Synthesis, Structural Characterization and Hirshfeld Surface Analysis of a 2D Coordination Polymer, [Co(4-dpds)(bdc)(H2O)2] 4-dpds

 ,
,
Abstract
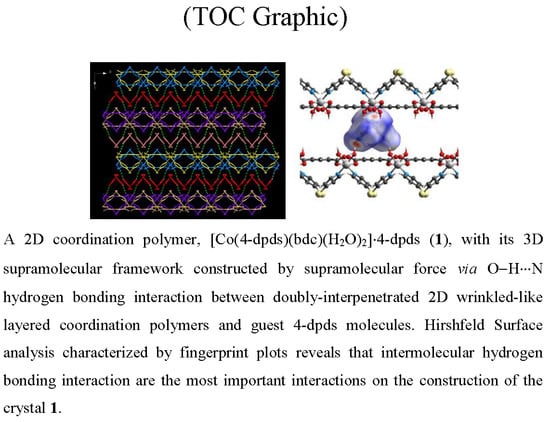
:
1. Introduction
2. Results and Discussion
2.1. Synthetic and Structural Description of (1)
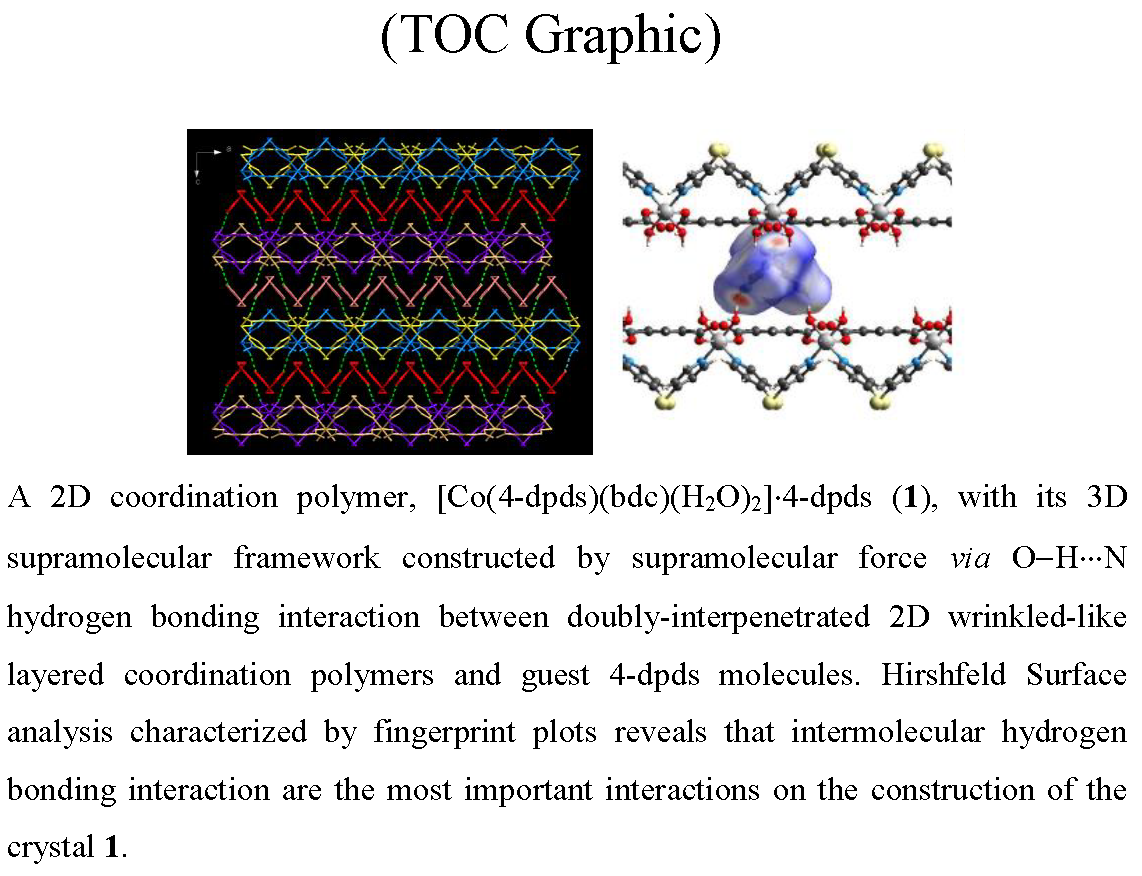
2.2. Thermal Stability of (1)
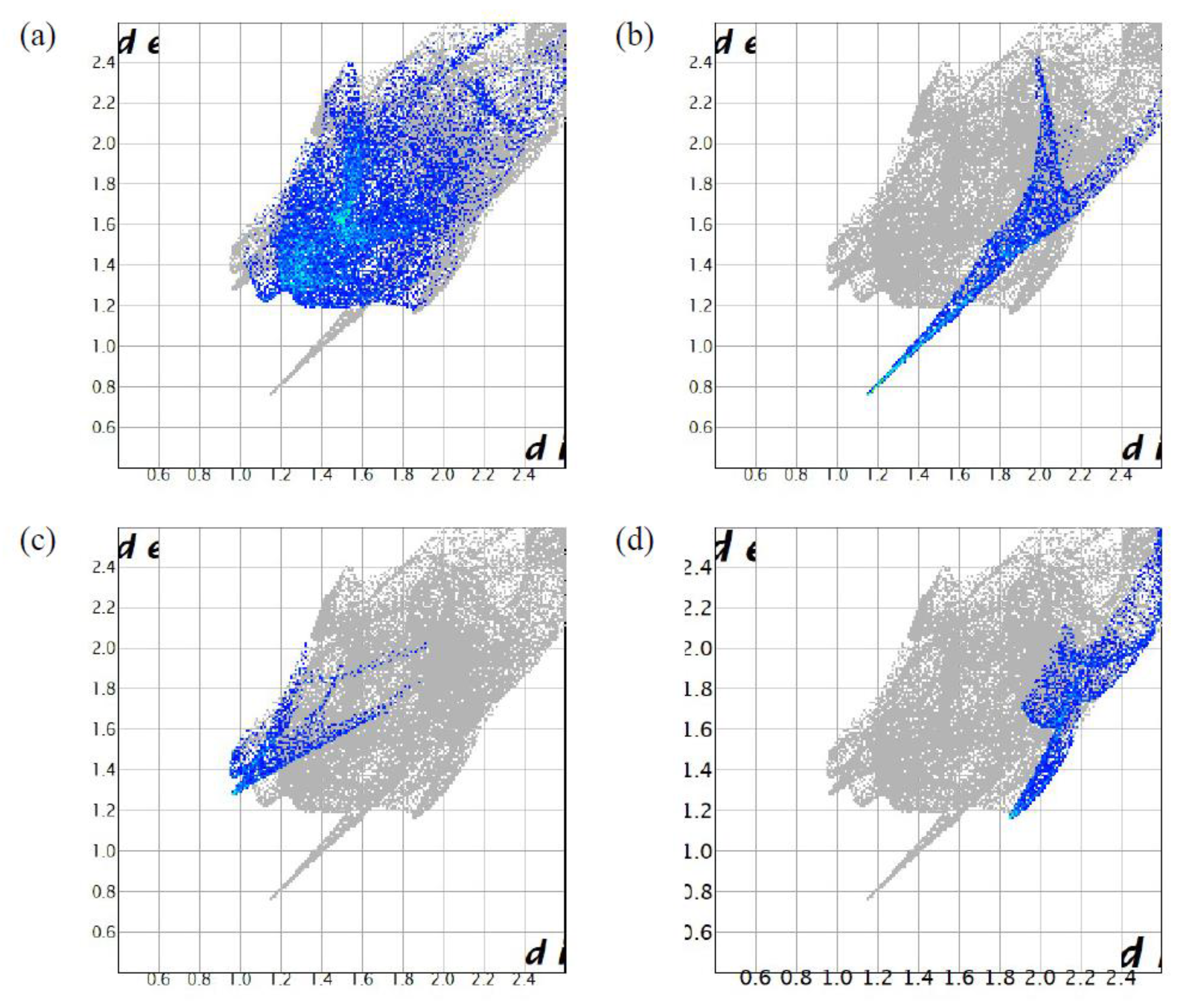
2.3. Hirdhfeld Surface Analysis of (1)
3. Experimental Section
3.1. Materials and Physical Techniques
3.2. Synthesis of [Co(4-dpds)(bdc)(H2O)2]·4-dpds (1)
3.3. Crystallographic Data Collection and Refinements
3.4. In Situ X-ray Powder Diffraction of 1
3.5. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Batten, S.R.; Champness, N.R.; Chen, X.M.; Garcia-Martinez, J.; Kitagawa, S.; Öhrström, L.; O’keeffe, M.; Suh, M.P.; Reedijk, J. Terminology of metal–organic frameworks and coordination polymers. Pure Appl. Chem. 2013, 85, 1715–1724. [Google Scholar] [CrossRef] [Green Version]
- Batten, S.R.; Champness, N.R.; Chen, X.M.; Garcia-Martinez, J.; Kitagawa, S.; Öhrström, L.; O’keeffe, M.; Suh, M.P.; Reedijk, J. Coordination polymers, metal–organic frameworks and the need for terminology guidelines. CrystEngComm 2012, 14, 3001–3004. [Google Scholar] [CrossRef] [Green Version]
- O’Keeffe, M.; Peskov, M.A.; Ramsden, S.J.; Yaghi, O.M. The Reticular Chemistry Structure Resource (RCSR) Database of, and Symbols for, Crystal Nets. Acc. Chem. Res. 2008, 41, 1782–1789. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.; Mahata, P. Metal-organic framework structures—How closely are they related to classical inorganic structures? Chem. Soc. Rev. 2009, 38, 2304–2318. [Google Scholar] [CrossRef] [PubMed]
