Injectable SN-38-embedded Polymeric Microparticles Promote Antitumor Efficacy against Malignant Glioma in an Animal Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. Fabrication of the SMPs
2.3. Fourier-Transform Infrared Spectroscopy
2.4. Thermal Analysis
2.5. Characterization of SMPs
2.6. In Vitro Elution Characteristics of SN-38
2.7. Surgical Procedure
2.8. In Vivo SN-38 Pharmacokinetics
2.9. Glioma Model Creation and Treatment
2.10. Statistical Analysis
3. Result
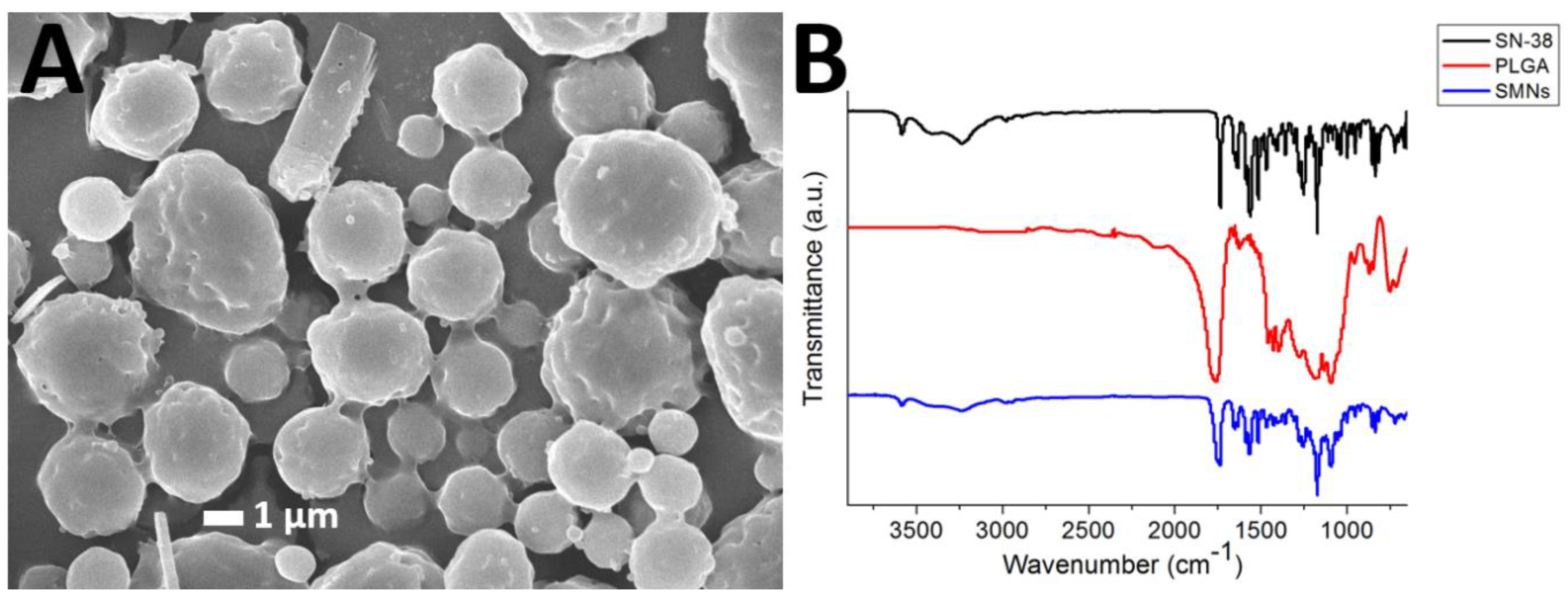
3.1. Morphology of the Microparticles
3.2. FTIR Spectroscopy
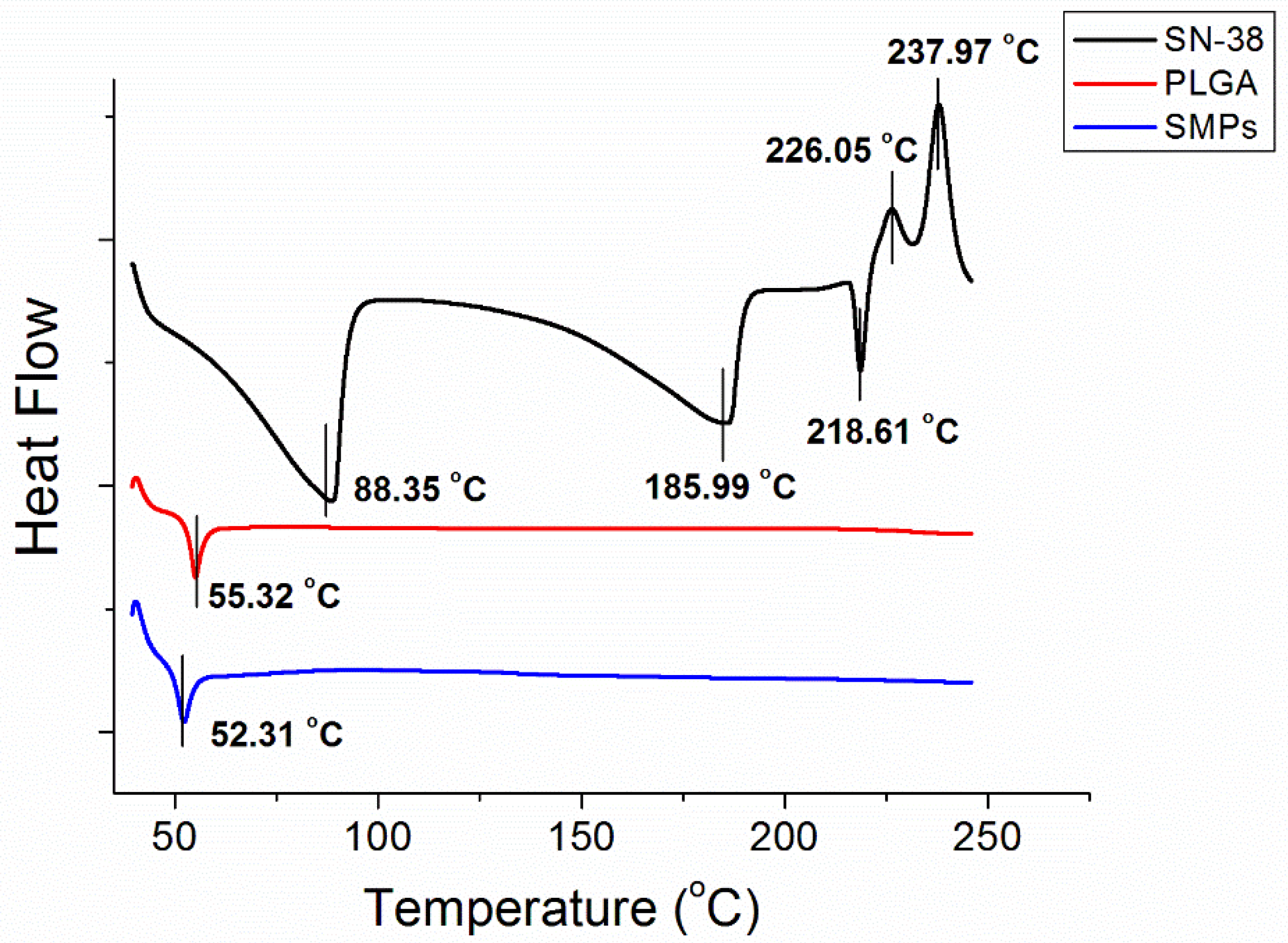
3.3. DSC Analysis
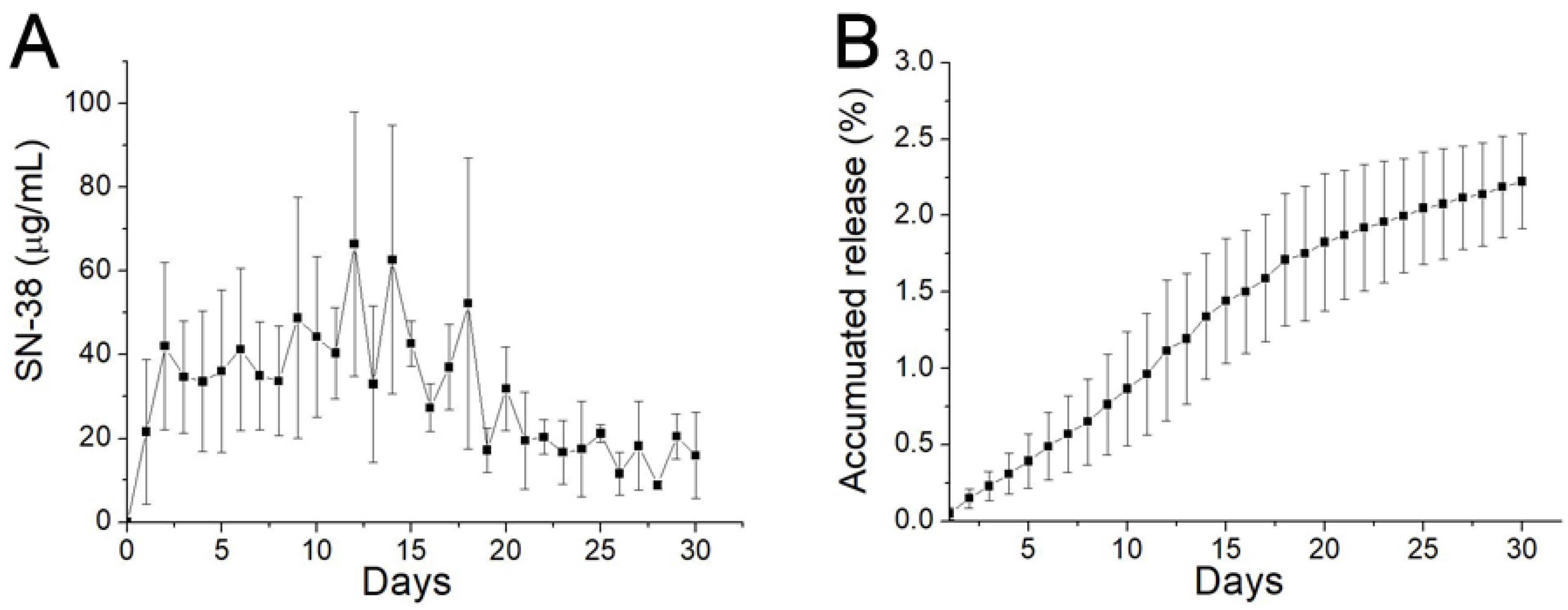
3.4. In Vitro Release Profiles of SN-38
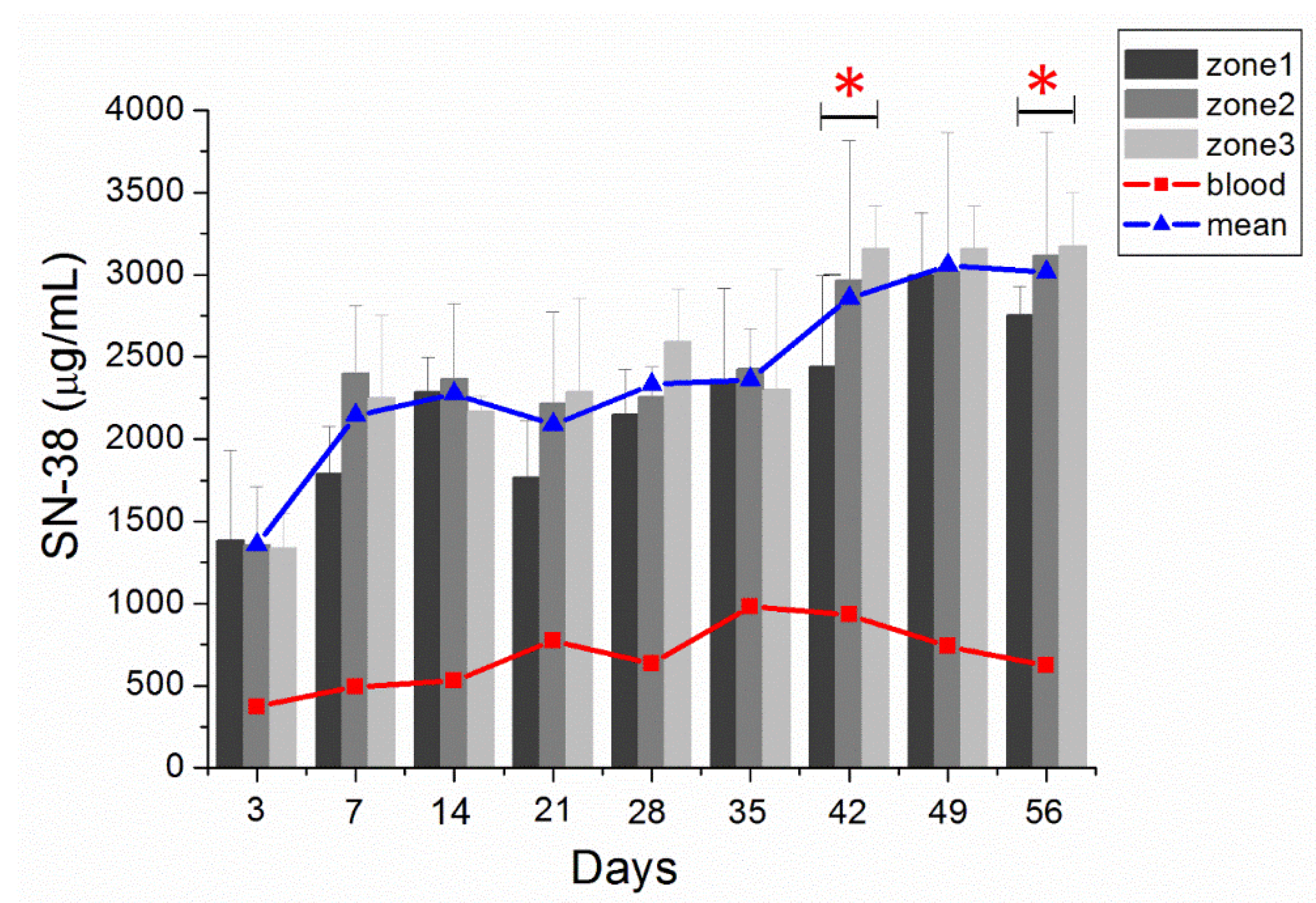
3.5. In Vivo Characteristics of SN-38 Release from SMPs
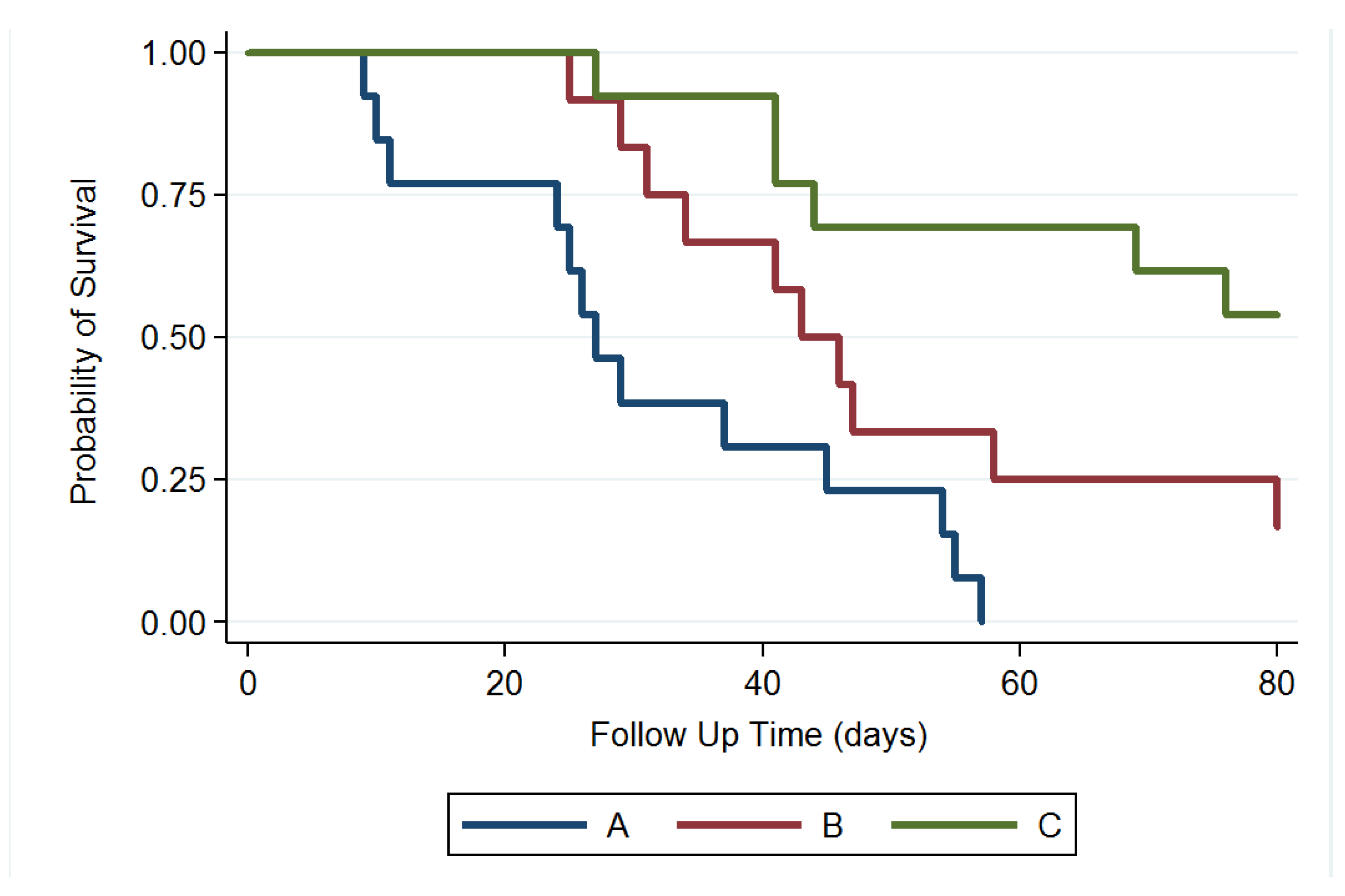
3.6. Survival Rate
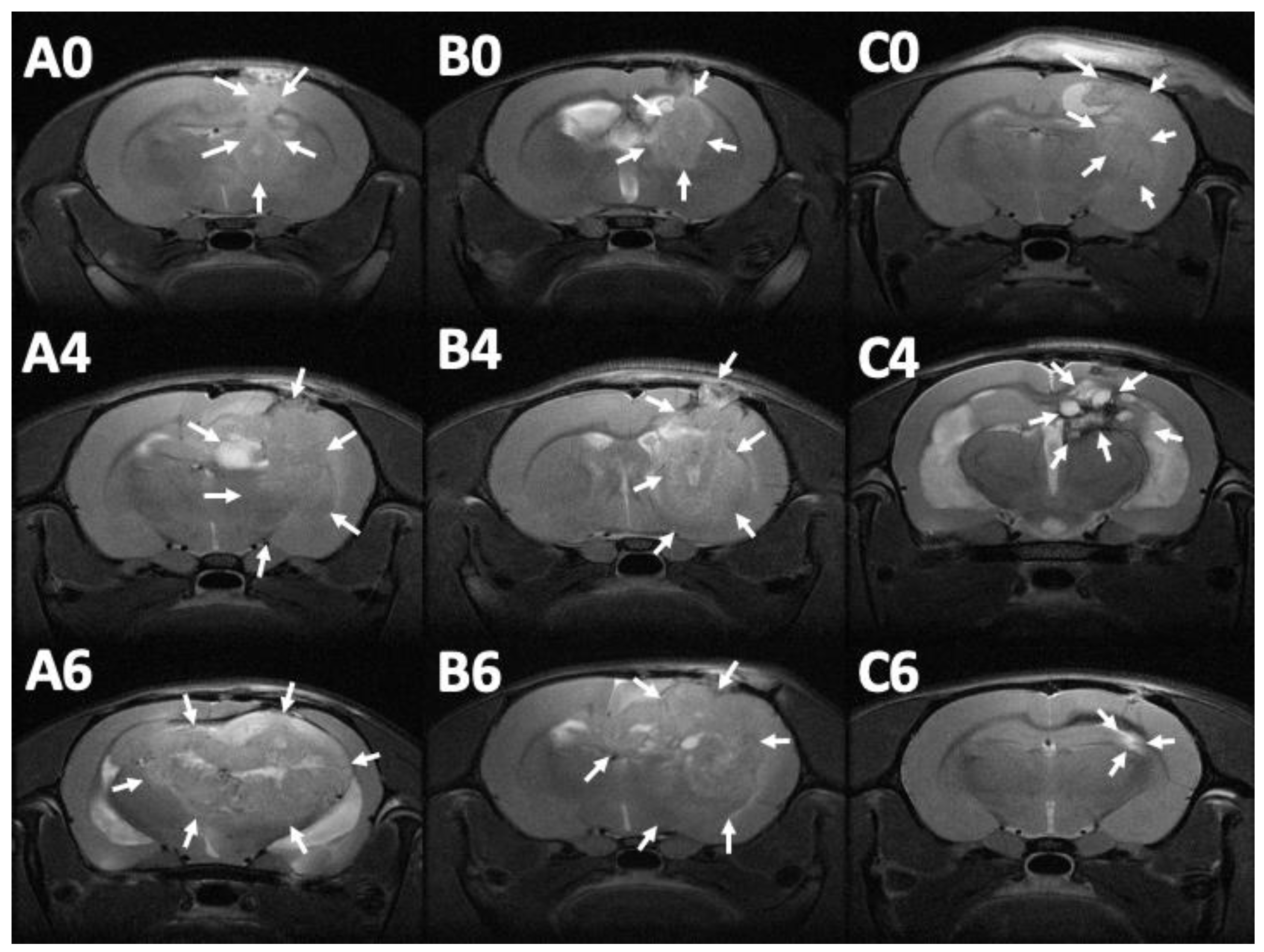
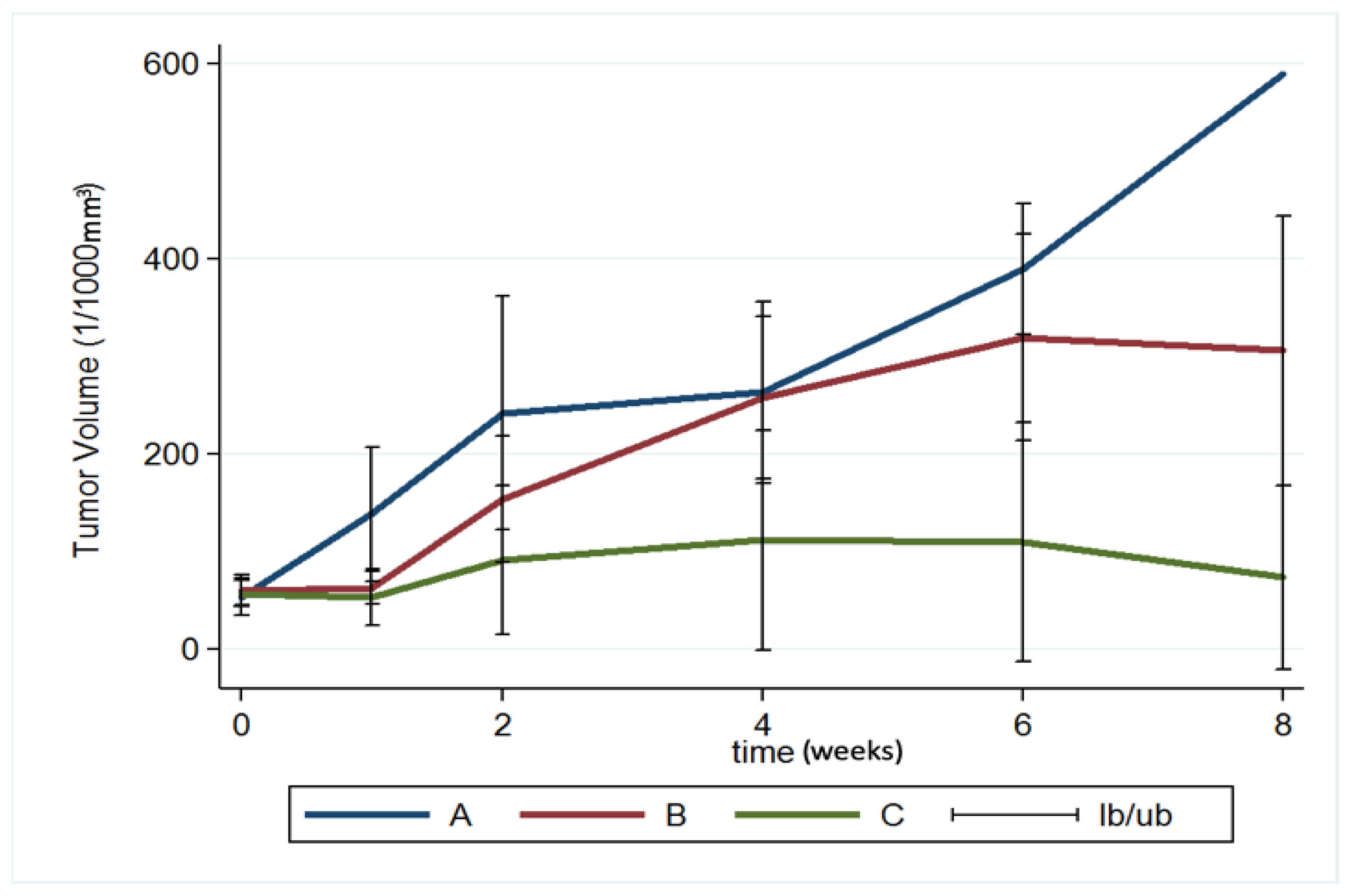
3.7. MRI and Tumor Volume
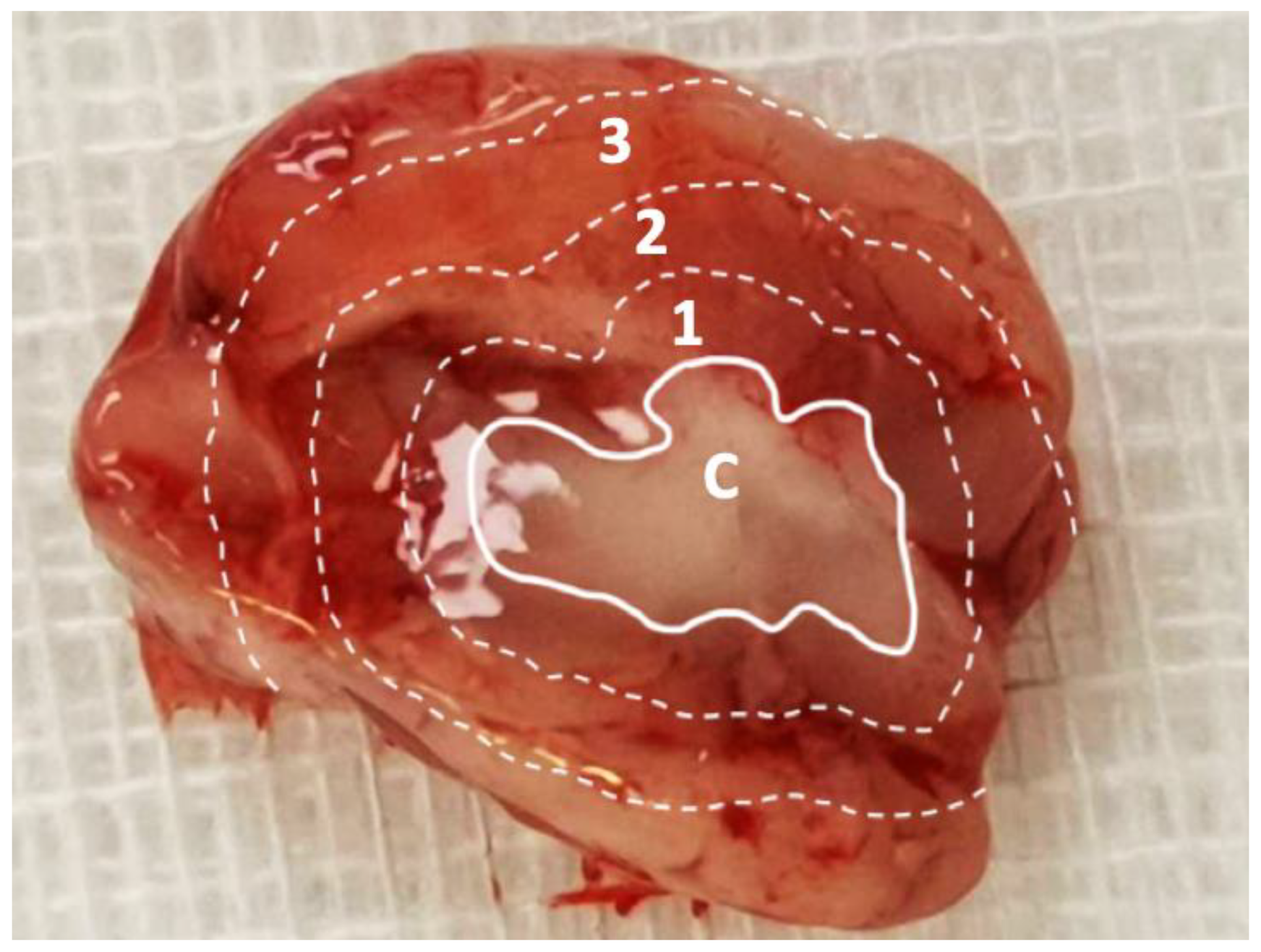
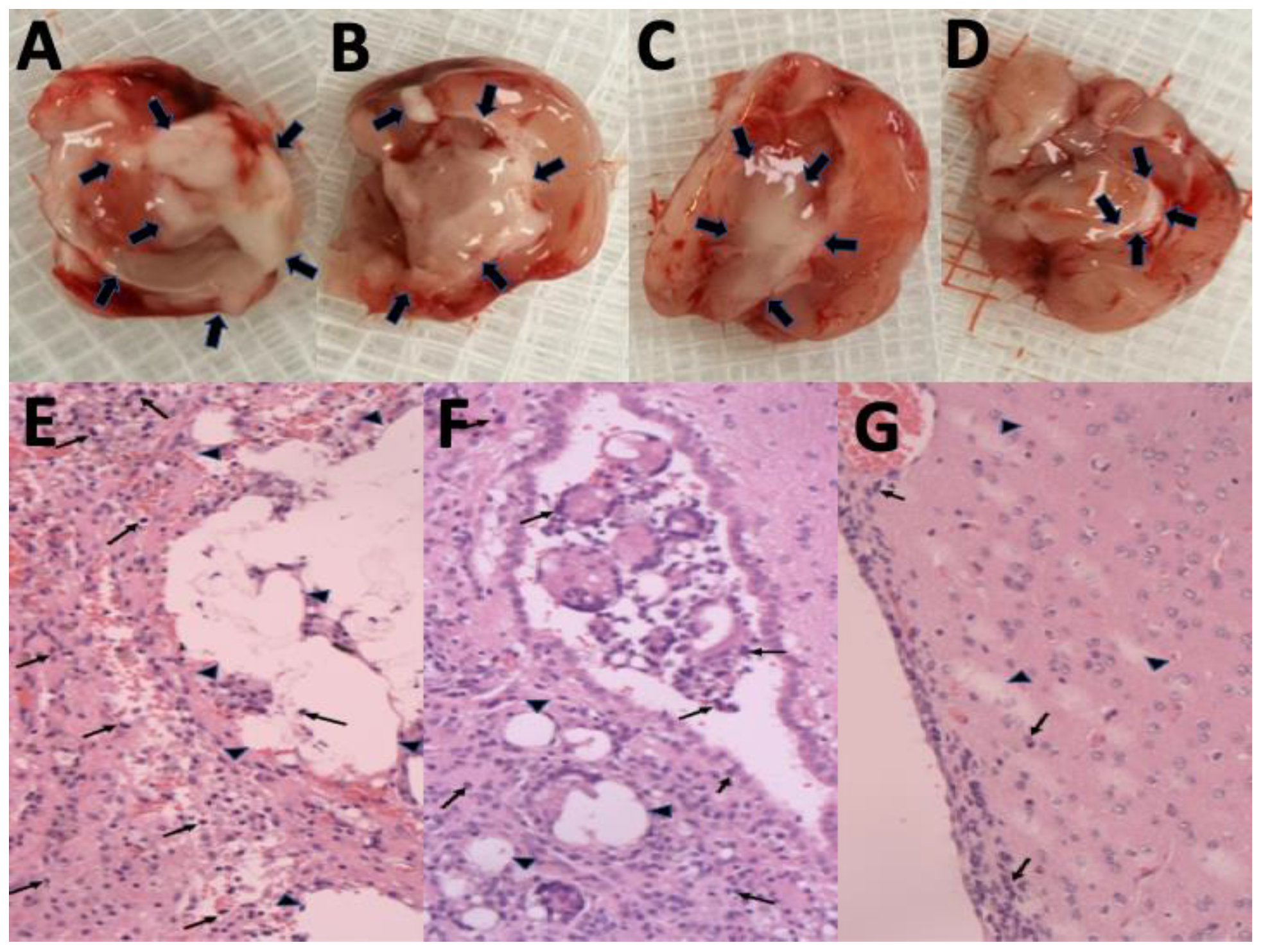
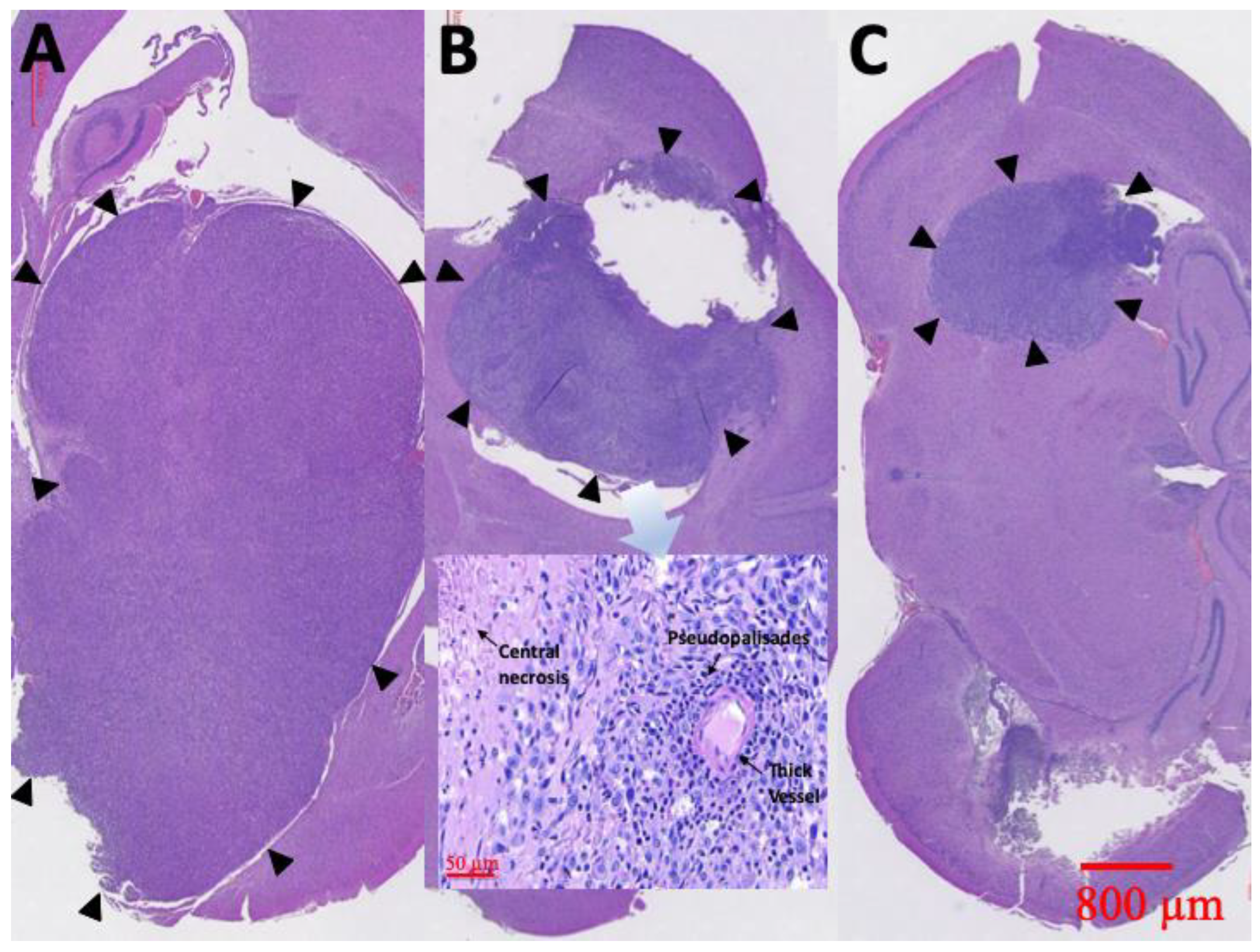
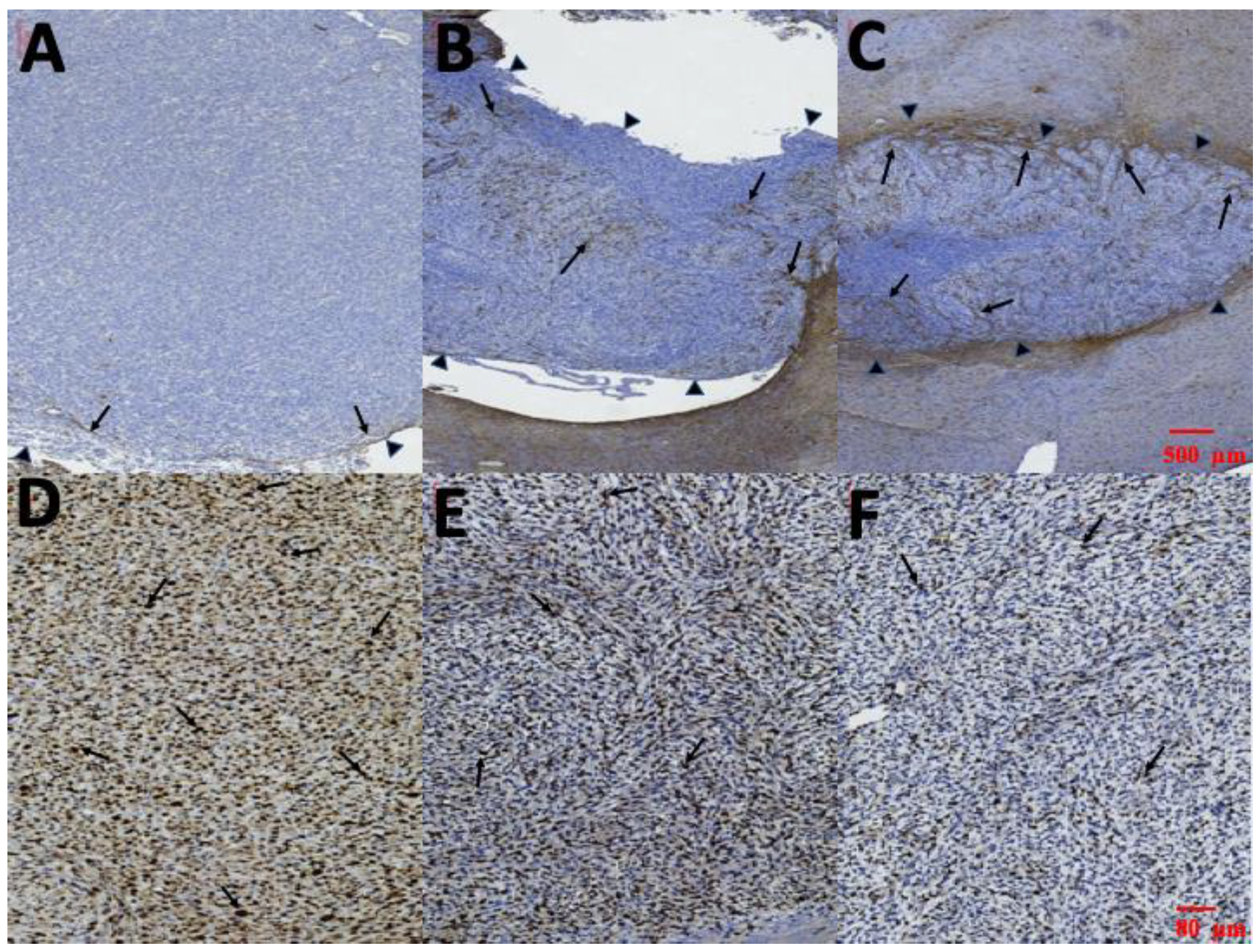
3.8. Pathological Finding
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brandes, A.A.; Tosoni, A.; Basso, U.; Reni, M.; Valduga, F.; Monfardini, S.; Amista, P.; Nicolardi, L.; Sotti, G.; Ermani, M. Second-line chemotherapy with irinotecan plus carmustine in glioblastoma recurrent or progressive after first-line temozolomide chemotherapy: A phase II study of the Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). J. Clin. Oncol. 2004, 22, 4779–4786. [Google Scholar] [CrossRef]
- Tseng, Y.Y.; Kau, Y.C.; Liu, S.J. Advanced interstitial chemotherapy for treating malignant glioma. Expert Opin. Drug Deliv. 2016, 13, 1533–1544. [Google Scholar] [CrossRef]
