Crucial Role of Lamin A/C in the Migration and Differentiation of MSCs in Bone


Stem Cells and Cell Therapy Laboratory, Biocruces Bizkaia Health Research Institute, Cruces University Hospital, Plaza de Cruces S/N, Barakaldo, 48903 Bizkaia, Spain
*
Authors to whom correspondence should be addressed.
Cells 2020, 9(6), 1330; https://doi.org/10.3390/cells9061330
Submission received: 29 April 2020
/
Revised: 22 May 2020
/
Accepted: 25 May 2020
/
Published: 26 May 2020
(This article belongs to the Collection Lamins and Laminopathies)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Lamin A/C, intermediate filament proteins from the nuclear lamina encoded by the LMNA gene, play a central role in mediating the mechanosignaling of cytoskeletal forces into nucleus. In fact, this mechanotransduction process is essential to ensure the proper functioning of other tasks also mediated by lamin A/C: the structural support of the nucleus and the regulation of gene expression. In this way, lamin A/C is fundamental for the migration and differentiation of mesenchymal stem cells (MSCs), the progenitors of osteoblasts, thus affecting bone homeostasis. Bone formation is a complex process regulated by chemical and mechanical cues, coming from the surrounding extracellular matrix. MSCs respond to signals modulating the expression levels of lamin A/C, and therefore, adapting their nuclear shape and stiffness. To promote cell migration, MSCs need soft nuclei with low lamin A content. Conversely, during osteogenic differentiation, lamin A/C levels are known to be increased. Several LMNA mutations present a negative impact in the migration and osteogenesis of MSCs, affecting bone tissue homeostasis and leading to pathological conditions. This review aims to describe these concepts by discussing the latest state-of-the-art in this exciting area, focusing on the relationship between lamin A/C in MSCs’ function and bone tissue from both, health and pathological points of view.
1. Introduction
The nuclear lamina, a thin protein network lining the inner surface of the nuclear envelope, is primarily composed of type V intermediate filaments known as A- and B-type lamins. In mammals, the two major isoforms of A-type lamins are generated by the alternative splicing of the LMNA gene: lamin A and lamin C, which are mainly expressed in differentiated cells. B-type lamins, lamin B1 and lamin B2, encoded by LMNB1 and LMNB2 respectively, are constitutively expressed in most cell types [1]. Soon after being synthesized, lamin A and B-type lamins undergo sequential post-translational modifications based on their C-terminal CaaX motif (C: Cys, a: an aliphatic residue, X: usually a Met) which functions as a substrate where farnesylation and carboxy-methylation take place. After this complex process, mature B-type lamins retain a farnesyl group at the C-terminal extreme, whereas mature lamin A loses it along with 15 amino acids of the C terminus [2]. This farnesyl group has a role in targeting newly synthesized cytoplasmic lamins to the nuclear envelope, by enhancing the hydrophobic interactions of lamins with the inner nuclear membrane [3]. However, this farnesylation is not always indispensable for the nuclear recruitment of lamins: lamin C is localized to the inner nuclear envelope although it does not contain the CaaX motif to be farnesylated [4]. Regarding the structural organization of lamins within mammalian nuclei, super-resolution microscopy techniques showed that lamin A and lamin B form independent but interacting filament networks adjacent to the inner nuclear membrane [5,6,7,8]. More recently, this observation has been tuned by two studies: not only has the existence of independent lamin A and lamin B networks been corroborated (showing only 18% of co-localization between the A- and B-type lamins), but also a distinct spatial organization of lamins. Thus, in mouse embryonic fibroblasts (MEFs) and human cells (HeLa, fibroblasts), lamin A and lamin B1 form concentric but overlapping networks. In this way, lamin B1, taking advantage of its farnesylated C-terminal group, shows a more peripheral localization, closest to the inner nuclear envelope [9,10].
The nuclear lamina has been shown to undertake two main cellular functions: (1) an essential structural role, providing the shape, and mechanical properties to the nucleus, and (2) as a regulator of gene expression, by modulating chromatin organization and the accessibility of signaling molecules and transcription factors to target promoters [1,11,12]. Recently, nuclear lamina has been shown to be an essential mediator of mechanosignaling, that is, the transduction of exterior physical forces into the nucleus to generate a biological response, which is essential to help the cells adapting to the continuously changing microenvironment [13]. Thus, nuclear lamina components have been shown to be the linkers between the mechanosignals transduced from the cytoskeleton to the nucleus, with lamin A/C executing an essential role in this process [14,15,16]. Indeed, this mechanosensing regulated by lamin A/C has been proposed to be the bridge integrating both the aforementioned structural and gene expression function mediated by lamin A/C [17]. Interestingly, the stoichiometry of the lamin A:B differs depending on the cell types, in fact the relative abundance of lamin A has been shown to scale with tissue and nuclei stiffness [18]. Thus, cells with a high content of A-type lamins exhibit high viscous and stiff nuclei [19], which hamper their migration capacity. On the other hand, cells expressing very low levels of lamin A and C, such as embryonic stem cells, display easily deformable nuclei [20]. Interestingly, bone tissue, which is of mesenchymal origin, has the highest rate of collagen content and thus the highest A:B ratio [18]. Mechanical signals and extracellular matrix (ECM) composition play an important role in bone homeostasis. Indeed, bones are known to respond to mechanical loading, such as exercise, to promote osteo-anabolic pathways [21]. Mesenchymal stem cells (MSCs) are the natural progenitors of osteoblasts, the bone forming cells. MSCs undergo the multi-step process of osteogenesis in response to different cues (of both biochemical and mechanical nature) coming mainly from the surrounding ECM [22,23]. Moreover, in the bone healing process, inflammatory mediators activate and mobilize tissue-resident, endogenous MSCs which migrate from their niche to the damaged site in order to facilitate bone tissue regeneration [24]. To achieve both migration and osteogenic differentiation, MSCs must reorganize their nuclear lamina shape and/or composition, with lamin A orchestrating this process. Thus, levels of lamin A are known to be increased during osteogenesis while they are decreased during adipogenesis of MSCs [18,25]. In the same way, there are pathological conditions affecting lamin A/C, such as physiological aging and LMNA mutations, which will have a negative impact in migration and osteogenesis of MSCs, affecting bone tissue homeostasis.
In this article, we will review the current knowledge about the crucial role of lamin A/C in regulating biological processes in MSCs which are critical to ensure a proper bone tissue homeostasis.
2. Lamin A/C and Bone Formation
Bone is a tissue in a constant remodeling, which is capable of self-repairing after an injury. Bone fracture healing can occur through two different mechanisms: intramembranous and endochondral bone formation. During intramembranous ossification, MSCs proliferate and condense into compact nodules called nidus. Here, MSCs differentiate into osteoprogenitor cells to finally become osteoblasts. Then, osteoblasts start secreting an extracellular matrix rich in type I collagen, which is able to bind calcium salts. Eventually, the extracellular matrix becomes calcified, forming the osteoid. Normally, osteoblasts are separated from the osteoid but some cells can be trapped into the calcified area, becoming osteocytes. The process continues with the growth of bone spicules from the osteoid. Lastly, the entire region is surrounded by compact mesenchymal cells to form the periosteum membrane. This ossification process occurs in flat bones of the face, most of the skull bones and clavicles.
On the other hand, endochondral bone formation requires the presence of cartilage and occurs in the tissue adjacent to the fracture and surrounding soft tissues. Through this mechanism, the long bones of the axial (spine and ribs) and appendicular (limbs) skeleton are formed. The beginning is similar to the previous process, with MSCs grouping together where the new bone will take shape. However unlike the former, cells differentiate into chondrocytes that will proliferate, synthesizing their proper extracellular matrix rich in type II collagen and proteoglycans. The cartilaginous tissue forms a soft callus that stabilizes the fracture area, serving as a mold for the newly forming bone. Subsequently, chondrocytes stop dividing and become hypertrophic chondrocytes. They modify the ECM secreting type X collagen and fibronectin, promoting the mineralization of the tissue by calcium deposition. At that point, the blood vessels invade the tissue in order to vascularize it, meanwhile hypertrophic chondrocytes begin to suffer apoptosis. Certain hypertrophic chondrocytes survive and can differentiate into osteoblasts [26]. The MSCs surrounding the area differentiate into osteoblasts, furthermore beginning to synthesize extracellular bone matrix rich in type I collagen. This process will proceed until all cartilaginous tissue is replaced by bone tissue.
Usually, both types of ossification are combined during fracture healing and bone repair, after bone injury [27]. In order to achieve an accurate bone remodeling, everything must be closely regulated by different factors, such as growth, differentiation and transcription factors [28,29]. Therefore, bone formation can be measured in vitro and in vivo by different markers, from transcription factors such as osteocalcin (OCN), osterix (OSX) and bone sialo-protein (BSP), to akaline phosphatase expression and calcium deposition by Alizarin Red staining. The influence of lamin A/C in bone formation has been assessed in vitro in several studies. In lamin A/C-inhibited MSCs and mature osteoblasts, a significant reduction in alkaline phosphatase and Alizarin red staining have been shown, confirming a significant reduction in osteoblast differentiation and mineralization. Additionally, significantly lower levels of OCN, OSX and BSP were measured in LMNA knocked-down cells [30]. Moreover, during bone remodeling, MSCs’ differentiation into osteoblasts (osteoblastogenesis) and induction and maturation of osteoclasts (osteoclastogenesis) were altered by the absence of lamin A/C in MSCs: while osteoblastogenesis was inhibited (measured by a reduction of osteocalcin secretion and alkaline phosphatase expression), osteoclastogenesis was enhanced [31]. In fact, lamin A/C modulates the expression of the two osteoclastogenesis-regulating proteins, receptor activator of nuclear factor κ-B ligand (RANKL) and osteoprotegerin (OPG): RANKL is expressed by immature osteoblasts, thus osteoblast differentiation arrest due to lamin A/C inhibition leads to increased expression of RANKL levels, unbalancing the RANKL/OPG ratio, inducing an osteoclastogenic environment. Conversely, lamin A overexpression in mouse pre-osteoblastic MC3T3-E1 cells has been shown to promote osteoblast differentiation and calcification by inducing the expression of: alkaline phosphatase, type I collagen, BSP, OCN and dentin matrix acidic phosphoprotein 1 (DMP-1), in the presence of bone morphogenetic protein 2 (BMP-2) growth factor [32]. Taken together, all this evidence reveals the crucial role lamin A/C plays in bone remodeling.
3. Lamin A/C and MSCs
Adult MSCs are spindle-shaped cells which use has been expanded to cell therapy, regenerative medicine and tissue repair, due to the beneficial properties that they present: self-renewable, multipotent, immunomodulation [33], easily accessible and culturally expandable in vitro, genomic stability and few ethical issues [34]. Moreover they have been isolated from many different sources [35], such as bone marrow and adipose tissue in addition to perinatal sources such as umbilical cord blood [36]. The International Society for Cellular Therapy has proposed minimum criteria to define MSCs [37]: They (i) should exhibits plastic adherence (ii) possess specific cell surface markers such as cluster of differentiation (CD)73, CD90, CD105 and lack expression of CD14, CD34, CD45 and human leucocyte antigen-DR (HLA-DR), besides, (iii) they should present the ability to differentiate in vitro into adipogenic, chondrogenic and osteogenic lineages.
These cells are gaining interest due to the benefits that are presenting when used in therapy, urging to delve into their potential and mechanisms of action [38]. They have been proposed to treat several diseases, showing promising results for repairing damaged tissues, both in animal models and in human clinical trials [39,40,41]. MSCs have demonstrated homing ability: the capacity to migrate into specific tissues and differentiate into chondrogenic, osteogenic or adipogenic lineage. Moreover, they secrete chemokines, cytokines and growth factors crucial in tissue regeneration [42]. The processes of MSCs’ migration to injured tissue and differentiation into specific lineage are paramount in bone formation and remodeling. These processes are highly determinate by lamin A/C implication for their correct accomplishment [43].
3.1. MSCs’ Migration
The process by which MSCs migrate to their target tissue has been deeply studied, since it is a vital requirement to later on differentiate and form new bone. Several studies have demonstrated that the migration of these cells is mainly affected by two types of factors: chemical and mechanical ones.
3.1.1. Chemical Factors Involved in MSCs’ Migration
The chemical factors are part of the microenvironment in which MSCs reside, playing a key role in the migration of these cells to injured tissue sites. The first step in bone formation requires MSCs to migrate and differentiate in the place where the injury was produced. MSCs migrate toward various signals, including growth factors, chemokines and cytokines [44], factors that are released by several immune cells (neutrophils, macrophages, lymphocytes and others) during the initial acute inflammatory phase of fracture healing [45]. Tan et al. [46] uncovered that platelet-derived growth factors (PDGFs) promoted endogenous MSCs’ migration to the fracture from remote sites. Specifically, PDGF-AA regulates MSCs’ migration and osteogenic differentiation via the BMP-Smad1/5/8 signaling pathway [47]. Secretion of tumor necrosis factor-α (TNF-α) by macrophages, immune inflammatory cells, and even by the MSCs present in the periosteum [48], has shown to induce osteogenic differentiation of MSCs in vitro [49]. Also, TNF-α-activated MSCs suppress inflammation by inducing Interleukin (IL)-10 production in macrophages [50], demonstrating the regulatory role of MSCs in the bone healing process. The downregulation of the pro-inflammatory response is fundamental for bone repair, since systemic and maintained inflammation is detrimental for fracture healing accomplishment [51].
During bone remodeling, transforming growth factor-β1 (TGF-β1) has been shown to play a role in the recruitment of MSCs to the bone resorption area. TGF-β1 is a cytokine present in bone extracellular matrix, normally in its inactive form through non-covalent binding to latency-associated protein. Thus, the receptor-binding domains of the active TGF-β1 are hidden by this union. This cytokine, crucial during the bone resorption process, is activated and released, favoring the migration of osteogenic MSCs through the SMAD signaling pathway to resorptive sites [52].
Another factor involved in bone remodeling is Sry-related high-mobility group box 9 (SOX9). This transcription factor regulates sex determination, chondrocyte differentiation and other developmental events. In fetal and juvenile growth plates, SOX9 is expressed in resting and proliferating chondrocytes, being maximal in pre-hypertrophic chondrocytes. However, in the hypertrophic zone, it disappears completely [53]. Hattori et al. demonstrated that SOX9 downregulation in the hypertrophic zone of the growth plate is a necessary event to allow cartilage vascularization, cartilage resorption and formation of trabecular bone [54].