- Blatov, V.A.; O’Keeffe, M.; Proserpio, D.M. Vertex-, face-, point-, Schläfli-, and Delaney-symbols in nets, polyhedra and tilings: Recommended terminology. CrystEngComm 2010, 12, 44–48. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, E.V.; Blatov, V.A.; Kochetkov, A.V.; Proserpio, D.M. Underlying nets in three-periodic coordination polymers: Topology, taxonomy and prediction from a computer-aided analysis of the Cambridge Structural Database. CrystEngComm 2011, 13, 3947–3958. [Google Scholar] [CrossRef]
- Baburin, I.A.; Blatov, V.A.; Carlucci, L.; Ciani, G.; Proserpio, D.M. Interpenetrated Three-Dimensional Networks of Hydrogen-Bonded Organic Species: A Systematic Analysis of the Cambridge Structural Database. Cryst. Growth Des. 2008, 8, 519–539. [Google Scholar] [CrossRef]
- O’Keeffe, M.; Yaghi, O.M. Deconstructing the Crystal Structures of Metal–Organic Frameworks and Related Materials into Their Underlying Nets. Chem. Rev. 2012, 112, 675–702. [Google Scholar] [CrossRef]
- Long, J.R.; Yaghi, O.M. The pervasive chemistry of metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1213–1314. [Google Scholar] [CrossRef]
- Zhou, H.C.; Long, J.R.; Yaghi, O.M. Introduction to Metal–Organic Frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef]
- Batten, S.R.; Neville, S.M.; Turner, D.R. Coordination Polymers: Design, Analysis and Application; Royal Society of Chemistry: Cambridge, UK, 2009. [Google Scholar]
- Farrusseng, D. Metal–Organic Frameworks Applications from Catalysis to Gas Storage; Wiley: Weinheim, Germany, 2011. [Google Scholar]
- Takamizawa, S.; Akatsuka, T.; Ueda, T. Gas-Conforming Transformability of an Ionic Single-Crystal HostConsisting of Discrete Charged Components. Angew. Chem. Int. Ed. 2008, 47, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, S.; Kohara, M.; Akatsuka, T.; Miyake, R. Gas-adsorbing ability of tris-ethylenediamine metal complexes (M = Co(III), Cr(III), Rh(III), Ir(III)) as transformable ionic single crystal hosts. New J. Chem. 2008, 32, 1782–1787. [Google Scholar] [CrossRef]