- Minniti, G.; Muni, R.; Lanzetta, G.; Marchetti, P.; Enrici, R.M. Chemotherapy for glioblastoma: Current treatment and future perspectives for cytotoxic and targeted agents. Anticancer Res. 2009, 29, 5171–5184. [Google Scholar]
- Tseng, Y.Y.; Huang, Y.C.; Yang, T.C.; Yang, S.T.; Liu, S.C.; Chang, T.M.; Kau, Y.C.; Liu, S.J. Concurrent Chemotherapy of Malignant Glioma in Rats by Using Multidrug-Loaded Biodegradable Nanofibrous Membranes. Sci. Rep. 2016, 6, 30630. [Google Scholar] [CrossRef] [Green Version]
- Affronti, M.L.; Heery, C.R.; Herndon, J.E., II; Rich, J.N.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Friedman, A.H.; Bigner, D.D.; Friedman, H.S. Overall survival of newly diagnosed glioblastoma patients receiving carmustine wafers followed by radiation and concurrent temozolomide plus rotational multiagent chemotherapy. Cancer 2009, 115, 3501–3511. [Google Scholar] [CrossRef]
- Tosi, G.; Costantino, L.; Ruozi, B.; Forni, F.; Vandelli, M.A. Polymeric nanoparticles for the drug delivery to the central nervous system. Expert Opin. Drug Deliv. 2008, 5, 155–174. [Google Scholar] [CrossRef]
- Tseng, Y.Y.; Su, C.H.; Yang, S.T.; Huang, Y.C.; Lee, W.H.; Wang, Y.C.; Liu, S.C.; Liu, S.J. Advanced interstitial chemotherapy combined with targeted treatment of malignant glioma in rats by using drug-loaded nanofibrous membranes. Oncotarget 2016, 7, 59902–59916. [Google Scholar] [CrossRef]
- Quinn, J.A.; Desjardins, A.; Weingart, J.; Brem, H.; Dolan, M.E.; Delaney, S.M.; Vredenburgh, J.; Rich, J.; Friedman, A.H.; Reardon, D.A.; et al. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J. Clin. Oncol. 2005, 23, 7178–7187. [Google Scholar] [CrossRef] [PubMed]
- Rhines, L.D.; Sampath, P.; Dolan, M.E.; Tyler, B.M.; Brem, H.; Weingart, J. O6-benzylguanine potentiates the antitumor effect of locally delivered carmustine against an intracranial rat glioma. Cancer Res. 2000, 60, 6307–6310. [Google Scholar] [PubMed]
