneous injections in vivo. Biopharm. Drug Dispos. 2007, 28, 403–407. [Google Scholar] [CrossRef]
- Takashima, T.; Kitamura, S.; Wada, Y.; Tanaka, M.; Shigihara, Y.; Ishii, H.; Ijuin, R.; Shiomi, S.; Nakae, T.; Watanabe, Y.; et al. PET imaging–based evaluation of hepatobiliary transport in humans with (15R)-11C-TIC-Me. J. Nucl. Med. 2012, 53, 741–748. [Google Scholar] [CrossRef] [Green Version]
- Hernández Lozano, I.; Karch, R.; Bauer, M.; Blaickner, M.; Matsuda, A.; Wulkersdorfer, B.; Hacker, M.; Zeitlinger, M.; Langer, O. Towards improved pharmacokinetic models for the analysis of transporter-mediated hepatic disposition of drug molecules with positron emission tomography. AAPS J. 2019, 21, 61. [Google Scholar] [CrossRef]
- Ørntoft, N.; Munk, O.; Frisch, K.; Ott, P.; Keiding, S.; Sørensen, M. Hepatobiliary transport kinetics of the conjugated bile acid tracer 11C-CSar quantified in healthy humans and patients by positron emission tomography. J. Hepatol. 2017, 67, 321–327. [Google Scholar] [CrossRef]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, M.; Shim, C. Pharmacokinetics of diltiazem and deacetyldiltiazem in rats. Int. J. Pharm. 1991, 76, 71–76. [Google Scholar] [CrossRef]
- Kusuhara, H. Imaging in the study of membrane transporters. Clin. Pharmacol. Ther. 2013, 94, 33–36. [Google Scholar] [CrossRef]
- Kenna, J.; Waterton, J.; Baudy, A.; Galetin, A.; Hines, C.; Hockings, P.; Patel, M.; Scotcher, D.; Sourbron, S.; Ziemian, S.; et al. Noninvasive preclinical and clinical imaging of liver transporter function relevant to drug-induced liver injury. In Drug-Induced Liver Toxicity; Chen, M., Will, Y., Eds.; Methods in Pharmacology and Toxicology; Springer: New York, NY, USA, 2018; pp. 627–651. ISBN 978-1-4939-7677-5. [Google Scholar]
- Woillard, J.; Debord, J.; Benz-de-Bretagne, I.; Saint-Marcoux, F.; Turlure, P.; Girault, S.; Abraham, J.; Choquet, S.; Marquet, P.; Barin-Le Guellec, C. A Time-dependent model describes methotrexate elimination and supports dynamic modification of MRP2/ABCC2 activity. Ther. Drug Monit. 2017, 39, 145–156. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.-M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Tournier, N.; Saba, W.; Cisternino, S.; Peyronneau, M.; Damont, A.; Goutal, S.; Dubois, A.; Dollé, F.; Scherrmann, J.; Valette, H.; et al. Effects of selected OATP and/or ABC transporter inhibitors on the brain and whole-body distribution of glyburide. AAPS J. 2013, 15, 1082–1090. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Yu, Y.; Prasad, B.; Link, J.; Miyaoka, R.; Chen, X.; Unadkat, J. PET imaging of oatp-mediated hepatobiliary transport of [11C]rosuvastatin in the rat. Mol. Pharm. 2014, 11, 2745–2754. [Google Scholar] [CrossRef]
- Doo, E.; Krishnamurthy, G.; Eklem, M.; Gilbert, S.; Brown, P. Quantification of hepatobiliary function as an integral part of imaging with technetium-99m-mebrofenin in health and disease. J. Nucl. Med. 1991, 32, 48–57. [Google Scholar]
- König, J.; Müller, F.; Fromm, M. Transporters and drug-drug interactions: Important determinants of drug disposition and effects. Pharmacol. Rev. 2013, 65, 944–966. [Google Scholar] [CrossRef] [Green Version]