- Morsali, A.; Masoomi, M.Y. Structures and properties of mercury(II) coordination polymers. Coord. Chem. Rev. 2009, 253, 1882–1905. [Google Scholar] [CrossRef]
- Masoomi, M.Y.; Morsali, A. Applications of metal-organic coordination polymers as precursors for preparation of nano-materials Authors. Coord. Chem. Rev. 2012, 256, 2921–2943. [Google Scholar] [CrossRef]
- Masoomi, M.Y.; Morsali, A. Morphological study and potential applications of nano metal–organic coordination polymer. RSC Adv. 2013, 3, 19191–19218. [Google Scholar] [CrossRef]
- Aslani, A.; Morsali, A.; Zeller, M. Dynamic crystal-to-crystal conversion of a 3D–3D coordination polymer by de- and re-hydration. Dalton Trans. 2008, 5173–5177. [Google Scholar] [CrossRef]
- Pan, Q.H.; Li, J.Y.; Chen, Q.; Han, Y.D.; Chang, Z.; Song, W.C.; Bu, X.H. [Co(en)3]1/3[In(ox)2] · 3.5H2O: A zeolitic metal-organic framework templated by Co(en)3Cl3. Microporous Mesoporous Mater. 2010, 132, 453–457. [Google Scholar] [CrossRef]
- Pan, Q.H.; Chen, Q.; Song, W.C.; Hu, T.L.; Bu, X.H. Template-directed synthesis of three new open-framework metal(II) oxalates using Co(III) complex as template. CrystEngComm 2010, 12, 4198–4204. [Google Scholar] [CrossRef]
- Bertani, R.; Sgarbossa, P.; Venzo, A.; Lelj, F.; Amati, M.; Resnati, G.; Pilati, T.; Metrangolo, P.; Terraneo, G. Halogen bonding in metal–organic–supramolecular networks. Coord. Chem. Rev. 2010, 254, 677–695. [Google Scholar] [CrossRef]
- Ingleson, M.J.; Bacsa, J.; Rosseinsky, M.J. Homochiral H-bonded proline based metal organic frameworks. Chem. Commun. 2007, 27, 3036–3038. [Google Scholar] [CrossRef]
- Desiraju, G.R. C–H⋅⋅⋅O and other weak hydrogen bonds. From crystal engineering to virtual screening. Chem. Commun. 2005, 25, 2995–3001. [Google Scholar] [CrossRef] [PubMed]