3.1.2. Mechanical Factors Involved in MSCs’ Migration
From a mechanical point of view, cells have to be able to deform in order to migrate into specific tissue to then differentiate and form new bone—a process highly regulated by the cytoskeleton [55]. Nevertheless, the nucleus is the largest, stiffest organelle in the cell; hence, it is the primary impediment to motion as cells squeeze through tight spaces. When the nucleus is too stiff to be pushed or pulled through a small opening, the migration is limited. Conversely, when nuclear softness and elasticity increases, cell migration is promoted. Therefore, a deformable nucleus facilitates MSCs’ migration as it accommodates motion of the cell through three-dimensional (3D) environments.
The nuclear shape and stiffness have been shown to be regulated by both lamin B1 and lamin A/C [9,56,57]. In the case of lamin A/C, several studies have demonstrated that cells which ectopically express lamin A/C have stiffer nuclei that resist deformation [56,58,59], limiting migration. Conversely, depletion of lamin A/C results in irregularly shaped nuclei and severely reduces nuclear stiffness [15,58,60,61,62]. Indeed, low lamin A levels increase MSCs’ migration, as it was demonstrated via lamin A knockdown by ∼50% [19]. However, not only the lamin A/C content, but also the lamin A/C to lamin B1 ratio will influence the migration capacity of cells. Indeed, MSCs have shown slower migration through two-dimensional (2D) and 3D microenvironments than other primary human cells derived from mesodermal origin, due to a high lamin A/C to lamin B1 ratio that increases nuclear rigidity [63].
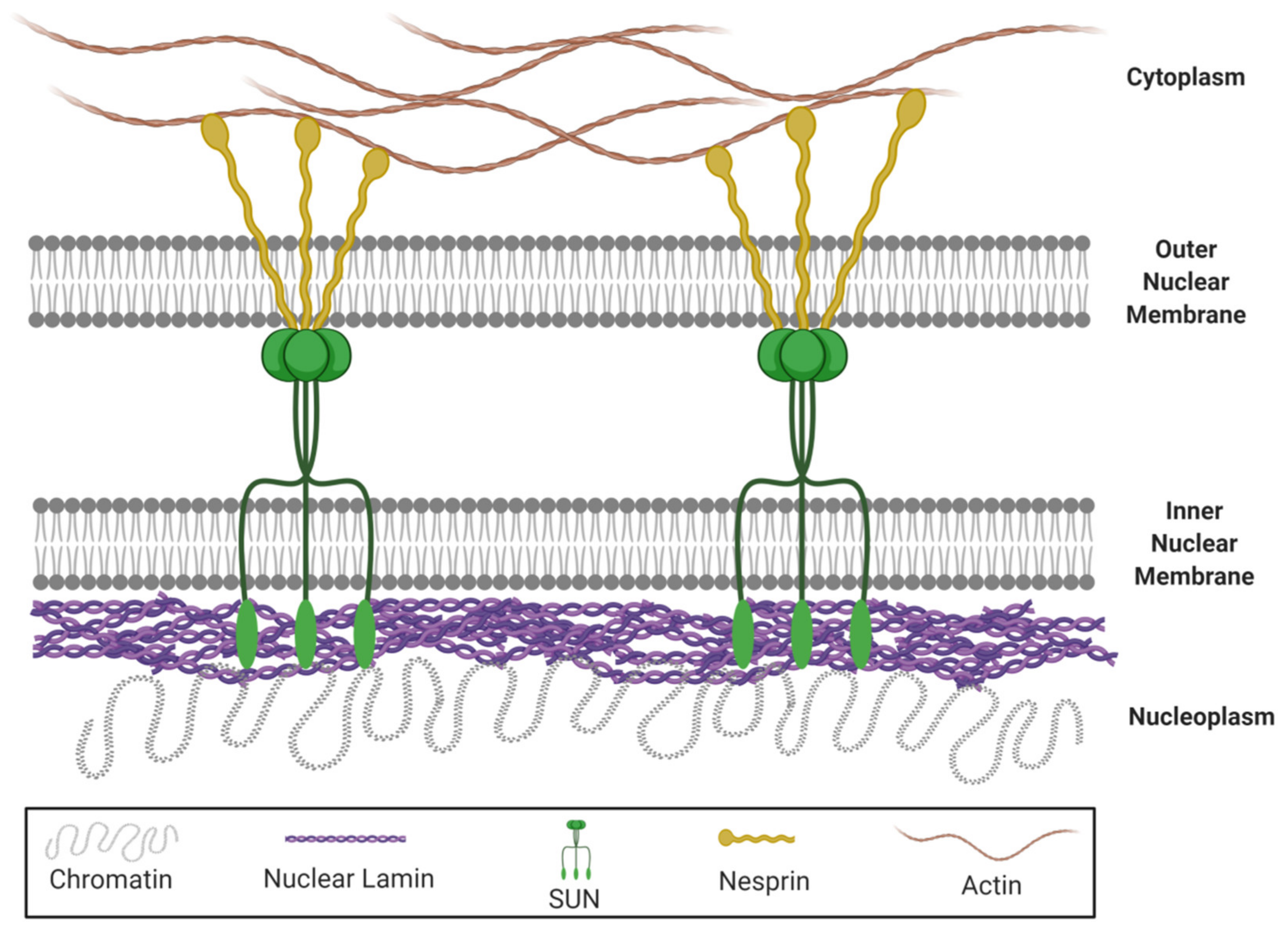
Besides, lamin A/C is implicated in the nuclear-cytoskeletal coupling, which plays a vital role in the nuclear mechanotransduction and determines the mechanical properties of nuclei [64,65]. In fact, mutations in lamin A/C disturb nuclear-cytoskeletal coupling in LMNA-/- mice [66]. The nucleus is mechanically connected to the rest of the cell, via Linker of Nucleus and Cytoskeleton (LINC) complex structures which are located in the nuclear envelope [67,68]. This complex consists of SUN (Sad1 and UNC-84) proteins anchored in the inner nuclear membrane and nuclear envelope spectrin-repeat-containing proteins (nesprins) anchored in the outer nuclear membrane (Figure 1). In that way, the LINC complex connects the cytoplasmic cytoskeleton with the inner nucleoskeletal lamin A/C network [68,69], allowing the reorganization of lamins in response to mechanical stress [70]. Therefore, it is responsible for limiting nuclear deformation [67].
Then, there are different mechanical factors that could affect the migration of MSCs through the influence of lamin A/C, such as mechanical strain, matrix stiffness [71], 3D geometry and gravity.
Different in vitro and in vivo studies have shown that the migration of MSCs is affected by mechanical strain. Mechanical strain has the ability to increase human MSCs migration, as it was demonstrated when static mechanical tension at 5% (for 6 h) [72] or 10% (for 12 h) [73] was applied. Moreover, cyclic mechanical stretching with 10% amplitude at 1 Hz frequency (for 8 h) promotes bone marrow MSCs’ (BMSCs) migration but reduces BMSCs’ invasion [74]. Cell invasion is a feature typical of malignant metastatic cells that actively invade a surrounding tissue, usually attracted by a chemoattractant gradient. This process, unlike cell migration, involves the secretion of specific proteases to destroy the ECM of the target tissue in order to facilitate its entrance, altering the structure of the receptor tissue. As we have previously reported, lamin A/C is closely implicated in the transmission of mechanical stress through the nucleus. Lamin A/C-deficient fibroblasts are characterized by defective nuclear mechanics and mechanotransduction under mechanical strain. That was reflected by increased nuclear deformations and nuclear fragility, attenuated expression of mechanosensitive genes, as well as impaired transcriptional activation leading to impaired viability of mechanically strained cells [60,75]. Overall, the involvement of lamin A/C in mechanical stress will influence MSCs’ migration.
Regarding matrix stiffness, MSCs are surrounded by an ECM that transmits complex biochemical [76] and biophysical signals [77]. The physical signals provided to MSCs could regulate their migration, so different studies have focused on the effect of matrix stiffness and elasticity [19,78]. Raab et al. demonstrated that MSCs migrate to stiffer portions of the substrates (from a soft matrix (1 kPa) to a stiff matrix (34 kPa)), by polarizing the cytoskeleton [77]. The assembled microtubule network was necessary for directed migration of MSCs [79]. Since lamin A/C is tightly linked to the cytoskeleton via the LINC complex, lamin A/C is also implicated in how cells respond to matrix stiffness [80]. In fact, lamin A/C levels control nuclear strain stiffening at large deformations, that is, during cell migration [81].
To deepen the understanding of the behavior of MSCs in tissue regeneration, it is mandatory to study how 3D geometry could influence it. It is known that the role of surface curvature is related with the migratory capacities of MSCs. Werner et al. demonstrated that cells present the ability to migrate faster on concave spherical surfaces, compared to flat and convex spherical surfaces, as a consequence of a reduced contact between the cell and the material surface [82]. Moreover, the nuclei of cells on convex surfaces present high lamin A levels, suggesting that these cells withstand high intracellular tensions, so the migration of MSCs is obstructed. These observations indicate that the 3D substrate curvature influences cell attachment morphology, thus modulating the cytoskeletal forces acting on the nucleus.
Another factor that affects MSCs’ migration is gravity. Little is known about the cellular reaction to simulated microgravity conditions in vitro. Mao et al. reported that stimulated microgravity (rotated at 10 rpm, approximately 1 × 10−3 g) inhibited the migration of rat BMSCs via reorganizing F-actin and increasing cell stiffness [83]. A recent report has studied how microgravity affects the migration of MSCs and the implications of lamin A/C. MSCs cultured under normal conditions present a stretched morphology and undergo unidirectional migration; on the contrary, cells cultured under simulated microgravity conditions undergo multidirectional migration, with higher frequency of directional changes of cell movement. Observing the cytoskeleton, cells grown under conventional culture conditions had longitudinal actin stress fibers along both the apical and basal sides. Alternatively, the cells grown under simulated microgravity conditions presented larger cortical actin fibers in the filopodia and lamellipodia regions and had less stress fibers located mainly along the basal side. Nevertheless, lamin A/C was mainly located on the apical side in cells under conventional culture conditions, indicating basal-to-apical polarization. Conversely, cells cultured under simulated microgravity conditions showed lamin A/C localization on both the apical and basal sides. Taken together, gravitational changes might induce alternations in the nuclear lamina organization and nuclear lamina–cytoskeleton interactions, inducing mechanotransduction changes in human MSCs [84].
All in all, MSCs’ migration to the target tissue is regulated by chemical factors such as growth factors, chemokines and cytokines. In addition, mechanical factors play a key role in MSCs’ migration. First of all, nucleus stiffness, which is determined by lamin A/C levels, is the main regulator during migration. Hence, the lamin A/C to lamin B1 ratio increases nuclear rigidity, thereby slowing down the migration of MSCs. Moreover, lamin A is implicated in the nuclear-cytoskeletal coupling via the LINC complex, allowing the reorganization of lamins in response to mechanical stress. There are different mechanical factors that could affect the migration of MSCs through the influence of lamin A/C. In summary, increased migration of MSCs is obtained with mechanical strain, concave spherical surfaces and under microgravity conditions.
3.2. MSCs’ Differentiation
Once MSCs have migrated to their target tissue, they differentiate into osteogenic, chondrogenic or adipogenic lineages. At this point, microenvironment is essential to determinate cell fate, and both biochemical and mechanical cues are involved in this process. Moreover, the nuclear envelope protein lamin A plays a key role in MSCs’ differentiation, since it interacts with different pathways involved. Different mutations in the LMNA gene alter the expression profile of MSCs during differentiation, in a mutation-specific manner. Thus, each LMNA mutation promotes a unique expression pattern of genes involved in a lineage-specific differentiation and this pattern is shared by the phenotype-specific mutations [85].
3.2.1. Response of MSCs to Biochemical Cues
Cellular and molecular signaling pathways, in addition to microenvironmental changes, have been studied in order to understand the role of cytokines, growth factors, extracellular matrix molecules and transcription factors [86] that regulate the differentiation of MSCs toward a specific cell type. Lamin A/C has the capacity to directly regulate the response of MSCs to biochemical cues, by interacting with those factors involved.
The Wnt/β-catenin signaling pathway plays a central regulatory role in differentiation of MSCs. In the canonical Wnt signaling pathway, Wnt binds to different receptors (Frizzled family receptors and low density lipoprotein (LDL)-receptor-related protein 5,6) to induce β-catenin stabilization. Subsequently, the stabilized β-catenin translocates to the nucleus and forms a complex with the DNA-binding transcription factors T cell factor/lymphoid enhancer factor to activate a Wnt-controlled gene expression program [87]. These genes are tightly related with MSCs’ differentiation, since the Wnt signaling pathway promotes osteogenic differentiation of MSCs [88]. β-catenin induces differentiation of osteoblasts and osteoblastic matrix production [89] and can also suppress the differentiation of MSCs into adipogenic and chondrogenic lineages [36,90]. Therefore, the Wnt/β-catenin pathway has an essential role in bone regeneration and repair [91]. Regarding the influence of lamin A/C, it physically interacts with β-catenin and facilitates its translocation into the nucleus and its transcriptional activity, thus inducing osteogenesis. In this way, Bermeo et al. demonstrated that overexpression of LMNA in human BMSCs leads to increased osteogenic and decreased adipogenic differentiation potential, through the regulation of the Wnt/β-catenin pathway [25].