- Patel, V.J.; Elion, G.B.; Houghton, P.J.; Keir, S.; Pegg, A.E.; Johnson, S.P.; Dolan, M.E.; Bigner, D.D.; Friedman, H.S. Schedule-dependent activity of temozolomide plus CPT-11 against a human central nervous system tumor-derived xenograft. Clin. Cancer Res. 2000, 6, 4154–4157. [Google Scholar] [PubMed]
- Oliva, C.R.; Nozell, S.E.; Diers, A.; McClugage, S.G., III; Sarkaria, J.N.; Markert, J.M.; Darley-Usmar, V.M.; Bailey, S.M.; Gillespie, G.Y.; Landar, A.; et al. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J. Biol. Chem. 2010, 285, 39759–39767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol. 2009, 11, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurita, A.; Kado, S.; Kaneda, N.; Onoue, M.; Hashimoto, S.; Yokokura, T. Modified irinotecan hydrochloride (CPT-11) administration schedule improves induction of delayed-onset diarrhea in rats. Cancer Chemother. Pharmacol. 2000, 46, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Coggins, C.A.; Elion, G.B.; Houghton, P.J.; Hare, C.B.; Keir, S.; Colvin, O.M.; Bigner, D.D.; Friedman, H.S. Enhancement of irinotecan (CPT-11) activity against central nervous system tumor xenografts by alkylating agents. Cancer Chemother. Pharmacol. 1998, 41, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Zeghari-Squalli, N.; Raymond, E.; Cvitkovic, E.; Goldwasser, F. Cellular pharmacology of the combination of the DNA topoisomerase I inhibitor SN-38 and the diaminocyclohexane platinum derivative oxaliplatin. Clin. Cancer Res. 1999, 5, 1189–1196. [Google Scholar]
- Voigt, W.; Matsui, S.; Yin, M.B.; Burhans, W.C.; Minderman, H.; Rustum, Y.M. Topoisomerase-I inhibitor SN-38 can induce DNA damage and chromosomal aberrations independent from DNA synthesis. Anticancer Res. 1998, 18, 3499–3505. [Google Scholar]