- García-Báez, E.V.; Martínez-Martínez, F.J.; Höpfl, H.; Padilla-Martínez, I.I. π-Stacking Interactions and CH···X (X = O, Aryl) Hydrogen Bonding as Directing Features of the Supramolecular Self-Association in 3-Carboxy and 3-Amido Coumarin Derivatives. Cryst. Growth Des. 2003, 3, 35–45. [Google Scholar] [CrossRef]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- Claessens, C.G.; Stoddart, J.F. π–π INTERACTIONS IN SELF-ASSEMBLY. J. Phys. Org. Chem. 1997, 10, 254–272. [Google Scholar] [CrossRef]
- Guo, H.D.; Guo, X.M.; Batten, S.R.; Song, J.F.; Song, S.Y.; Dang, S.; Zhang, G.L.; Tang, J.K.; Zhang, H.J. Hydrothermal Synthesis, Structures, and Luminescent Properties of Seven d10 Metal–Organic Frameworks Based on 9,9-Dipropylfluorene-2,7-Dicarboxylic Acid (H2DFDA). Cryst. Growth Des. 2009, 9, 1394–1401. [Google Scholar] [CrossRef]
- Reger, D.L.; Horger, J.J.; Smith, M.D.; Long, G.J.; Grandjean, F. Homochiral, Helical Supramolecular Metal–Organic Frameworks Organized by Strong π···π Stacking Interactions: Single-Crystal to Single-Crystal Transformations in Closely Packed Solids. Inorg. Chem. 2011, 50, 686–704. [Google Scholar] [CrossRef] [PubMed]
- Horikoshi, R.; Mochida, T. Metal complexes of 4, 4′-dipyridyldisulfide—Structural diversity derived from a twisted ligand with axial chirality. Coord. Chem. Rev. 2006, 250, 2595–2609. [Google Scholar] [CrossRef]
- Lang, J.P.; Xu, Q.F.; Zhang, W.H.; Li, H.X.; Ren, Z.G.; Chen, J.X.; Zhang, Y. Mo(W)/Cu/S Cluster-Based Supramolecular Arrays Assembled from Preformed Clusters [Et4N]4[WS4Cu4I6] and [(n-Bu)4N]2[MoOS3Cu3X3] (X = I, SCN) with Flexible Ditopic Ligands. Inorg. Chem. 2006, 45, 10487–10496. [Google Scholar] [CrossRef]