Notch signaling is a highly conserved pathway that regulates cell fate decisions and skeletal development [92]. In the canonical Notch pathway, the single-transmembrane cell surface receptors undergo sequential proteolytic cleavage upon binding of their ligand. Following binding, the Notch intracellular domain is released from the plasma membrane and translocated into the nucleus, where it interacts with transcription factors to activate transcription of target genes [93,94]. Notch stimulates differentiation of MSCs into osteoblasts [95] and activation of Notch promotes osteogenic differentiation in a tissue-specific, dose-dependent manner [96]. Conversely, its inhibition promotes adipogenic differentiation [97]. In this way, lamin A/C interacts with Notch signaling [98], thereby influencing MSCs’ differentiation. The lamin A–Notch interaction can be realized both through the chromatin regulatory mechanism and through direct structural interactions, for example, via emerin-dependent suppression of Notch signaling [99,100]. Many studies have been reported analyzing the effect of different LMNA mutations in the Notch pathway, and thereby in differentiation of human MSCs and mesenchymal origin cells. Bogdanova et al. reported that mutations in the LMNA gene may affect the function of Notch signaling in human MSCs, suggesting that the interaction of lamin A/C with Notch signaling components may be one of the mechanisms regulating MSCs’ differentiation. LMNA mutations have different effects on the efficiency of MSCs’ osteogenic differentiation and on the expression of specific osteogenic markers [98]. Therefore, the Notch pathway may be involved in the modified cell capacity for differentiation caused by LMNA mutations. In addition, Perepelina et al. reported that a specific mutation in LMNA (R482L, associated with Dunningan-type familial partial lipodystrophy) contributes to the downregulation of Notch activation in MSCs and decreases adipogenic differentiation when Notch is activated [101]. This mutation leads to nuclear blebbing and alterations in nuclear morphology in fibroblasts. Moreover, emerin binding to mutated lamin was impaired [102], suggesting that interaction with Notch might be mediated by emerin. A recent study has explored the influence of specific LMNA mutations (R527C and R471C) on the pro-osteogenic response of human cells of mesenchymal origin, and the interaction of LMNA with the Notch pathway. The outcomes obtained have shown a broad range in expression level of Notch-related and pro-osteogenic genes between the different cellular types used in the study, suggesting that the effect of a LMNA mutation might be influenced by the intrinsic molecular context of a cell lineage [103]. In conclusion, these studies demonstrate that lamin A/C mutations could have a critical effect on osteogenic differentiation depending on Notch activation and cell type.
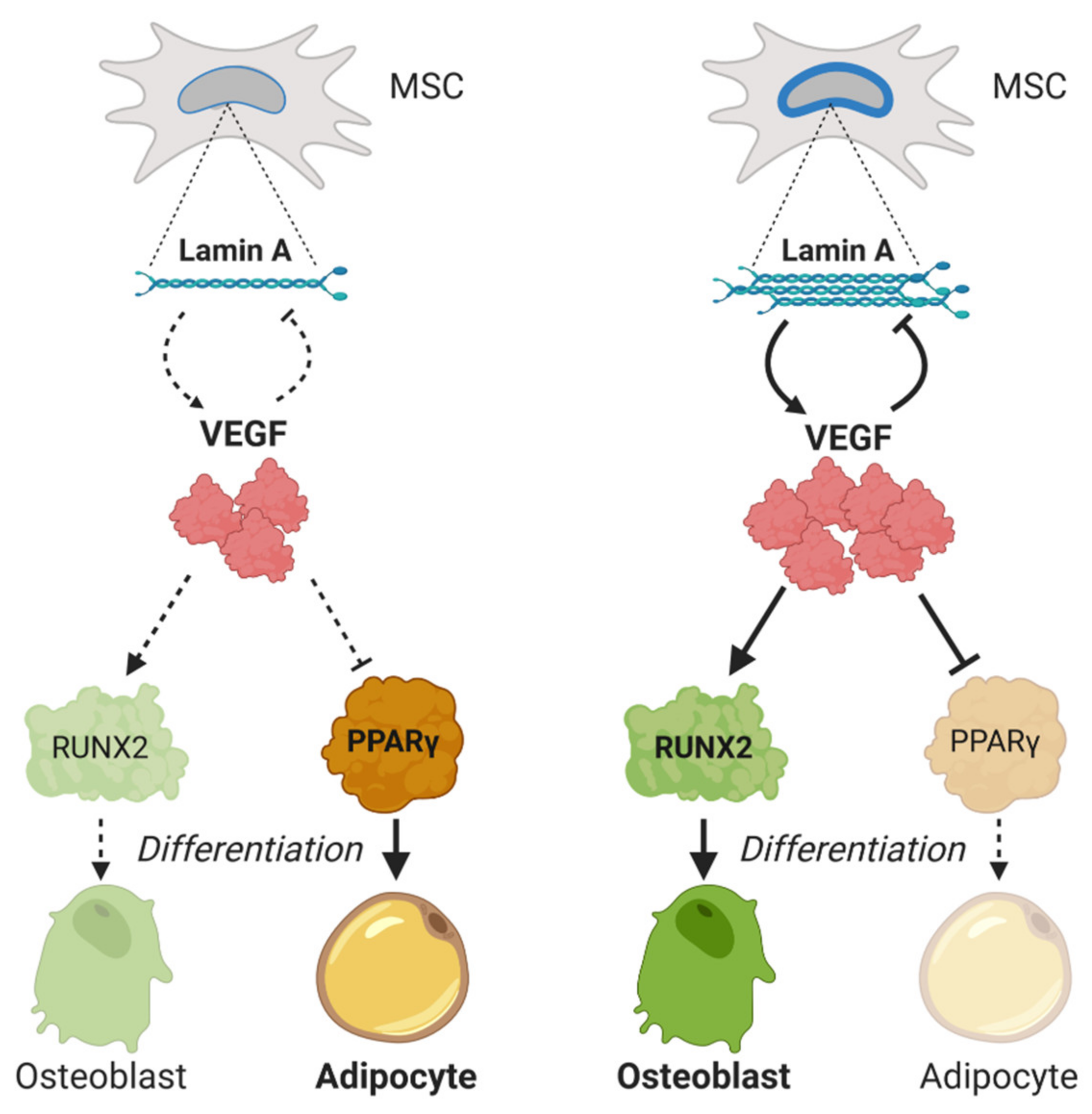
Vascular endothelial growth factor A (VEGFA) is a key regulator in MSCs’ differentiation. Its transcription is downregulated by SOX9, by interaction with regulatory SRY elements in the VEGFA gene [54]. Some studies show a significant decrease of VEGF levels in MSCs with age [104,105]. Accordingly, postmenopausal women with high/low production of VEGF polymorphisms have higher/lower lumbar spine bone mineral density, respectively [106]. Furthermore, the effect of VEGF on osteoblastogenesis seems to be mediated by a functional interaction between VEGF and lamin A/C [107,108]. Heterozygous lamin A-deficient mice express reduced levels of VEGF and Runt-related transcription factor (RUNX2), along with higher levels of peroxisome proliferator-activated receptor (PPAR)-γ (PPARγ). In addition, VEGF knock-down MSCs show the same expression profile, with low RUNX2 and increased peroxisome proliferator-activated receptor (PPAR)-γ. Interestingly lamin A levels are increased when VEGF levels are low, meaning a reciprocal interaction between both molecules (Figure 2).
RUNX2 is also a known downstream transcription factor in Wnt/β-catenin signaling. RUNX2 induces the differentiation of MSCs into immature osteoblasts [109], promoting the expression of osteogenesis-related genes [110], so it is considered an essential transcription factor in osteoblastogenesis. Lamin A/C interacts with RUNX2, the essential transcription factor for osteoblast differentiation [111]. When lamin A/C was knocked down in mature osteoblasts, reduced expression levels of RUNX2 were reported [31]. Moreover, downregulation of lamin A/C in MSCs reduces nuclear binding of RUNX2 to osteogenic promoters [30]. The absence of lamin A/C would allow proteins of the nuclear membrane such as inner nuclear membrane protein (MAN-1) to antagonize osteogenic proteins, affecting RUNX2 mobility and activation. MAN-1 is a protein of the nuclear envelope that is closely regulated by lamin A through a direct physical interaction [112]. Moreover, MAN-1 colocalizes with RUNX2. An in vivo study of LMNA null mice has associated absence of lamin A/C with an abnormal interaction between RUNX2 and MAN-1 [113]. In the absence of lamin A/C, expression of MAN-1 increases, thus reducing the availability of RUNX2 and its DNA binding. To sum up, the presence of lamin A/C is necessary for correct osteogenesis and consequent bone formation, guaranteeing the correct function of the RUNX2 pathway.
The nuclear lamina is known to serve as a resting platform for transcription factors, thus restricting their access to chromatin and therefore limiting their activity as activators or repressors of gene expression [114]. In MSCs, alterations in lamin A protein have been described to disrupt nuclear organization, leading to the sequestration of transcription factors to the nuclear periphery [115,116,117,118]. This pathological entrapment of transcription factors leads to an impaired homeostasis in MSCs, affecting their fate [116,119]. This is the case of PPARs, which are essential regulators of MSCs’ differentiation [120]. PPARγ acts as a positive regulator of adipocyte differentiation [121] and its overexpression enhances and accelerates the adipogenic differentiation of MSCs, both in vitro and in vivo [122]. These receptors are also regulated by lamin A/C. PPARγ ligands are normally trapped at the nuclear periphery by lamin A/C, so the lack of lamin facilitates its release, promoting PPARγ activation and inducing adipogenesis. Thus, lamin A/C acts as an inhibitor of adipocyte differentiation, affecting PPARγ signaling [123].
3.2.2. Response of MSCs to Mechanical Cues
Recent studies have reported that the effects of physical/mechanical cues of the microenvironment play an important role in regulating MSCs’ fate [124,125], suggesting that the mechanical properties [126] and the differentiation ability of MSCs are closely linked [127,128]. In the same way as MSCs’ migration, the nucleus of the cell is the main regulator of cell fate, since it regulates gene expression and its capacity to respond to external stimuli. Nuclear shape and stiffness changes during MSCs’ differentiation, until the type of cell required for each specific tissue is obtained. These two properties of the nucleus are closely regulated by lamin A/C [129]. During cell differentiation, nuclear stiffness increases, along with the LMNA expression [130]. Conversely, knockdown of the LMNA gene in differentiated cells decreases nuclear stiffness [65].
The nucleus and the nuclear envelope protein lamin A/C also play a crucial role in cellular mechanotransduction, which are needed to transmit mechanical signals and respond to mechanical forces [131] during differentiation. As we have mentioned before, LINC complex structures mechanically connect the cytoplasmic cytoskeleton with the inner lamin A/C network. This ability is important to respond to external mechanical cues [132], such as mechanical strain, matrix stiffness [133] and 3D geometry. In addition, chromatin is arranged on the internal nuclear scaffold that lamin makes up. So, external mechanical stimuli could regulate heterochromatization [134] via LINC and lamin A mechanotransduction [135], thus regulating the expression of genes involved in MSCs’ differentiation [136]. Moreover, during development, lamin A levels increase to mechano-protect the genome [137].
MSCs’ migration is in part regulated by mechanical cues, such as mechanical strain, matrix stiffness and 3D geometry, and the same happens with MSCs’ differentiation. To demonstrate the influence of mechanical strain in MSCs’ fate, MSCs were cultured in highly adipogenic medium and mechanical strain was applied for 6 h daily. Under mechanical strain, expression of adipogenic markers was inhibited and β-catenin nuclear translocation was enhanced, suggesting an induction of osteoblast lineage differentiation [138,139]. Moreover, the effects of mechanical strain on osteogenic and adipogenic differentiation of cultured MSCs were also studied. In MSCs subjected to strain stimulation, levels of osteogenic markers (RUNX2 and OSX) gradually increased, while levels of adipogenic markers (PPARγ-2) and the emergence of lipid droplets decreased. Thus, mechanical strain promotes differentiation of MSCs into osteoblasts and impedes differentiation into adipocytes, clarifying the mechanisms underlying the effects of exercise on bone repair and reconstruction [140]. Since lamin A/C is implicated in mechanotransduction, mechanical strain that MSCs support would influence their differentiation through the envelope protein lamin A/C.
Matrix stiffness could determine MSCs’ fate, since nuclear shape changes depending on the substrate that is surrounding the cell. On soft substrate, the nuclear envelope is wrinkled and relaxed, whereas, on stiff substrate, the nucleus is flattened by stress fibers and appears tense and smooth [141]. So, remodeling of the nucleus (based on the substrate) could have significant regulatory effects on cell differentiation and function [135]. In this way, lamin A/C levels will be tightly related with matrix stiffness: lamin A/C level increases with tissue stiffness and decreases in soft matrix. Finally, lamin A levels and matrix stiffness regulate MSCs’ differentiation [18]. For adipogenic differentiation, a soft matrix and low lamin A/C levels (or LMNA knockdown [142]) are needed. Conversely, high lamin A levels and a hard matrix (Elasticity = 25–40 kPa, at least as pre-calcified bone [22]) is needed for osteoblast differentiation [30]. As mentioned before, this occurs because matrix stiffness tenses the nucleus, increasing lamin A/C content, thus suppressing soft tissue phenotypes such as adipose tissue [143].
As occurred in MSCs migration, surface curvature also regulates differentiation, since focal adhesions have the ability to sense the mechanical cues and regulate signaling pathway responses [144]. It has been demonstrated that convex spherical surfaces induce osteogenic marker expression, and thus osteogenic differentiation of MSCs, compared to flat and concave spherical structures. This effect was observed both in cells cultured in expansion medium and in osteogenic medium. Furthermore, equally, F-actin and osteocalcin intensity levels increased with increasing curvature, indicating that cells can also sense and respond to the magnitude of curvature. The level of lamin A/C was quantified in different curvatures: on convex substrates, lamin A/C levels were 2.5× higher compared to concave surfaces, and 1.4× higher compared to flat surfaces. High lamin A/C levels suggest that high intracellular tensions are exerted on the nuclei of cells on convex surfaces. These results show that in addition to substrate stiffness, 3D surface curvature is a further relevant parameter that can change the stress fiber forces on the nucleus, and thus nuclear morphology and lamin A expression [82].
3.2.3. Response of MSCs to Other Mechanosensitive Pathways
New mechanisms by which nuclear lamin A/C mediates cell differentiation have been evaluated [145]: the interaction with other mechanosensitive pathways that tightly regulate differentiation of MSCs. The retinoic acid (RA) pathway plays a role in development and regeneration, in addition to participating in MSCs’ lineage specification [146]. Concerning osteogenesis, it has been demonstrated that RA inhibits differentiation and mineralization of pre-osteoblasts by downregulation of the Wnt/β-Catenin signaling pathway [147]. Lamin A is strongly linked with vitamin A/RA, since lamin A transcription, dephosphorylation and subsequent stabilization are regulated by the RA pathway [18]. How antagonists and agonists of RA receptors (RAR) could regulate lamin A levels and MSCs differentiation has been analyzed. While RAR-specific antagonist increases the expression of lamin A in rigid matrix, RAR-agonist represses its expression. Therefore, RAR-antagonist promotes lamin A-dependent osteogenesis on rigid substrates, with pretreated xenografts calcifying in vivo to a similar extent as native bone. In conclusion, lamin A and the RA pathway are involved in MSCs, sensing substrate stiffness and lineage determination [148].