- Sapra, P.; Zhao, H.; Mehlig, M.; Malaby, J.; Kraft, P.; Longley, C.; Greenberger, L.M.; Horak, I.D. Novel delivery of SN38 markedly inhibits tumor growth in xenografts, including a camptothecin-11-refractory model. Clin. Cancer Res. 2008, 14, 1888–1896. [Google Scholar] [CrossRef] [Green Version]
- Xuan, T.; Zhang, J.A.; Ahmad, I. HPLC method for determination of SN-38 content and SN-38 entrapment efficiency in a novel liposome-based formulation, LE-SN38. J. Pharm. Biomed. Anal. 2006, 41, 582–588. [Google Scholar] [CrossRef]
- Zhang, R.; Saito, R.; Mano, Y.; Sumiyoshi, A.; Kanamori, M.; Sonoda, Y.; Kawashima, R.; Tominaga, T. Convection-enhanced delivery of SN-38-loaded polymeric micelles (NK012) enables consistent distribution of SN-38 and is effective against rodent intracranial brain tumor models. Drug Deliv. 2016, 23, 2780–2786. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.K. Liposomes for Enhanced Bioavailability of Water-Insoluble Drugs: In Vivo Evidence and Recent Approaches. Pharmaceutics 2020, 12, 264. [Google Scholar] [CrossRef] [Green Version]
- Di Costanzo, A.; Angelico, R. Formulation Strategies for Enhancing the Bioavailability of Silymarin: The State of the Art. Molecules 2019, 24, 2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, A.; Preet, S.; Kumar, V.; Kumar, R.; Kumar, R. Synergetic effect of vancomycin loaded silver nanoparticles for enhanced antibacterial activity. Colloids Surf. B Biointerfaces 2019, 176, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Vangara, K.K.; Ali, H.I.; Lu, D.; Liu, J.L.; Kolluru, S.; Palakurthi, S. SN-38-cyclodextrin complexation and its influence on the solubility, stability, and in vitro anticancer activity against ovarian cancer. AAPS PharmSciTech 2014, 15, 472–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Hegi, M.E.; Gilbert, M.R.; Chakravarti, A. Chemoradiotherapy in malignant glioma: Standard of care and future directions. J. Clin. Oncol. 2007, 25, 4127–4136. [Google Scholar] [CrossRef] [PubMed]