- Ma, L.F.; Wang, L.Y.; Du, M. A novel 3D Mn(II) coordination polymer involving 4,4′-dipyridylsulfide and 4,4′-dipyridyltrisulfide obtained by in situ ligand formation from 4,4′-dipyridyldisulfide. CrystEngComm 2009, 11, 2593–2596. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Blatov, V.A.; Shevchenko, A.P.; Serezhkin, V.N. TOPOS3.2: A new version of the program package for multipurpose crystal-chemical analysis. J. Appl. Crystallogr. 2000, 33, 1193. [Google Scholar] [CrossRef] [Green Version]
- Blatov, V.A.; Carlucci, L.; Ciani, G.; Proserpio, D.M. Interpenetrating metal–organic and inorganic 3D networks: A computer-aided systematic investigation. Part I. Analysis of the Cambridge structural database. CrystEngComm 2004, 6, 377–395. [Google Scholar] [CrossRef]
- SMART V 4.043 Software for CCD Detector System; Siemens Analytical Instruments Division: Madison, WI, USA, 1995.
- SAINT V 4.035 Software for CCD Detector System; Siemens Analytical Instruments Division: Madison, WI, USA, 1995.
- Sheldrick, G.M. Program for the Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1993. [Google Scholar]
- SHELXTL 5.03 (PC-Version), Program Liberary for Structure Solution and Molecular Graphics. Siemens Analytical Instruments Division: Madison, WI, USA, 1995.
- Cambridge Crystallographic Data Centre. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 29 April 2020).
- Toby, B.H.; Von Dreele, R.B. GSAS-II: The genesis of a modern open-source all purpose crystallography software package. J. Appl. Crystallogr. 2013, 46, 544–549. [Google Scholar] [CrossRef]
- Larson, A.C.; Von Dreele, R.B. General Structure Analysis System (GSAS); Los Alamos National Laboratory Report LAUR 86-748; Los Alamos National Laboratory: Los Alamos, NM, USA, 2000. [Google Scholar]
- Putz, H.; Brandenburg, K. Match—Phase Identification from Powder Diffraction; Crystal Impact: Bonn, Germany, 2015; Available online: http://www.crystalimpact.com/match (accessed on 29 April 2020).