Another pathway that modulates cell fate determination of MSCs is the Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ). These proteins are key regulators in cells, perceiving their microenvironments [149] and then determining MSCs’ fate. In response to stiff substrates, YAP1 expression increases, is translocated into the nucleus and osteogenesis in MSCs is enhanced [150]. In this way, the YAP/TAZ pathway plays a crucial role in human adipo-osteogenic differentiation induced by ECM stiffness [151]. Increasing YAP expression enhances osteogenic differentiation but suppresses differentiation to adipocytes. On the contrary, low YAP levels promote adipogenic differentiation but inhibit osteogenic differentiation. However, how lamin A/C affects the YAP/TAZ pathway is still not fully understood. Evidence suggests that mechanosensing through YAP depends on forces transmitted through the cytoskeleton and to the nucleus [152]. In this way, lamin A/C might have crosstalk with the YAP/TAZ pathway mediated by actin filaments. Bertrand et al. demonstrated that human myoblasts with LMNA mutations have mechanosensing defects through a YAP-dependent pathway [153]. Moreover, lamin A overexpression in MSCs on stiff matrix produces a decrease in both total YAP1 levels and nuclear localization, but LMNA knockdown also resulted in decreased YAP1 levels [18]. Therefore, the results obtained until now do not have a consensus, besides being cell-type-dependent.
Finally, the serum response factor (SRF)/megakaryoblastic leukaemia 1(MKL1) pathway senses mechanical signals via actin polymerization [154]. Regulation of the transcriptional coactivator MKL1 by actin cytoskeleton dynamics decreases adipocyte differentiation by the inhibition of PPARγ [155]. The same result was obtained with the SRF transcription factor [156]. Moreover, SRF-deficient adult mice presented a marked decrease in bone mineral density and bone formation, whereas SFR-deficient osteoblasts exhibited reduced cell differentiation and mineralization in vitro. The main reason is that SRF deficiency decreased the transcriptional activity of RUNX2, pointing out the relevance of SRF in osteoblast differentiation. On the other hand, SRF indirectly regulates lamin A by nuclear actin binding [157]. It has been reported that lamin A/C promotes SRF/Mkl1-dependent transcription: LMNA-deficient mutant cells have shown impaired nuclear translocation and downstream signaling of the mechanosensitive transcription factor MKL1 [158]. Moreover, LMNA knockdown suppress the transcription of several components of the SRF pathway [159] as well as enhances adipogenesis [18].
3.3. MSCs’ Migration versus Differentiation
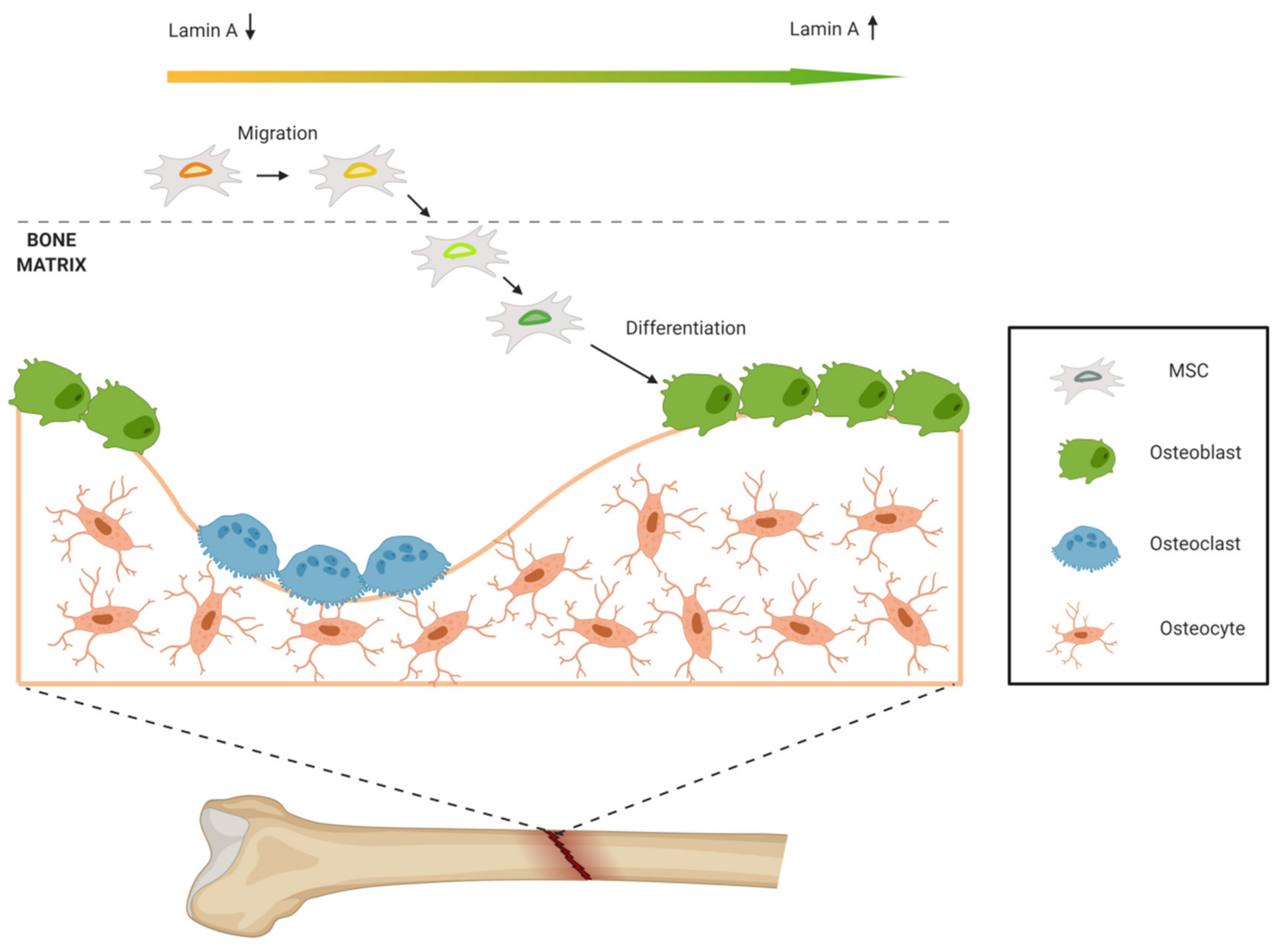
It is known that cells with higher migration ability tend to have enhanced differentiation potential, but the optimum amount of lamin A/C is different for MSCs’ migration and differentiation [43]. During bone formation, MSCs migrate until they reach the bone. For MSCs’ migration, soft nucleus allows cell deformation and motion through tight spaces, so low lamin A/C levels are required. Conversely, to promote osteogenic differentiation, high lamin A/C levels are needed. This contrary phenomenon may be the result of mechanical microenvironment modulation. When MSCs reach the bone, they adhere to bone tissue and mechanical signaling changes: the 3D geometry is different, and the rigidity of the matrix is higher. In this way, lamin A/C levels increase, promoting osteogenic differentiation and forming new bone. So, MSCs with high migration ability may be more sensitive to force and able to upregulate lamin A/C in mechanical microenvironment for later osteogenic differentiation (Figure 3).
4. Lamin A/C Dysfunction in MSCs, Aging and Bone Disease
4.1. Lamin A/C Levels Alterations
As mentioned above, levels of lamin A/C expression fluctuate depending on MSCs’ fate, being higher under osteogenesis conditions, and therefore in osteoblasts [18], and lower in adipogenesis and adipocytes [130]. Subsequently, overexpression of lamin A/C is known to promote osteogenesis differentiation [25], and on the contrary, depletion of lamin A/C levels is associated to a shift to adipogenesis at the expense of osteogenesis by MSCs [30,113,160]. The need of a tight regulation of lamin A/C levels to maintain cellular homeostasis appears evident in the context of aging. Thus, aged mice were shown to have less lamin A/C levels in osteoblasts [161]. This is in agreement with the observation that in aging, the LMNA dysregulation is among the mechanisms leading to MSCs’ fate imbalance, increasing adipogenesis at the expense of osteogenesis, a hallmark of age-related bone pathologies [162]. Closely linked to aging, frailty syndrome is usually suffered by older adults, characterized by exhaustion, unintentional weight loss and a multisystemic function decline. Interestingly, frailty patients have been shown to possess low percentages of circulating osteoprogenitors (COPs) [163], cells with characteristics of MSCs [164]. Moreover, a later study proposed lamin A/C as a potential biomarker for this condition, since lamin A/C levels were found to be decreased in COPs from aged individuals suffering frailty [165]. In light of these last evidences, frailty is starting to be considered as a mesenchymal disease and therefore, there are clinical trials addressing MSCs-based therapies to counteract it [166,167].
4.2. Premature Aging Syndromes
According to the Universal Mutation Database (www.umd.be/LMNA/), there are over 500 mutations reported on the LMNA gene, which lead to at least 15 different laminopathies, a group of rare diseases mainly affecting mesenchymal tissues [168]. Initially, laminopathies were divided into two groups: as tissue-specific disorders, such as striated muscle diseases, lipodystrophic syndromes and peripheral neuropathies, or as multisystem diseases, which affect several tissues, of accelerated aging. However, the observation that there were overlapping clinical phenotypes among different laminopathies [169] suggested that they should be a continuum of related disorders sharing common mechanisms rather than different, independent entities.
Interestingly, bone pathologies such as skeletal deformities and/or osteoporosis have been described in different LMNA-linked accelerating aging syndromes, which differ between them in the time of onset and in the severity of the progeroid phenotypes. Since these premature aging syndromes share an altered lamin A/C protein, it has been proposed that mutated lamin A/C has a negative impact on the homeostasis of MSCs, the natural progenitors of osteoblasts, leading to the observed abnormal bone phenotypes. Next, we will summarize the pathological bone tissue findings in the context of these premature aging laminopathies:
4.2.1. Hutchinson-Gilford Progeria Syndrome (HGPS)
HGPS is a devastating genetic disease, characterized by multiple features of premature aging which mainly affects tissues of mesenchymal origin [170]. Affected children appear healthy at birth, but soon after, exhibit severe growth retardation, lipodystrophy, alopecia and skin changes, among others. Cardiovascular dysfunction usually leads to patients’ death in early teenage years [170]. HGPS is caused by a de novo point mutation in the LMNA gene, which activates a splice donor site, resulting in a smaller protein lacking 50 amino acids, among them the cleavage site for the zinc metalloproteinase ZMPSTE24. As a result, the truncated lamin A protein, named as progerin, is permanently farnesylated and therefore tightly associated with the nuclear envelope, leading to nucleus abnormalities [171,172]. Indeed, the nuclear blebbings observed in cells from HGPS patients is considered to be a cellular hallmark of the disease, and to a greater extent, of other laminopathies. HGPS patients develop low bone mass and atypical skeletal geometry, and HGPS is thus considered to be a skeletal dysplasia [173]. Since osteoblasts, the bone forming cells, come from MSCs’ differentiation, an impaired MSCs osteogenesis could be a possible explanation for this bone phenotype in progeria. In fact, MSCs exhibited the higher expression of progerin among different cell types differentiated from progeria-MSCs [174]. Several studies have confirmed this hypothesis: Scaffidi and Misteli were the first addressing this possibility, by the use of human immortalized MSCs stably expressing progerin [175]. Unexpectedly, and taking into account the bone phenotypes of HGPS patients, such as osteoporosis, they showed an increased osteogenesis in MSCs due to progerin accumulation. This result has been later corroborated by other groups using primary human MSCs derived from progeria-iPSCs [176,177]. Interestingly, secretome from MSCs which accumulate prelamin A, another farnesylated precursor of lamin A, whose accumulation is associated with premature aging and bone pathological phenotypes [178], has been shown to be pro-osteogenic, enhancing the osteogenesis of MSCs [179]. The high bone turnover that HGPS patients show has been proposed as an explanation of this “premature osteogenesis” induced by accumulation of unprocessed lamin A precursors in MSCs.
4.2.2. Mandibuloacral Dysplasia Type A (MADA)
There are about 15 different LMNA mutations leading to MADA [180], a progeroid laminopathy with mildly accelerating aging and a special affectation of bone and adipose tissue [181]. Regarding bone phenotypes, affected patients usually exhibit osteolysis and osteoporosis, suggesting an increased bone turnover and a defective osteogenesis of MSCs as causative altered processes; however, the studies with primary cells from MADA patients to address this question are scarce. Pre-osteoblasts isolated from one MADA patient carrying a homozygous mutation R527H in the LMNA gene showed no impairment in osteogenic differentiation [182]. However, the secretome composition from these MADA-pre-osteoblasts was altered, showing an increased expression of osteoprotegerin and TGF-β2. The MADA-secretome enhanced osteoclast differentiation, which could explain an increased bone turnover and therefore the osteolysis phenotype seen in these patients.
4.2.3. Atypical Progeroid Syndromes (APS)
Over 20 mutations in the LMNA gene lead to APS (www.umd.be/LMNA/). Patients affected by atypical progeroid syndromes have variable manifestations of progeroid features, becoming evident in the second or third decade of life. Bone pathologies related to aging have also been described in these patients, such as osteoporosis [183,184,185], and thinning of cortical bone together with femoral fractures in different reported cases of APS [185]. LMNA mutations leading to APS do not affect lamin A processing, and therefore, cells isolated from patients have been shown to not accumulate either of the two pathological unprocessed forms of lamin A: progerin or prelamin A [186]. However, these mutations seem to have a role in MSCs’ fate, thus hampering osteogenesis and leading to the described bone phenotypes, but unfortunately, there are not any studies addressing this question. Currently, the underlying mechanisms leading to APS are unknown, but it is likely that the mutation has a negative impact in the dimerization of mutant lamin A/C, and therefore in the proper formation of lamin A/C filament meshwork. This, in turn, would hamper the proper formation of nuclear lamina, disturbing lamin A/C interactions with the other nuclear envelope proteins as well as with transcription factors and chromatin.