- Barbu, E.; Molnar, E.; Tsibouklis, J.; Gorecki, D.C. The potential for nanoparticle-based drug delivery to the brain: Overcoming the blood-brain barrier. Expert Opin. Drug Deliv. 2009, 6, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.Y.; Liao, J.Y.; Chen, W.A.; Kao, Y.C.; Liu, S.J. Sustainable release of carmustine from biodegradable poly[((d,l))-lactide-co-glycolide] nanofibrous membranes in the cerebral cavity: In vitro and in vivo studies. Expert Opin. Drug Deliv. 2013, 10, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Hare, C.B.; Elion, G.B.; Houghton, P.J.; Houghton, J.A.; Keir, S.; Marcelli, S.L.; Bigner, D.D.; Friedman, H.S. Therapeutic efficacy of the topoisomerase I inhibitor 7-ethyl-10-(4-[1-piperidino]-1-piperidino)-carbonyloxy-camptothecin against pediatric and adult central nervous system tumor xenografts. Cancer Chemother. Pharmacol. 1997, 39, 187–191. [Google Scholar] [CrossRef]
- Reardon, D.A.; Friedman, H.S.; Powell, J.B., Jr.; Gilbert, M.; Yung, W.K. Irinotecan: Promising activity in the treatment of malignant glioma. Oncology 2003, 17, 9–14. [Google Scholar]
- Friedman, H.S.; Keir, S.T.; Houghton, P.J. The emerging role of irinotecan (CPT-11) in the treatment of malignant glioma in brain tumors. Cancer 2003, 97, 2359–2362. [Google Scholar] [CrossRef]
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E., II; Marcello, J.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Sampson, J.; et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J. Clin. Oncol. 2007, 25, 4722–4729. [Google Scholar] [CrossRef] [Green Version]