- Boultif, A.; Louer, D. Program for the Automatic Indexing of Powder Diffraction Patterns by the Successive Dichotomy Method. J. Appl. Cryst. 2004, 37, 724–731. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K.; van de Streek, J.; Pidcock, E.; Motherwell, W.D.S.; Cole, J.C. DASH: A program for crystal structures determination from powder diffraction data. J. Appl. Cryst. 2006, 39, 910–915. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Crawley, Australia, 2017; Available online: http://hirshfeldsurface.net (accessed on 29 April 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Co(1)–O(1) | 2.071(1) | Co(1)–O(1)i | 2.071(1) |
| Co(1)–O(3) | 2.123(1) | Co(1)–O(3)i | 2.123(1) |
| Co(1)–N(1)i | 2.176(2) | Co(1)–N(1) | 2.176(2) |
| O(1)–Co(1)–O(1)i | 175.28(6) | O(1)–Co(1)–O(3)i | 90.52(4) |
| O(1)–Co(1)–O(3) | 86.18(4) | O(1)i–Co(1)–O(3)i | 86.18(4) |
| O(1)i–Co(1)–O(3) | 90.52(4) | O(3)–Co(1)–O(3)i | 91.53(6) |
| O(1)–Co(1)–N(1)i | 93.76(4) | O(3)i–Co(1)–N(1)i | 86.33(4) |
| O(1)i–Co(1)–N(1)i | 89.40(4) | O(1)–Co(1)–N(1) | 89.40(4) |
| O(3)–Co(1)–N(1)i | 177.86(4) | O(1)i–Co(1)–N(1) | 93.76(4) |
| O(3)–Co(1)–N(1) | 86.33(4) | O(3)i–Co(1)–N(1) | 177.86(4) |
| N(1)i–Co(1)–N(1) | 95.81(6) |
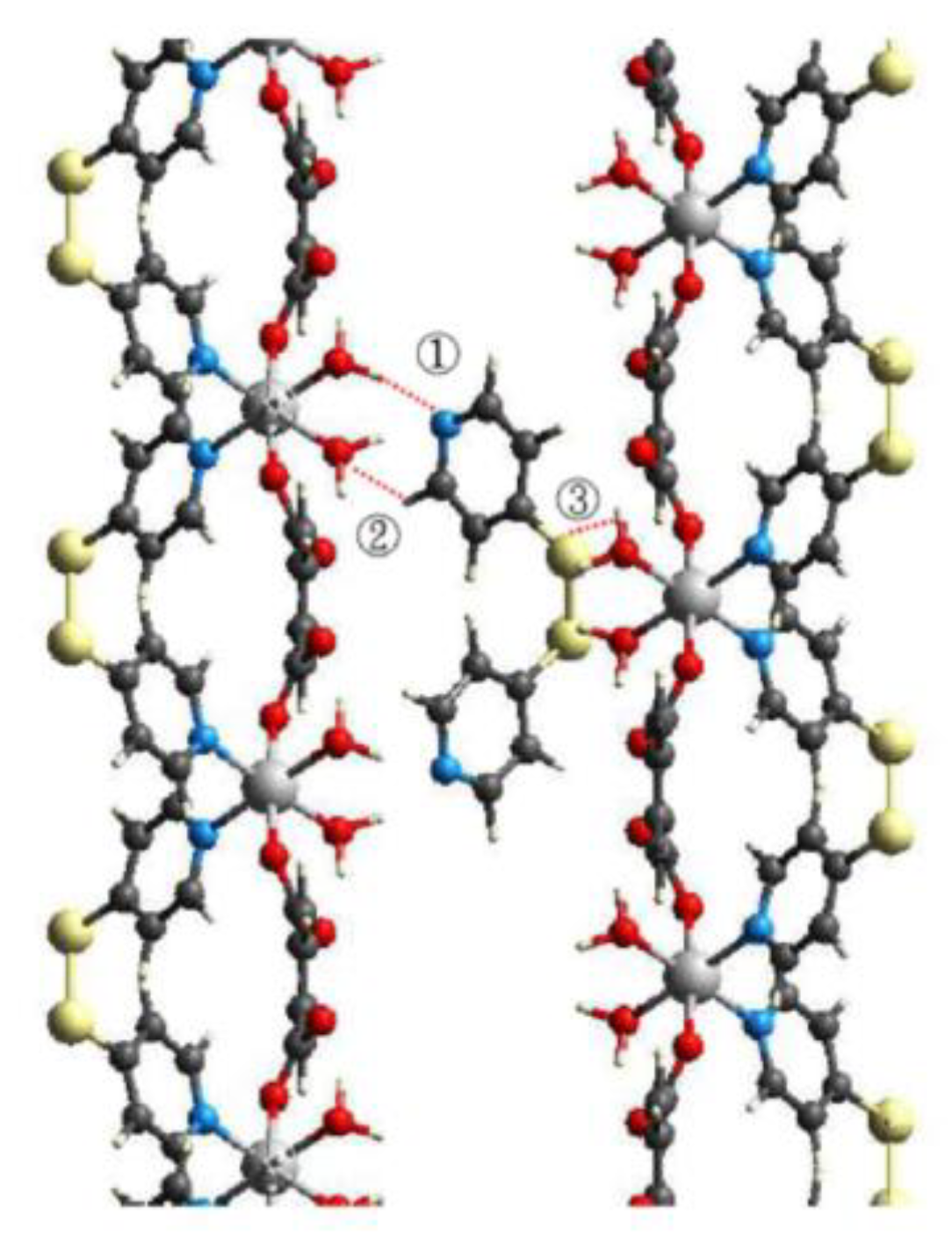
| D–H⋅⋅⋅A | D–H (Å) | H⋅⋅⋅A (Å) | D⋅⋅⋅A (Å) | ∠ D–H⋅⋅⋅A (°) |
|---|---|---|---|---|
| O(3)–H(3A)⋅⋅⋅N(2) | 0.821(5) | 2.045(5) | 2.848(2) | 166.0(1) |
| O(3)–H(3B)⋅⋅⋅O(2) | 0.806(5) | 1.920(5) | 2.684(2) | 157.9(1) |