5. Conclusions
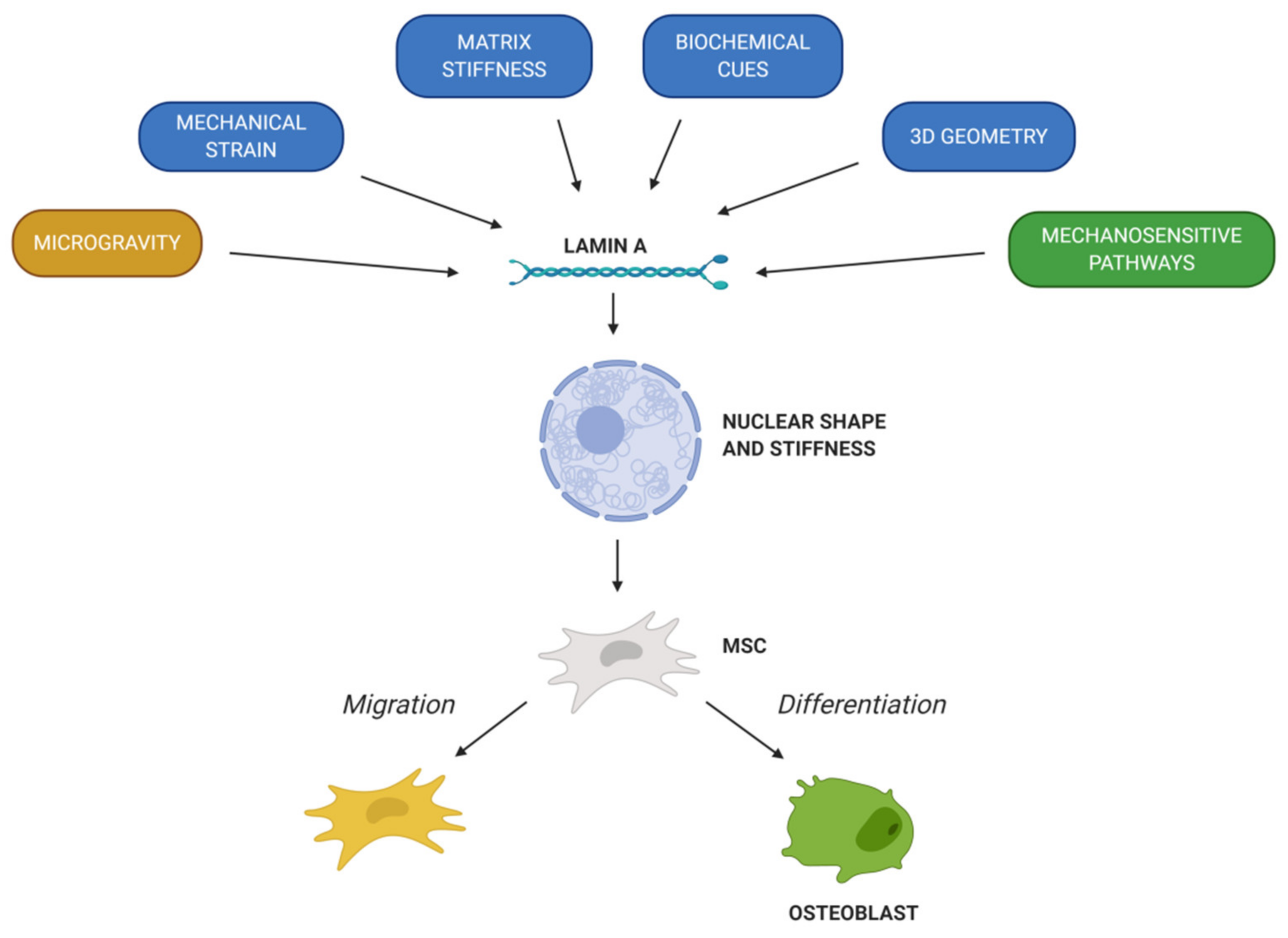
The different characteristics of MSCs pinpoint them as crucial tools to elucidate the mechanisms governing bone repair and disorders. Likewise, as MSCs present many beneficial properties, they are under extensive investigation for their potential in bone therapy. MSCs have demonstrated the capacity to migrate into specific tissues and differentiate into chondrogenic, osteogenic or adipogenic lineage. However, studies in vivo have shown that the migration and differentiation capacity of MSCs is limited, so the expected engraftment is not usually obtained. In this way, factors involved in MSCs’ migration and osteogenic differentiation must be widely studied. Chemical factors (chemokines, cytokines and growth factors) are the ones that induce MSCs’ migration to injured tissue and determinate lineage specification. In addition, mechanical cues play a key role in MSCs’ migration and differentiation. In fact, the nuclear envelope protein lamin A/C regulates the response of MSCs to external factors. First of all, lamin A/C levels determinate the nuclear shape and stiffness of the cell, which is essential to promote MSCs’ migration and cell fate. Then, lamin A/C is linked with the LINC complex, thus connecting cytoskeleton with the nucleus and playing a vital role in mechanotransduction. In summary, mechanical strain, matrix stiffness, 3D geometry and microgravity will regulate MSCs’ migration and differentiation into osteoblasts, thus promoting bone formation and remodeling (Figure 4). Moreover, LMNA mutations and level alterations could disrupt MSCs’ fate. Indeed, several LMNA mutations affect bone tissue homeostasis and lead to a pathological condition, such as premature aging syndromes. All in all, it is necessary to further delve into the knowledge of lamin A in MSCs’ migration and osteogenic differentiation to understand its implication in bone formation and remodeling; that way, to develop new potential therapies to address bone disorders.
Author Contributions
N.A.-S., I.M. and A.I., writing—original draft; A.I. and C.I.R., revising and editing of the manuscript; C.I.R., funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by: Instituto de Salud Carlos III through the projects No. PI15/00820 and PI18/00202 (Co-funded by European Regional Development Fund/European Social Fund; “A way to make Europe”/“Investing in your future”), Basque Country government under the ELKARTEK program, No. kk-2018/00031/BC and No. kk-2019/00093/BC and Fundación Mutua Madrileña, No AP165892017.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the collection, analyses or interpretation of data; in the writing of the manuscript, or in the decision to publish the review.
Abbreviations
| Definition: | Abbreviation: |
| Two-dimensional | 2D |
| Three-dimensional | 3D |
| Atypical progeroid syndromes | APS |
| Bone marrow mesenchymal stem cells | BMSCs |
| Bone morphogenetic protein 2 | BMP-2 |
| Bone sialo-protein | BSP |
| Circulating osteoprogenitors | COPs |
| Cluster of differentiation | CD |
| Dentin matrix acidic phosphoprotein 1 | DMP-1 |
| Extracellular Matrix | ECM |
| Human leucocyte antigen-DR | HLA-DR |
| Hutchinson-Gildford progeria syndrome | HGPS |
| Interleukin | IL |
| Linker of nucleus and cytoskeletonLow density lipoprotein | LINCLDL |
| Mandibuloacral dysplasia type A | MADA |
| Inner nuclear membrane proteinMegakaryoblastic leukaemia 1 | MAN-1MKL1 |
| Mesenchymal stem cells | MSCs |
| Mouse embryonic fibroblasts | MEFs |
| Nuclear envelope spectrin-repeat-containing proteins | Nesprins |
| Osteocalcin | OCN |
| Osteoprotegerin | OPG |
| Osterix | OSX |
| Peroxisome proliferator-activated receptor | PPAR |
| Platelet-derived growth factors | PDGFs |
| Receptor activator of nuclear factor k-B ligand | RANKL |
| Retinoic acid | RA |
| Retinoic acid receptors | RAR |
| Run-t related transcription factor | RUNX2 |
| Serum response factor | SRF |
| Sry-related high-mobility group box 9 | SOX9 |
| Transcriptional coactivator with PDZ-binding motif | TAZ |
| Transforming growth factor-β1 | TGF-β1 |
| Tumor necrosis factor-α | TNF-α |
| Vascular endothelial growth factor A | VEGFA |
| Yes-associated protein | YAP |
References
- Burke, B.; Stewart, C.L. The nuclear lamins: Flexibility in function. Nat. Rev. Mol. Cell. Biol. 2013, 14, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.A.; Hosick, T.J.; Sinensky, M. Isoprenylation is required for the processing of the lamin A precursor. J. Cell. Biol. 1990, 110, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtz, D.; Tanaka, R.A.; Hartwig, J.; McKeon, F. The CaaX motif of lamin A functions in conjunction with the nuclear localization signal to target assembly to the nuclear envelope. Cell 1989, 59, 969–977. [Google Scholar] [CrossRef]
- Fong, L.G.; Ng, J.K.; Lammerding, J.; Vickers, T.A.; Meta, M.; Cote, N.; Gavino, B.; Qiao, X.; Chang, S.Y.; Young, S.R.; et al. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Investig. 2006, 116, 743–752. [Google Scholar] [CrossRef]
- Shimi, T.; Pfleghaar, K.; Kojima, S.; Pack, C.G.; Solovei, I.; Goldman, A.E.; Adam, S.A.; Shumaker, D.K.; Kinjo, M.; Cremer, T.; et al. The A- and B-type nuclear lamin networks: Microdomains involved in chromatin organization and transcription. Genes Dev. 2008, 22, 3409–3421. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Chojnowski, A.; Boudier, T.; Lim, J.S.; Ahmed, S.; Ser, Z.; Stewart, C.; Burke, B. A-type Lamins Form Distinct Filamentous Networks with Differential Nuclear Pore Complex Associations. Curr. Biol. 2016, 26, 2651–2658. [Google Scholar] [CrossRef] [Green Version]
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.E.; Adam, S.A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell 2015, 26, 4075–4086. [Google Scholar] [CrossRef]
- Turgay, Y.; Medalia, O. The structure of lamin filaments in somatic cells as revealed by cryo-electron tomography. Nucleus 2017, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Nmezi, B.; Xu, J.; Fu, R.; Armiger, T.J.; Rodriguez-Bey, G.; Powell, J.S.; Ma, H.; Sullivan, M.; Tu, Y.; Chen, N.Y.; et al. Concentric organization of A- and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc. Natl. Acad. Sci. USA 2019, 116, 4307–4315. [Google Scholar] [CrossRef] [Green Version]
- Figueiras, E.; Silvestre, O.F.; Ihalainen, T.O.; Nieder, J.B. Phasor-assisted nanoscopy reveals differences in the spatial organization of major nuclear lamina proteins. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118530. [Google Scholar] [CrossRef]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechat, T.; Adam, S.A.; Taimen, P.; Shimi, T.; Goldman, R.D. Nuclear lamins. Cold Spring Harb. Perspect. Biol. 2010, 2, a000547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, F.; Perestrelo, A.R.; Vinarsky, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Jaalouk, D.E.; Lammerding, J. Novel insights into the disease etiology of laminopathies. Rare Dis. 2013, 1, e27002. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Osborne, L.D.; Van Landeghem, L.; Sharek, L.; Superfine, R.; Garcia-Mata, R.; Burridge, K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell. Biol. 2014, 16, 376–381. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Dechat, T.; Foisner, R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015, 29, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Osmanagic-Myers, S.; Foisner, R. The structural and gene expression hypotheses in laminopathic diseases-not so different after all. Mol. Biol. Cell 2019, 30, 1786–1790. [Google Scholar] [CrossRef]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [Green Version]
- Harada, T.; Swift, J.; Irianto, J.; Shin, J.W.; Spinler, K.R.; Athirasala, A.; Diegmiller, R.; Dingal, P.C.; Ivanovska, I.L.; Discher, D.E. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 2014, 204, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Lammerding, J. Mechanics of the nucleus. Compr. Physiol. 2011, 1, 783–807. [Google Scholar]
- Ozcivici, E.; Luu, Y.K.; Adler, B.; Qin, Y.X.; Rubin, J.; Judex, S.; Rubin, C.T. Mechanical signals as anabolic agents in bone. Nat. Rev. Rheumatol. 2010, 6, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muncie, J.M.; Weaver, V.M. The Physical and Biochemical Properties of the Extracellular Matrix Regulate Cell Fate. Curr. Top. Dev. Biol. 2018, 130, 1–37. [Google Scholar] [PubMed]
- Medhat, D.; Rodriguez, C.I.; Infante, A. Immunomodulatory Effects of MSCs in Bone Healing. Int. J. Mol. Sci. 2019, 20, 5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermeo, S.; Vidal, C.; Zhou, H.; Duque, G. Lamin A/C Acts as an Essential Factor in Mesenchymal Stem Cell Differentiation Through the Regulation of the Dynamics of the Wnt/beta-Catenin Pathway. J. Cell. Biocehm. 2015, 116, 2344–2353. [Google Scholar] [CrossRef]
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 12097–12102. [Google Scholar] [CrossRef] [Green Version]
- Su, P.; Tian, Y.; Yang, C.; Ma, X.; Wang, X.; Pei, J.; Qian, A. Mesenchymal Stem Cell Migration during Bone Formation and Bone Diseases Therapy. Int. J. Mol. Sci. 2018, 19, 2343. [Google Scholar] [CrossRef] [Green Version]
- Fakhry, M.; Hamade, E.; Badran, B.; Buchet, R.; Magne, D. Molecular mechanisms of mesenchymal stem cell differentiation towards osteoblasts. World J. Stem Cells 2013, 5, 136–148. [Google Scholar] [CrossRef]
- Berendsen, A.D.; Olsen, B.R. Bone development. Bone 2015, 80, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Akter, R.; Rivas, D.; Geneau, G.; Drissi, H.; Duque, G. Effect of lamin A/C knockdown on osteoblast differentiation and function. J. Bone Miner. Res. 2009, 24, 283–293. [Google Scholar] [CrossRef]
- Rauner, M.; Sipos, W.; Goettsch, C.; Wutzl, A.; Foisner, R.; Pietschmann, P.; Hofbauer, L.C. Inhibition of lamin A/C attenuates osteoblast differentiation and enhances RANKL-dependent osteoclastogenesis. J. Bone Miner. Res. 2009, 24, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Tsukune, N.; Naito, M.; Kubota, T.; Ozawa, Y.; Nagao, M.; Ohashi, A.; Sato, S.; Takahashi, T. Lamin A overexpression promotes osteoblast differentiation and calcification in the MC3T3-E1 preosteoblastic cell line. Biophys. Res. Commun. 2017, 488, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of Action of Living, Apoptotic, and Dead MSCs. Front. Immunol. 2019, 10, 1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, A. Mesenchymal stromal cells. Curr. Opin. Hematol. 2006, 13, 419–425. [Google Scholar] [CrossRef]
- Wagner, W.; Wein, F.; Seckinger, A.; Frankhauser, M.; Wirkner, U.; Krause, U.; Blake, J.; Schwager, C.; Eckstein, V.; Ansorge, W.; et al. Comparative characteristics of mesenchymal stem cells from human bone marrow, adipose tissue, and umbilical cord blood. Exp. Hematol. 2005, 33, 1402–1416. [Google Scholar] [CrossRef]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells—current trends and future prospective. Biosci. Rep. 2015, 35, e00191. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Discher, D.E.; Péault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.J.; Lee, H.W.; Kalimuthu, S.; Kim, T.J.; Kim, H.M.; Baek, S.H.; Zhu, L.; Oh, J.M.; Son, S.H.; Chung, H.Y.; et al. In Vivo migration of mesenchymal stem cells to burn injury sites and their therapeutic effects in a living mouse model. J. Control Release 2018, 279, 79–88. [Google Scholar] [CrossRef]
- Kawai, T.; Katagiri, W.; Osugi, M.; Sugimura, Y.; Hibi, H.; Ueda, M. Secretomes from bone marrow-derived mesenchymal stromal cells enhance periodontal tissue regeneration. Cytotherapy 2015, 17, 369–381. [Google Scholar] [CrossRef]
- Nakamura, Y.; Ishikawa, H.; Kawai, K.; Tabata, Y.; Suzuki, S. Enhanced wound healing by topical administration of mesenchymal stem cells transfected with stromal cell-derived factor-1. Biomaterials 2013, 34, 9393–9400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnecchi, M.; Danieli, P.; Malpasso, G.; Ciuffreda, M.C. Paracrine Mechanisms of Mesenchymal Stem Cells in Tissue Repair. Methods Mol. Biol. 2016, 1416, 123–146. [Google Scholar]