- Friedman, H.S.; Petros, W.P.; Friedman, A.H.; Schaaf, L.J.; Kerby, T.; Lawyer, J.; Parry, M.; Houghton, P.J.; Lovell, S.; Rasheed, K.; et al. Irinotecan therapy in adults with recurrent or progressive malignant glioma. J. Clin. Oncol. 1999, 17, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C. Salvage chemotherapy with CPT-11 for recurrent glioblastoma multiforme. J. Neurooncol. 2002, 56, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ghandi, A.; Liebes, L.; Louie, S.G.; Hofman, F.M.; Schonthal, A.H.; Chen, T.C. Effective conversion of irinotecan to SN-38 after intratumoral drug delivery to an intracranial murine glioma model in vivo. Laboratory investigation. J. Neurosurg. 2011, 114, 689–694. [Google Scholar] [CrossRef] [Green Version]
- Nakatsu, S.; Kondo, S.; Kondo, Y.; Yin, D.; Peterson, J.W.; Kaakaji, R.; Morimura, T.; Kikuchi, H.; Takeuchi, J.; Barnett, G.H. Induction of apoptosis in multi-drug resistant (MDR) human glioblastoma cells by SN-38, a metabolite of the camptothecin derivative CPT-11. Cancer Chemother. Pharmacol. 1997, 39, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Vejjasilpa, K.; Nasongkla, N.; Manaspon, C.; Larbcharoensub, N.; Boongird, A.; Hongeng, S.; Israsena, N. Antitumor efficacy and intratumoral distribution of SN-38 from polymeric depots in brain tumor model. Exp. Biol. Med. 2015, 240, 1640–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivory, L.P.; Chatelut, E.; Canal, P.; Mathieu-Boue, A.; Robert, J. Kinetics of the in vivo interconversion of the carboxylate and lactone forms of irinotecan (CPT-11) and of its metabolite SN-38 in patients. Cancer Res. 1994, 54, 6330–6333. [Google Scholar] [PubMed]
- Fike, J.R.; Gobbel, G.T.; Mesiwala, A.H.; Shin, H.J.; Nakagawa, M.; Lamborn, K.R.; Seilhan, T.M.; Elliott, P.J. Cerebrovascular effects of the bradykinin analog RMP-7 in normal and irradiated dog brain. J. Neurooncol. 1998, 37, 199–215. [Google Scholar] [CrossRef]
- Nittayacharn, P.; Manaspon, C.; Hongeng, S.; Nasongkla, N. HPLC analysis and extraction method of SN-38 in brain tumor model after injected by polymeric drug delivery system. Exp. Biol. Med. 2014, 239, 1619–1629. [Google Scholar] [CrossRef]