- Patilea-Vrana, G.; Unadkat, J. Transport vs. metabolism: What determines the pharmacokinetics (PK) and pharmacodynamics (PD) of drugs? Insights from the extended clearance model. Clin. Pharmacol. Ther. 2016, 100, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Muzi, M.; Mankoff, D.A.; Link, J.M.; Shoner, S.; Collier, A.C.; Sasongko, L.; Unadkat, J.D. Imaging of cyclosporine inhibition of P-Glycoprotein activity using 11C-verapamil in the brain: Studies of healthy humans. J. Nucl. Med. 2009, 50, 1267–1275. [Google Scholar] [CrossRef] [Green Version]
- Kreisl, W.; Boellaard, R.; Morse, C.; Woock, A.; Zoghbi, S.; Shetty, H.; Pike, V.; Innis, R. Increased permeability-glycoprotein inhibition at the human blood-brain barrier can be safely achieved by performing PET during peak plasma concentrations of tariquidar. J. Nucl. Med. 2015, 56, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Tournier, N.; Bauer, M.; Pichler, V.; Nics, L.; Klebermass, E.-M.; Bamminger, K.; Matzneller, P.; Weber, M.; Karch, R.; Caille, F.; et al. Impact of P-glycoprotein function on the brain kinetics of the weak substrate 11C-metoclopramide assessed with PET imaging in humans. J. Nucl. Med. 2019, 60, 985–991. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | AUC0–30 min Blood ([cps/MBq].min) | AUC0–30 min Liver ([cps/MBq].min) | Xmax Liver (cps/MBq) | Tmax Live (min) | AUC0–30 min Intestine ([cps/MBq].min) |
|---|---|---|---|---|---|
| Control (n = 5) | 51 ± 14 | 461 ± 109 | 26.5 ± 5.1 | 4.3 ± 1.7 | 424 ± 85 |
| RIF (n = 5) | 109 ± 12 * | 385 ± 28 | 16.0 ± 0.9 * | 9.6 ± 0.8 * | 223 ± 54 * |
| DTZ 10 mg/kg (n = 5) | 47 ± 4 | 520 ± 61 | 30.1 ± 3.2 | 4.3 ± 0.7 | 271 ± 30 * |
| DTZ 20 mg/kg (n = 5) | 72 ± 23 | 562 ± 96 | 31.1 ± 5.8 | 5.4 ± 1.6 | 286 ± 102 * |
| DTZ 40 mg/kg (n = 5) | 84 ± 30 * | 543 ± 95 | 24.3 ± 3.0 | 6.4 ± 0.8 | 238 ± 76 * |
| CsA 0.01 mg/kg (n = 6) | 71 ± 9 | 660 ± 61 * | 34.6 ± 3.0 * | 4.0 ± 0.6 | 327 ± 61 |
| CsA 0.1 mg/kg (n = 5) | 72 ± 11 | 601 ± 105 * | 27.9 ± 4.0 | 5.7 ± 1.9 | 223 ± 26 * |
| CsA 0.5 mg/kg (n = 6) | 97 ± 9 * | 668 ± 48 * | 28.9 ± 2.1 | 7.6 ± 2.1 * | 239 ± 64 * |
| CsA 1 mg/kg (n = 6) | 124 ± 9 * | 695 ± 40 * | 29.9 ± 2.5 | 7.4 ± 1.3 * | 275 ± 38 * |
| CsA 5 mg/kg (n = 6) | 188 ± 18 * | 648 ± 66 * | 27.6 ± 2.1 | 9.1 ± 1.3 * | 314 ± 30 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marie, S.; Hernández-Lozano, I.; Breuil, L.; Saba, W.; Novell, A.; Gennisson, J.-L.; Langer, O.; Truillet, C.; Tournier, N. Validation of Pharmacological Protocols for Targeted Inhibition of Canalicular MRP2 Activity in Hepatocytes Using [99mTc]mebrofenin Imaging in Rats. Pharmaceutics 2020, 12, 486. https://doi.org/10.3390/pharmaceutics12060486
Marie S, Hernández-Lozano I, Breuil L, Saba W, Novell A, Gennisson J-L, Langer O, Truillet C, Tournier N. Validation of Pharmacological Protocols for Targeted Inhibition of Canalicular MRP2 Activity in Hepatocytes Using [99mTc]mebrofenin Imaging in Rats. Pharmaceutics. 2020; 12(6):486. https://doi.org/10.3390/pharmaceutics12060486
Chicago/Turabian StyleMarie, Solène, Irene Hernández-Lozano, Louise Breuil, Wadad Saba, Anthony Novell, Jean-Luc Gennisson, Oliver Langer, Charles Truillet, and Nicolas Tournier. 2020. "Validation of Pharmacological Protocols for Targeted Inhibition of Canalicular MRP2 Activity in Hepatocytes Using [99mTc]mebrofenin Imaging in Rats" Pharmaceutics 12, no. 6: 486. https://doi.org/10.3390/pharmaceutics12060486