- Chen, L.; Jiang, F.; Qiao, Y.; Li, H.; Wei, Z.; Huang, T.; Lan, J.; Xia, Y.; Li, J. Nucleoskeletal stiffness regulates stem cell migration and differentiation through lamin A/C. J. Cell Physiol. 2018, 233, 5112–5118. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.; Liu, D.D.; Thakor, A.S. Mesenchymal Stromal Cell Homing: Mechanisms and Strategies for Improvement. iScience 2019, 15, 421–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loi, F.; Cordova, L.A.; Pajarinen, J.; Lin, T.H.; Yao, Z.; Goodman, S.B. Inflammation, fracture and bone repair. Bone 2016, 86, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.B.; Giannoudis, P.V.; Boxall, S.A.; McGonagle, D.; Jones, E. The systemic influence of platelet-derived growth factors on bone marrow mesenchymal stem cells in fracture patients. BMC Med. 2015, 13, 6. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Xia, X.; Yeh, J.; Kua, H.; Liu, H.; Mishina, Y.; Hao, A.; Li, B. PDGF-AA promotes osteogenic differentiation and migration of mesenchymal stem cell by down-regulating PDGFRalpha and derepressing BMP-Smad1/5/8 signaling. PLoS ONE 2014, 9, e113785. [Google Scholar]
- Kon, T.; Cho, T.J.; Aizawa, T.; Yamazaki, M.; Nooh, N.; Graves, D.; Gerstenfeld, L.C.; Einhorn, T.A. Expression of osteoprotegerin, receptor activator of NF-kappaB ligand (osteoprotegerin ligand) and related proinflammatory cytokines during fracture healing. J. Bone Miner. Res. 2001, 16, 1004–1014. [Google Scholar] [CrossRef]
- Cho, H.H.; Kyoung, K.M.; Seo, M.J.; Kim, Y.J.; Bae, Y.C.; Jung, J.S. Overexpression of CXCR4 increases migration and proliferation of human adipose tissue stromal cells. Stem Cells Dev. 2006, 15, 853–864. [Google Scholar] [CrossRef]
- Putra, A.; Ridwan, F.B.; Putridewi, A.I.; Kustiyah, A.R.; Wirastuti, K.; Sadyah, N.A.C.; Rosdiana, I.; Munir, D. The Role of TNF-alpha induced MSCs on Suppressive Inflammation by Increasing TGF-beta and IL-10. Open Access Maced. J. Med. Sci. 2018, 6, 1779–1783. [Google Scholar] [CrossRef] [Green Version]
- Claes, L.; Recknagel, S.; Ignatius, A. Fracture healing under healthy and inflammatory conditions. Nat. Rev. Rheumatol. 2012, 8, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Eberspaecher, H.; Lefebvre, V.; De Crombrugghe, B. Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev. Dyn. 1997, 209, 377–386. [Google Scholar] [CrossRef]
- Hattori, T.; Muller, C.; Gebhard, S.; Bauer, E.; Pausch, F.; Schlund, B.; Bosl, M.R.; Hess, A.; Surmann-Schmitt, C.; von der Mark, H.; et al. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development 2010, 137, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Bear, J.E.; Haugh, J.M. Directed migration of mesenchymal cells: Where signaling and the cytoskeleton meet. Curr. Opin. Cell Biol. 2014, 30, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Wolf, K.; Lammerding, J. Nuclear mechanics during cell migration. Curr. Opin. Cell Biol. 2011, 23, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Wintner, O.; Hirsch-Attas, N.; Schlossberg, M.; Brofman, F.; Friedman, R.; Kupervaser, M.; Kitsberg, D.; Buxboim, A. A Unified Linear Viscoelastic Model of the Cell Nucleus Defines the Mechanical Contributions of Lamins and Chromatin. Adv. Sci. 2020, 7, 1901222. [Google Scholar] [CrossRef] [Green Version]
- Davidson, P.M.; Denais, C.; Bakshi, M.C.; Lammerding, J. Nuclear deformability constitutes a rate-limiting step during cell migration in 3-D environments. Cell. Mol. Bioeng. 2014, 7, 293–306. [Google Scholar] [CrossRef] [Green Version]
- Denais, C.; Lammerding, J. Nuclear mechanics in cancer. Adv. Exp. Med. Biol. 2014, 773, 435–470. [Google Scholar]
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Investig. 2004, 113, 370–378. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Hale, C.M.; Panorchan, P.; Khatau, S.B.; George, J.P.; Tseng, Y.; Stewart, C.L.; Hodzic, D.; Wirtz, D. Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophys. J. 2007, 93, 2542–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins A and C but not lamin B1 regulate nuclear mechanics. J. Biol. Chem. 2006, 281, 25768–25780. [Google Scholar] [CrossRef] [Green Version]
- Dorland, Y.L.; Cornelissen, A.S.; Kuijk, C.; Tol, S.; Hoogenboezem, M.; van Buul, J.D.; Nolte, M.A.; Voermans, C.; Huveneers, S. Nuclear shape, protrusive behaviour and in vivo retention of human bone marrow mesenchymal stromal cells is controlled by Lamin-A/C expression. Sci. Rep. 2019, 9, 14401. [Google Scholar] [CrossRef] [PubMed]
- Dahl, K.N.; Kalinowski, A. Nucleoskeleton mechanics at a glance. J. Cell. Sci. 2011, 124 Pt 5, 675–678. [Google Scholar] [CrossRef] [Green Version]
- Pajerowski, J.D.; Dahl, K.N.; Zhong, F.L.; Sammak, P.J.; Discher, D.E. Physical plasticity of the nucleus in stem cell differentiation. Proc. Natl. Acad. Sci. USA 2007, 104, 15619–15624. [Google Scholar] [CrossRef] [PubMed]
- Méjat, A.; Decostre, V.; Li, J.; Renou, L.; Kesari, A.; Hantaï, D.; Stewart, C.L.; Xiao, X.; Hoffman, E.; Bonne, G.; et al. Lamin A/C-mediated neuromuscular junction defects in Emery-Dreifuss muscular dystrophy. J. Cell Biol. 2009, 184, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Belaadi, N.; Aureille, J.; Guilluy, C. Under Pressure: Mechanical Stress Management in the Nucleus. Cells 2016, 5, 27. [Google Scholar] [CrossRef]
- Bouzid, T.; Kim, E.; Riehl, B.D.; Esfahani, A.M.; Rosenbohm, J.; Yang, R.; Duan, B.; Lim, J.Y. The LINC complex, mechanotransduction, and mesenchymal stem cell function and fate. J. Biol. Eng. 2019, 13, 68. [Google Scholar] [CrossRef]
- Haque, F.; Lloyd, D.J.; Smallwood, D.T.; Dent, C.L.; Shanahan, C.M.; Fry, A.M.; Trembath, R.C.; Shackleton, S. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol. Cell. Biol. 2006, 26, 3738–3751. [Google Scholar] [CrossRef] [Green Version]
- Philip, J.T.; Dahl, K.N. Nuclear mechanotransduction: Response of the lamina to extracellular stress with implications in aging. J. Biomech. 2008, 41, 3164–3170. [Google Scholar] [CrossRef]
- Fu, X.; Liu, G.; Halim, A.; Ju, Y.; Luo, Q.; Song, A.G. Mesenchymal Stem Cell Migration and Tissue Repair. Cells 2019, 8, 784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Huang, X.; Zhou, Y.; Jin, R.; Li, Q. Mechanical Stretching Promotes Skin Tissue Regeneration via Enhancing Mesenchymal Stem Cell Homing and Transdifferentiation. Stem Cells Transl. Med. 2016, 5, 960–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.B.; Wang, J.; Chiang, C.A.; Sheng, L.L.; Li, Q.F. Mechanical stretch upregulates SDF-1α in skin tissue and induces migration of circulating bone marrow-derived stem cells into the expanded skin. Stem Cells 2013, 31, 2703–2713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Luo, Q.; Chen, Z.; Sun, J.; Xu, B.; Ju, Y.; Song, G. Cyclic mechanical stretching promotes migration but inhibits invasion of rat bone marrow stromal cells. Stem Cell Res. 2015, 14, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammerding, J.; Lee, R.T. The nuclear membrane and mechanotransduction: Impaired nuclear mechanics and mechanotransduction in lamin A/C deficient cells. Novartis Found Symp. 2005, 264, 264–273. [Google Scholar]
- Vincent, L.G.; Choi, Y.S.; Alonso-Latorre, B.; del Álamo, J.C.; Engler, A.J. Mesenchymal stem cell durotaxis depends on substrate stiffness gradient strength. Biotechnol. J. 2013, 8, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.; Swift, J.; Dingal, P.C.; Shah, P.; Shin, J.W.; Discher, D.E. Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain. J. Cell. Biol. 2012, 199, 669–683. [Google Scholar] [CrossRef] [Green Version]
- Isermann, P.; Lammerding, J. Nuclear mechanics and mechanotransduction in health and disease. Curr. Biol. 2013, 23, R1113–R1121. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.; Discher, D.E. Matrix rigidity regulates microtubule network polarization in migration. Cytoskeleton 2017, 74, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Luo, Q.; Sun, J.; Song, G. Nucleus and nucleus-cytoskeleton connections in 3D cell migration. Exp. Cell. Res. 2016, 348, 56–65. [Google Scholar] [CrossRef]
- Stephens, A.D.; Banigan, E.J.; Adam, S.A.; Goldman, R.D.; Marko, J.F. Chromatin and lamin A determine two different mechanical response regimes of the cell nucleus. Mol. Biol. Cell 2017, 28, 1984–1996. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Blanquer, S.B.; Haimi, S.P.; Korus, G.; Dunlop, J.W.; Duda, G.N.; Grijpma, D.W.; Petersen, A. Surface Curvature Differentially Regulates Stem Cell Migration and Differentiation via Altered Attachment Morphology and Nuclear Deformation. Adv. Sci. 2017, 4, 1600347. [Google Scholar] [CrossRef]
- Mao, X.; Chen, Z.; Luo, Q.; Zhang, B.; Song, G. Simulated microgravity inhibits the migration of mesenchymal stem cells by remodeling actin cytoskeleton and increasing cell stiffness. Cytotechnology 2016, 68, 2235–2243. [Google Scholar] [CrossRef] [PubMed]
- Koaykul, C.; Kim, M.H.; Kawahara, Y.; Yuge, L.; Kino-Oka, M. Alterations in Nuclear Lamina and the Cytoskeleton of Bone Marrow-Derived Human Mesenchymal Stem Cells Cultured Under Simulated Microgravity Conditions. Stem Cells Dev. 2019, 28, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Malashicheva, A.; Bogdanova, M.; Zabirnyk, A.; Smolina, N.; Ignatieva, E.; Freilikhman, O.; Fedorov, A.; Dmitrieva, R.; Sjoberg, G.; Sejersen, T.; et al. Various lamin A/C mutations alter expression profile of mesenchymal stem cells in mutation specific manner. Mol. Genet. Metab. 2015, 115, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Almalki, S.G.; Agrawal, D.K. Key transcription factors in the differentiation of mesenchymal stem cells. Differentiation 2016, 92, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.A.; Choi, H.K.; Kim, T.M.; Leem, S.H.; Oh, I.H. Regulation of mesenchymal stromal cells through fine tuning of canonical Wnt signaling. Stem Cell Res. 2015, 14, 356–368. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.C.; Fu, W.M.; Wang, Y.B.; Sun, Y.X.; Xu, L.L.; Wong, C.W.; Chan, K.M.; Li, G.; Waye, M.M.; Zhang, J.F. H19 activates Wnt signaling and promotes osteoblast differentiation by functioning as a competing endogenous RNA. Sci. Rep. 2016, 6, 20121. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, X.; Bikle, D.D. Osteogenic Differentiation of Periosteal Cells during Fracture Healing. J. Cell Physiol. 2017, 232, 913–921. [Google Scholar] [CrossRef] [Green Version]
- Case, N.; Rubin, J. Beta-catenin--a supporting role in the skeleton. J. Cell Biochem. 2010, 110, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Houschyar, K.S.; Tapking, C.; Borrelli, M.R.; Popp, D.; Duscher, D.; Maan, Z.N.; Chelliah, M.P.; Li, J.; Harati, K.; Wallner, C.; et al. Wnt Pathway in Bone Repair and Regeneration—What Do We Know So Far. Front. Cell Dev. Biol. 2018, 6, 170. [Google Scholar] [CrossRef]
- Zanotti, S.; Canalis, E. Notch Signaling and the Skeleton. Endocr. Rev. 2016, 37, 223–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrique, D.; Schweisguth, F. Mechanisms of Notch signaling: A simple logic deployed in time and space. Development 2019, 146, dev172148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Wei, Y.; Lian, J.; Yang, L.; Zhang, X.; Xie, J.; Liu, Q.; Luo, J.; He, B.; Tang, M. Notch signaling pathway promotes osteogenic differentiation of mesenchymal stem cells by enhancing BMP9/Smad signaling. Int. J. Mol. Med. 2017, 40, 378–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenova, D.; Bogdanova, M.; Kostina, A.; Golovkin, A.; Kostareva, A.; Malashicheva, A. Dose-dependent mechanism of Notch action in promoting osteogenic differentiation of mesenchymal stem cells. Cell Tissue Res. 2020, 379, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Song, B.Q.; Chi, Y.; Li, X.; Du, W.J.; Han, Z.B.; Tian, J.J.; Li, J.J.; Chen, F.; Wu, H.H.; Han, L.X.; et al. Inhibition of Notch Signaling Promotes the Adipogenic Differentiation of Mesenchymal Stem Cells Through Autophagy Activation and PTEN-PI3K/AKT/mTOR Pathway. Cell. Physiol. Biochem. 2015, 36, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, M.A.; Gudkova, A.; Zabirnik, A.S.; Ignat’eva, E.V.; Dmitrieva, R.I.; Smolina, N.A.; Kostareva, A.A.; Malashicheva, A.B. Nuclear lamins regulate osteogenic differentiation of mesenchymal stem cells. Tsitologiia 2014, 56, 260–267. [Google Scholar] [CrossRef]