- Zhu, X.; Ni, S.; Xia, T.; Yao, Q.; Li, H.; Wang, B.; Wang, J.; Li, X.; Su, W. Anti-Neoplastic Cytotoxicity of SN-38-Loaded PCL/Gelatin Electrospun Composite Nanofiber Scaffolds against Human Glioblastoma Cells In Vitro. J. Pharm. Sci. 2015, 104, 4345–4354. [Google Scholar] [CrossRef]
- Manaspon, C.; Nasongkla, N.; Chaimongkolnukul, K.; Nittayacharn, P.; Vejjasilpa, K.; Kengkoom, K.; Boongird, A.; Hongeng, S. Injectable SN-38-loaded Polymeric Depots for Cancer Chemotherapy of Glioblastoma Multiforme. Pharm. Res. 2016, 33, 2891–2903. [Google Scholar] [CrossRef]
- Kuroda, J.; Kuratsu, J.; Yasunaga, M.; Koga, Y.; Saito, Y.; Matsumura, Y. Potent antitumor effect of SN-38-incorporating polymeric micelle, NK012, against malignant glioma. Int. J. Cancer 2009, 124, 2505–2511. [Google Scholar] [CrossRef] [PubMed]
- Essa, S.; Daoud, J.; Lafleur, M.; Martel, S.; Tabrizian, M. SN-38 active loading in poly(lactic-co-glycolic acid) nanoparticles and assessment of their anticancer properties on COLO-205 human colon adenocarcinoma cells. J. Microencapsul. 2015, 32, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B., III; Ajani, J.A.; Catalano, R.B.; Engelking, C.; Kornblau, S.M.; Martenson, J.A., Jr.; McCallum, R.; Mitchell, E.P.; O’Dorisio, T.M.; Vokes, E.E.; et al. Recommended guidelines for the treatment of cancer treatment-induced diarrhea. J. Clin. Oncol. 2004, 22, 2918–2926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, D.E.; Boers-Doets, C.B.; Bensadoun, R.J.; Herrstedt, J.; Committee, E.G. Management of oral and gastrointestinal mucosal injury: ESMO Clinical Practice Guidelines for diagnosis, treatment, and follow-up. Ann. Oncol. 2015, 26 (Suppl. 5), v139–v151. [Google Scholar] [CrossRef] [Green Version]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Preat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Bota, D.A.; Desjardins, A.; Quinn, J.A.; Affronti, M.L.; Friedman, H.S. Interstitial chemotherapy with biodegradable BCNU (Gliadel) wafers in the treatment of malignant gliomas. Ther. Clin. Risk Manag. 2007, 3, 707–715. [Google Scholar]