- Sakaki, M.; Koike, H.; Takahashi, N.; Sasagawa, N.; Tomioka, S.; Arahata, K.; Ishiura, S. Interaction between emerin and nuclear lamins. J. Biochem. 2001, 129, 321–327. [Google Scholar] [CrossRef]
- Lee, B.; Lee, T.H.; Shim, J. Emerin suppresses Notch signaling by restricting the Notch intracellular domain to the nuclear membrane. Biochim. Biophys. Acta. Mol. Cell Res. 2017, 1864, 303–313. [Google Scholar] [CrossRef]
- Perepelina, K.; Dmitrieva, R.; Ignatieva, E.; Borodkina, A.; Kostareva, A.; Malashicheva, A. Lamin A/C mutation associated with lipodystrophy influences adipogenic differentiation of stem cells through interaction with Notch signaling. Biochem. Cell Biol. 2018, 96, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Capanni, C.; Cenni, V.; Mattioli, E.; Sabatelli, P.; Ognibene, A.; Columbaro, M.; Parnaik, V.K.; Wehnert, M.; Maraldi, N.M.; Squarzoni, S.; et al. Failure of lamin A/C to functionally assemble in R482L mutated familial partial lipodystrophy fibroblasts: Altered intermolecular interaction with emerin and implications for gene transcription. Exp. Cell. Res. 2003, 291, 122–134. [Google Scholar] [CrossRef]
- Perepelina, K.; Klauzen, P.; Kostareva, A.; Malashicheva, A. Tissue-Specific Influence of Lamin A Mutations on Notch Signaling and Osteogenic Phenotype of Primary Human Mesenchymal Cells. Cells 2019, 8, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.; Shehadeh, L.A.; Yu, H.; Webster, K.A. Age-related molecular genetic changes of murine bone marrow mesenchymal stem cells. BMC Genom. 2010, 11, 229. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Kh Haider, H.; Ahmed, R.P.; Idris, N.M.; Salim, A.; Ashraf, M. Transcriptional profiling of young and old mesenchymal stem cells in response to oxygen deprivation and reparability of the infarcted myocardium. J. Mol. Cell. Cardiol. 2008, 44, 582–596. [Google Scholar] [CrossRef] [Green Version]
- Costa, N.; Paramanathan, S.; Mac Donald, D.; Wierzbicki, A.S.; Hampson, G. Factors regulating circulating vascular endothelial growth factor (VEGF): Association with bone mineral density (BMD) in post-menopausal osteoporosis. Cytokine 2009, 46, 376–381. [Google Scholar] [CrossRef]
- Berendsen, A.D.; Olsen, B.R. Regulation of adipogenesis and osteogenesis in mesenchymal stem cells by vascular endothelial growth factor A. J. Inter. Med. 2015, 277, 674–680. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Berendsen, A.D.; Jia, S.; Lotinun, S.; Baron, R.; Ferrara, N.; Olsen, B.R. Intracellular VEGF regulates the balance between osteoblast and adipocyte differentiation. J. Clin. Investig. 2012, 122, 3101–3113. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. Regulation of osteoblast differentiation by Runx2. Adv. Exp. Med. Biol. 2010, 658, 43–49. [Google Scholar]
- Xu, J.; Li, Z.; Hou, Y.; Fang, W. Potential mechanisms underlying the Runx2 induced osteogenesis of bone marrow mesenchymal stem cells. Am. J. Transl. Res. 2015, 7, 2527–2535. [Google Scholar]
- Komori, T. Regulation of Proliferation, Differentiation and Functions of Osteoblasts by Runx2. Int. J. Mol. Sci. 2019, 20, 1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostlund, C.; Sullivan, T.; Stewart, C.L.; Worman, H.J. Dependence of diffusional mobility of integral inner nuclear membrane proteins on A-type lamins. Biochemistry 2006, 45, 1374–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yeo, L.S.; Vidal, C.; McCorquodale, T.; Herrmann, M.; Fatkin, D.; Duque, G. Decreased bone formation and osteopenia in lamin a/c-deficient mice. PLoS ONE 2011, 6, e19313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heessen, S.; Fornerod, M. The inner nuclear envelope as a transcription factor resting place. EMBO Rep. 2007, 8, 914–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capanni, C.; Mattioli, E.; Columbaro, M.; Lucarelli, E.; Parnaik, V.K.; Novelli, G.; Wehnert, M.; Cenni, V.; Maraldi, N.M.; Squarzoni, S.; et al. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum. Mol. Genet. 2005, 14, 1489–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz de Eguino, G.; Infante, A.; Schlangen, K.; Aransay, A.M.; Fullaondo, A.; Soriano, M.; Garcia-Verdugo, J.M.; Martin, A.G.; Rodriguez, C.I. Sp1 transcription factor interaction with accumulated prelamin a impairs adipose lineage differentiation in human mesenchymal stem cells: Essential role of sp1 in the integrity of lipid vesicles. Stem Cells Transl. Med. 2012, 1, 309–321. [Google Scholar] [CrossRef]
- Infante, A.; Gago, A.; de Eguino, G.R.; Calvo-Fernandez, T.; Gomez-Vallejo, V.; Llop, J.; Schlangen, K.; Fullaondo, A.; Aransay, A.M.; Martin, A.; et al. Prelamin A accumulation and stress conditions induce impaired Oct-1 activity and autophagy in prematurely aged human mesenchymal stem cell. Aging 2014, 6, 264–280. [Google Scholar] [CrossRef] [Green Version]
- Infante, A.; Rodriguez, C.I. Pathologically Relevant Prelamin A Interactions with Transcription Factors. Methods Enzymol. 2016, 569, 485–501. [Google Scholar]
- Casciaro, F.; Beretti, F.; Zavatti, M.; McCubrey, J.A.; Ratti, S.; Marmiroli, S.; Follo, M.Y.; Maraldi, T. Nuclear Nox4 interaction with prelamin A is associated with nuclear redox control of stem cell aging. Aging 2018, 10, 2911–2934. [Google Scholar] [CrossRef]
- Zhuang, H.; Zhang, X.; Zhu, C.; Tang, X.; Yu, F.; Shang, G.W.; Cai, X. Molecular Mechanisms of PPAR-gamma Governing MSC Osteogenic and Adipogenic Differentiation. Curr. Stem Cell Res. Ther. 2016, 11, 255–264. [Google Scholar] [CrossRef]
- Moerman, E.J.; Teng, K.; Lipschitz, D.A.; Lecka-Czernik, B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: The role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell 2004, 3, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.M.; Guo, Y.; Wang, Q.; Xu, Y.; Wang, M.; Chen, H.N.; Shen, W.M. Over-expression of PPAR-gamma2 gene enhances the adipogenic differentiation of hemangioma-derived mesenchymal stem cells in vitro and in vivo. Oncotarget 2017, 8, 115817–115828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boguslavsky, R.L.; Stewart, C.L.; Worman, H.J. Nuclear lamin A inhibits adipocyte differentiation: Implications for Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2006, 15, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.K.; Chen, C.S. Cell adhesion and mechanical stimulation in the regulation of mesenchymal stem cell differentiation. J. Cell. Mol. Med. 2013, 17, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Zhang, Y.; Jing, D.; Shen, Y.; Tang, G.; Huang, S.; Zhao, Z. Mechanobiology of mesenchymal stem cells: Perspective into mechanical induction of MSC fate. Acta Biomater. 2015, 20, 1–9. [Google Scholar] [CrossRef]
- Uzer, G.; Rubin, C.T.; Rubin, J. Cell Mechanosensitivity is Enabled by the LINC Nuclear Complex. Curr. Mol. Biol. Rep. 2016, 2, 36–47. [Google Scholar] [CrossRef] [Green Version]
- McBeath, R.; Pirone, D.M.; Nelson, C.M.; Bhadriraju, K.; Chen, C.S. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 2004, 6, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Bacil, E.D.; Mazzardo Junior, O.; Rech, C.R.; Legnani, R.F.; de Campos, W. Physical activity and biological maturation: A systematic review. Rev. Paul. Pediatr. 2015, 33, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.Y.; Lammerding, J. Lamins at a glance. J. Cell Sci. 2012, 125 Pt 9, 2087–2093. [Google Scholar] [CrossRef] [Green Version]
- Constantinescu, D.; Gray, H.L.; Sammak, P.J.; Schatten, G.P.; Csoka, A.B. Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells 2006, 24, 177–185. [Google Scholar] [CrossRef]
- Fedorchak, G.R.; Kaminski, A.; Lammerding, J. Cellular mechanosensing: Getting to the nucleus of it all. Prog. Biophys. Mol. Biol. 2014, 115, 76–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.; Cho, S.; Discher, D.E. Mechanosensing of matrix by stem cells: From matrix heterogeneity, contractility, and the nucleus in pore-migration to cardiogenesis and muscle stem cells in vivo. Semin. Cell Dev. Biol. 2017, 71, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Chi, G.; Li, P.; Lv, S.; Xu, J.; Xu, Z.; Xia, Y.; Tan, Y.; Xu, J.; Li, L.; et al. Effects of Matrix Stiffness on the Morphology, Adhesion, Proliferation and Osteogenic Differentiation of Mesenchymal Stem Cells. Int. J. Med. Sci. 2018, 15, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Miroshnikova, Y.A.; Nava, M.M.; Wickstrom, S.A. Emerging roles of mechanical forces in chromatin regulation. J. Cell Sci. 2017, 130, 2243–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, R.P.; Finan, J.D.; Guilak, F.; Lee, D.A. Mechanical regulation of nuclear structure and function. Annu. Rev. Biomed. Eng. 2012, 14, 431–455. [Google Scholar] [CrossRef] [Green Version]
- Rubin, J.; Styner, M.; Uzer, G. Physical Signals May Affect Mesenchymal Stem Cell Differentiation via Epigenetic Controls. Exerc. Sport Sci. Rev. 2018, 46, 42–47. [Google Scholar] [CrossRef]
- Cho, S.; Vashisth, M.; Abbas, A.; Majkut, S.; Vogel, K.; Xia, Y.; Ivanovska, I.L.; Irianto, J.; Tewari, M.; Zhu, K.; et al. Mechanosensing by the Lamina Protects against Nuclear Rupture, DNA Damage, and Cell-Cycle Arrest. Dev. Cell 2019, 49, 920–935. [Google Scholar] [CrossRef]
- Sen, B.; Xie, Z.; Case, N.; Ma, M.; Rubin, C.; Rubin, J. Mechanical strain inhibits adipogenesis in mesenchymal stem cells by stimulating a durable beta-catenin signal. Endocrinology 2008, 149, 6065–6075. [Google Scholar] [CrossRef] [Green Version]
- Styner, M.; Meyer, M.B.; Galior, K.; Case, N.; Xie, Z.; Sen, B.; Thompson, W.R.; Pike, J.W.; Rubin, J. Mechanical strain downregulates C/EBPbeta in MSC and decreases endoplasmic reticulum stress. PLoS ONE 2012, 7, e51613. [Google Scholar] [CrossRef]
- Li, R.; Liang, L.; Dou, Y.; Huang, Z.; Mo, H.; Wang, Y.; Yu, B. Mechanical strain regulates osteogenic and adipogenic differentiation of bone marrow mesenchymal stem cells. BioMed Res. Int. 2015, 2015, 873251. [Google Scholar] [CrossRef]
- Swift, J.; Discher, D.E. The nuclear lamina is mechano-responsive to ECM elasticity in mature tissue. J. Cell Sci. 2014, 127 Pt 14, 3005–3015. [Google Scholar] [CrossRef] [Green Version]
- Naito, M.; Omoteyama, K.; Mikami, Y.; Takagi, M.; Takahashi, T. Suppression of lamin A/C by short hairpin RNAs promotes adipocyte lineage commitment in mesenchymal progenitor cell line, ROB-C26. Histochem. Cell Biol. 2012, 137, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Buxboim, A.; Irianto, J.; Swift, J.; Athirasala, A.; Shin, J.W.; Rehfeldt, F.; Discher, D.E. Coordinated increase of nuclear tension and lamin-A with matrix stiffness outcompetes lamin-B receptor that favors soft tissue phenotypes. Mol. Biol. Cell 2017, 28, 3333–3348. [Google Scholar] [CrossRef] [PubMed]
- Sastry, S.K.; Burridge, K. Focal adhesions: A nexus for intracellular signaling and cytoskeletal dynamics. Exp. Cell Res. 2000, 261, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Yang, Y.; Keyimu, R.; Hao, J.; Zhao, Z.; Ye, R. The role of lamin A/C in mesenchymal stem cell differentiation. J. Physiol. Biochem. 2019, 75, 11–18. [Google Scholar] [CrossRef]
- Gudas, L.J.; Wagner, J.A. Retinoids regulate stem cell differentiation. J. Cell. Physiol. 2011, 226, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Roa, L.A.; Bloemen, M.; Carels, C.E.L.; Wagener, F.; Von den Hoff, J.W. Retinoic acid disrupts osteogenesis in pre-osteoblasts by down-regulating WNT signaling. Int. J. Biochem. Cell Biol. 2019, 116, 105597. [Google Scholar] [CrossRef]
- Ivanovska, I.L.; Swift, J.; Spinler, K.; Dingal, D.; Cho, S.; Discher, D.E. Cross-linked matrix rigidity and soluble retinoids synergize in nuclear lamina regulation of stem cell differentiation. Mol. Biol. Cell 2017, 28, 2010–2022. [Google Scholar] [CrossRef]