- Wilhelmsson, U.; Eliasson, C.; Bjerkvig, R.; Pekny, M. Loss of GFAP expression in high-grade astrocytomas does not contribute to tumor development or progression. Oncogene 2003, 22, 3407–3411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.J.; Yang, S.T.; Chen, S.M.; Huang, Y.C.; Lee, W.H.; Ho, J.; Chen, Y.C.; Tseng, Y.Y. Novel multi-drugs incorporating hybrid-structured nanofibers enhance alkylating agent activity in malignant gliomas. Ther. Adv. Med. Oncol. 2019, 11, 1758835919875555. [Google Scholar] [CrossRef] [Green Version]
- Thotakura, M.; Tirumalasetti, N.; Krishna, R. Role of Ki-67 labeling index as an adjunct to the histopathological diagnosis and grading of astrocytomas. J. Cancer Res. Ther. 2014, 10, 641–645. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tseng, Y.-Y.; Yang, T.-C.; Chen, S.-M.; Yang, S.-T.; Tang, Y.-L.; Liu, S.-J. Injectable SN-38-embedded Polymeric Microparticles Promote Antitumor Efficacy against Malignant Glioma in an Animal Model. Pharmaceutics 2020, 12, 479. https://doi.org/10.3390/pharmaceutics12050479
Tseng Y-Y, Yang T-C, Chen S-M, Yang S-T, Tang Y-L, Liu S-J. Injectable SN-38-embedded Polymeric Microparticles Promote Antitumor Efficacy against Malignant Glioma in an Animal Model. Pharmaceutics. 2020; 12(5):479. https://doi.org/10.3390/pharmaceutics12050479
Chicago/Turabian StyleTseng, Yuan-Yun, Tao-Chieh Yang, Shu-Mei Chen, Shun-Tai Yang, Ya-Ling Tang, and Shih-Jung Liu. 2020. "Injectable SN-38-embedded Polymeric Microparticles Promote Antitumor Efficacy against Malignant Glioma in an Animal Model" Pharmaceutics 12, no. 5: 479. https://doi.org/10.3390/pharmaceutics12050479