- Dupont, S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Lorthongpanich, C.; Thumanu, K.; Tangkiettrakul, K.; Jiamvoraphong, N.; Laowtammathron, C.; Damkham, N.; Yaowalak, U.P.; Issaragrisil, S. YAP as a key regulator of adipo-osteogenic differentiation in human MSCs. Stem Cell Res. Ther. 2019, 10, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driscoll, T.P.; Cosgrove, B.D.; Heo, S.J.; Shurden, Z.E.; Mauck, R.L. Cytoskeletal to Nuclear Strain Transfer Regulates YAP Signaling in Mesenchymal Stem Cells. Biophys. J. 2015, 108, 2783–2793. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, A.T.; Ziaei, S.; Ehret, C.; Duchemin, H.; Mamchaoui, K.; Bigot, A.; Mayer, M.; Quijano-Roy, S.; Desguerre, I.; Laine, J.; et al. Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J. Cell Sci. 2014, 127 Pt 13, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Baarlink, C.; Wang, H.; Grosse, R. Nuclear actin network assembly by formins regulates the SRF coactivator MAL. Science 2013, 340, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Nobusue, H.; Onishi, N.; Shimizu, T.; Sugihara, E.; Oki, Y.; Sumikawa, Y.; Chiyoda, T.; Akashi, K.; Saya, H.; Kano, K. Regulation of MKL1 via actin cytoskeleton dynamics drives adipocyte differentiation. Nat. Commun. 2014, 5, 3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenwald, M.; Efthymiou, V.; Opitz, L.; Wolfrum, C. SRF and MKL1 Independently Inhibit Brown Adipogenesis. PLoS ONE 2017, 12, e0170643. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.N.; Zastrow, M.S.; Wilson, K.L. Direct actin binding to A- and B-type lamin tails and actin filament bundling by the lamin A tail. Nucleus 2010, 1, 264–272. [Google Scholar] [CrossRef]
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013, 497, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Buxboim, A.; Swift, J.; Irianto, J.; Spinler, K.R.; Dingal, P.C.; Athirasala, A.; Kao, Y.R.; Cho, S.; Harada, T.; Shin, J.W.; et al. Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr. Biol. 2014, 24, 1909–1917. [Google Scholar] [CrossRef]
- Tong, J.; Li, W.; Vidal, C.; Yeo, L.S.; Fatkin, D.; Duque, G. Lamin A/C deficiency is associated with fat infiltration of muscle and bone. Mech. Ageing Dev. 2011, 132, 552–559. [Google Scholar] [CrossRef]
- Duque, G.; Rivas, D. Age-related changes in lamin A/C expression in the osteoarticular system: Laminopathies as a potential new aging mechanism. Mech. Ageing Dev. 2006, 127, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Infante, A.; Rodriguez, C.I. Osteogenesis and aging: Lessons from mesenchymal stem cells. Stem Cell Res. Ther. 2018, 9, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunawardene, P.; Bermeo, S.; Vidal, C.; Al-Saedi, A.; Chung, P.; Boersma, D.; Phu, S.; Pokorski, I.; Suriyaarachchi, P.; Demontiero, O.; et al. Association Between Circulating Osteogenic Progenitor Cells and Disability and Frailty in Older Persons: The Nepean Osteoporosis and Frailty Study. J. Gerontol. Ser. A Biomed. Sci. Med Sci. 2016, 71, 1124–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feehan, J.; Nurgali, K.; Apostolopoulos, V.; Al Saedi, A.; Duque, G. Circulating osteogenic precursor cells: Building bone from blood. EBioMedicine 2019, 39, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Al Saedi, A.; Gunawardene, P.; Bermeo, S.; Vogrin, S.; Boersma, D.; Phu, S.; Singh, L.; Suriyaarachchi, P.; Duque, G. Lamin A expression in circulating osteoprogenitors as a potential biomarker for frailty: The Nepean Osteoporosis and Frailty (NOF) Study. Exp. Gerontol. 2018, 102, 69–75. [Google Scholar] [CrossRef]
- Golpanian, S.; DiFede, D.L.; Khan, A.; Schulman, I.H.; Landin, A.M.; Tompkins, B.A.; Heldman, A.W.; Miki, R.; Goldstein, B.J.; Mushtaq, M.; et al. Allogeneic Human Mesenchymal Stem Cell Infusions for Aging Frailty. J. Gerontol. Ser. A 2017, 72, 1505–1512. [Google Scholar] [CrossRef]
- Tompkins, B.A.; DiFede, D.L.; Khan, A.; Landin, A.M.; Schulman, I.H.; Pujol, M.V.; Heldman, A.W.; Miki, R.; Goldschmidt-Clermont, P.J.; Goldstein, B.J.; et al. Allogeneic Mesenchymal Stem Cells Ameliorate Aging Frailty: A Phase II Randomized, Double-Blind, Placebo-Controlled Clinical Trial. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 72, 1513–1522. [Google Scholar] [CrossRef]
- Ho, R.; Hegele, R.A. Complex effects of laminopathy mutations on nuclear structure and function. Clin. Genet. 2019, 95, 199–209. [Google Scholar] [CrossRef]
- Capell, B.C.; Collins, F.S. Human laminopathies: Nuclei gone genetically awry. Nat. Rev. Genet. 2006, 7, 940–952. [Google Scholar] [CrossRef]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.M.; Gordon, L.B.; Snyder, B.D.; Nazarian, A.; Quinn, N.; Huh, S.; Giobbie-Hurder, A.; Neuberg, D.; Cleveland, R.; Kleinman, M.; et al. Hutchinson-Gilford progeria is a skeletal dysplasia. J. Bone Min. Res. 2011, 26, 1670–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaffidi, P.; Misteli, T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008, 10, 452–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondel, S.; Egesipe, A.L.; Picardi, P.; Jaskowiak, A.L.; Notarnicola, M.; Ragot, J.; Tournois, J.; Le Corf, A.; Brinon, B.; Poydenot, P.; et al. Drug screening on Hutchinson Gilford progeria pluripotent stem cells reveals aminopyrimidines as new modulators of farnesylation. Cell Death Dis. 2016, 7, e2105. [Google Scholar] [CrossRef] [Green Version]
- Lo Cicero, A.; Jaskowiak, A.L.; Egesipe, A.L.; Tournois, J.; Brinon, B.; Pitrez, P.R.; Ferreira, L.; de Sandre-Giovannoli, A.; Levy, N.; Nissan, X. A High Throughput Phenotypic Screening reveals compounds that counteract premature osteogenic differentiation of HGPS iPS-derived mesenchymal stem cells. Sci. Rep. 2016, 6, 34798. [Google Scholar] [CrossRef]
- Bergo, M.O.; Gavino, B.; Ross, J.; Schmidt, W.K.; Hong, C.; Kendall, L.V.; Mohr, A.; Meta, M.; Genant, H.; Jiang, Y.; et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl. Acad. Sci. USA 2002, 99, 13049–13054. [Google Scholar] [CrossRef] [Green Version]
- Infante, A.; Rodriguez, C.I. Secretome analysis of in vitro aged human mesenchymal stem cells reveals IGFBP7 as a putative factor for promoting osteogenesis. Sci. Rep. 2018, 8, 4632. [Google Scholar] [CrossRef] [Green Version]
- Cenni, V.; D’Apice, M.R.; Garagnani, P.; Columbaro, M.; Novelli, G.; Franceschi, C.; Lattanzi, G. Mandibuloacral dysplasia: A premature ageing disease with aspects of physiological ageing. Ageing Res. Rev. 2018, 42, 1–13. [Google Scholar] [CrossRef]
- Novelli, G.; Muchir, A.; Sangiuolo, F.; Helbling-Leclerc, A.; D’Apice, M.R.; Massart, C.; Capon, F.; Sbraccia, P.; Federici, M.; Lauro, R.; et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am. J. Hum. Genet. 2002, 71, 426–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avnet, S.; Pallotta, R.; Perut, F.; Baldini, N.; Pittis, M.G.; Saponari, A.; Lucarelli, E.; Dozza, B.; Greggi, T.; Maraldi, N.M.; et al. Osteoblasts from a mandibuloacral dysplasia patient induce human blood precursors to differentiate into active osteoclasts. Biochim. Biophys. Acta 2011, 1812, 711–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renard, D.; Fourcade, G.; Milhaud, D.; Bessis, D.; Esteves-Vieira, V.; Boyer, A.; Roll, P.; Bourgeois, P.; Levy, N.; De Sandre-Giovannoli, A. Novel LMNA mutation in atypical Werner syndrome presenting with ischemic disease. Stroke 2009, 40, e11–e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motegi, S.; Yokoyama, Y.; Uchiyama, A.; Ogino, S.; Takeuchi, Y.; Yamada, K.; Hattori, T.; Hashizume, H.; Ishikawa, Y.; Goto, M.; et al. First Japanese case of atypical progeroid syndrome/atypical Werner syndrome with heterozygous LMNA mutation. J. Dermatol. 2014, 41, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Jiajue, R.; Feng, K.; Wang, R.; Xia, W. Recurrent Femoral Fractures in a Boy with an Atypical Progeroid Syndrome: A Case Report. Calcif. Tissue Int. 2020, 106, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Subramanyam, L.; Agarwal, A.K.; Simha, V.; Levine, B.; D’Apice, M.R.; Novelli, G.; Crow, Y. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J. Clin. Endocrinol. Metab. 2009, 94, 4971–4983. [Google Scholar] [CrossRef]
Figure 1.
Linker of Nucleus and Cytoskeleton (LINC) complex. This structure consists of SUN (Sad1 and UNC-84) proteins anchored in the inner nuclear membrane and nesprins anchored in the outer nuclear membrane. While nesprins interact with different proteins of the cytoskeleton such as actin, SUN proteins are linked with lamins that surround the nuclear envelope.
Figure 1.
Linker of Nucleus and Cytoskeleton (LINC) complex. This structure consists of SUN (Sad1 and UNC-84) proteins anchored in the inner nuclear membrane and nesprins anchored in the outer nuclear membrane. While nesprins interact with different proteins of the cytoskeleton such as actin, SUN proteins are linked with lamins that surround the nuclear envelope.

Figure 2.
Mesenchymal stem cells’ (MSCs) fate is regulated by lamin A and vascular endothelial growth factor (VEGF) interaction. High levels of lamin A induce the expression of VEGF, which in turn induces the Runt-related transcription factor (RUNX2) transcription factor synthesis, responsible for osteoblast differentiation, inhibiting the adipocyte differentiation. However, low levels of VEGF lead to activation of the peroxisome proliferator-activated receptor (PPAR)-γ transcription factor, responsible of adipogenesis. Dashed lines represent the routes that are downregulated or compromised.
Figure 2.
Mesenchymal stem cells’ (MSCs) fate is regulated by lamin A and vascular endothelial growth factor (VEGF) interaction. High levels of lamin A induce the expression of VEGF, which in turn induces the Runt-related transcription factor (RUNX2) transcription factor synthesis, responsible for osteoblast differentiation, inhibiting the adipocyte differentiation. However, low levels of VEGF lead to activation of the peroxisome proliferator-activated receptor (PPAR)-γ transcription factor, responsible of adipogenesis. Dashed lines represent the routes that are downregulated or compromised.

Figure 3.
MSCs’ migration and differentiation. Lamin A/C levels in MSCs vary according to the microenvironment. Low lamin A/C levels are needed for migration, while high levels are required for osteogenic differentiation.
Figure 3.
MSCs’ migration and differentiation. Lamin A/C levels in MSCs vary according to the microenvironment. Low lamin A/C levels are needed for migration, while high levels are required for osteogenic differentiation.

Figure 4.
Chemical and mechanical factors that regulate MSCs’ migration and differentiation. These factors regulate lamin A/C levels, thus affecting nuclear shape and stiffness. In this way, MSCs’ migration and/or differentiation into osteogenic lineage depend on lamin A/C expression.
Figure 4.
Chemical and mechanical factors that regulate MSCs’ migration and differentiation. These factors regulate lamin A/C levels, thus affecting nuclear shape and stiffness. In this way, MSCs’ migration and/or differentiation into osteogenic lineage depend on lamin A/C expression.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Alcorta-Sevillano, N.; Macías, I.; Rodríguez, C.I.; Infante, A. Crucial Role of Lamin A/C in the Migration and Differentiation of MSCs in Bone. Cells 2020, 9, 1330. https://doi.org/10.3390/cells9061330
AMA Style
Alcorta-Sevillano N, Macías I, Rodríguez CI, Infante A. Crucial Role of Lamin A/C in the Migration and Differentiation of MSCs in Bone. Cells. 2020; 9(6):1330. https://doi.org/10.3390/cells9061330
Chicago/Turabian StyleAlcorta-Sevillano, Natividad, Iratxe Macías, Clara I. Rodríguez, and Arantza Infante. 2020. "Crucial Role of Lamin A/C in the Migration and Differentiation of MSCs in Bone" Cells 9, no. 6: 1330. https://doi.org/10.3390/cells9061330
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.