| Ring(i) a → Ring(j) a | Slip Angle (i,j)/° | Interplanar (i,j) Distance/Å | Horizontal Shift between the (i,j) Ring Centroids/Å | Distance between the (i,j) Ring Centroids/Å |
|---|---|---|---|---|
| R(1)→R(2) | 0.0 | 3.604(5) | 0.0 | 3.604(5) |
| Compound 1 | a | b | c | Vol |
|---|---|---|---|---|
| Single crystal | 15.3322(4) | 16.5790(4) | 45.5861(12) | 11587.6(5) |
| RT | 15.426(1) | 16.587(1) | 46.059(2) | 11785.5(9) |
| 90 °C | 15.4520(6) | 16.5776(7) | 46.228(1) | 11841.6(5) |
| 120 °C | 15.4603(7) | 16.5736(9) | 46.341(2) | 11874.0(6) |
| 150 °C | 15.4686(6) | 16.5702(8) | 46.450(2) | 11905.9(6) |
| Empirical Formula | C28H24 CoN4O6S4 | Formula Mass (g mol−1) | 699.68 |
|---|---|---|---|
| crystal system | Orthorhombic | space group | Fddd |
| a/Å | 15.3322(4) | α (°) | 90 |
| b/Å | 16.5790(4) | β (°) | 90 |
| c/Å | 45.5861(12) | γ (°) | 90 |
| V/Å3 | 11587.6(5) | Z | 16 |
| Dcalcd (g cm−3) | 1.604 | θ range (deg) | 2.252–29.998 |
| μ/mm−1 | 0.932 | T (K) | 150(2) |
| total no. of data collected | 23999 | no. of unique data | 4231 |
| R1, wR21 (I > 2σ(I)) | 0.0283, 0.0681 | R1, wR21 (all data) | 0.0344, 0.0728 |
| GOF 2 | 1.028 | refine params | 203 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-C.; Huang, Z.-L.; Tseng, Y.-Y.; Sheu, G.-B.; Lu, S.-I.; Lee, G.-H.; Sheu, H.-S. Synthesis, Structural Characterization and Hirshfeld Surface Analysis of a 2D Coordination Polymer, [Co(4-dpds)(bdc)(H2O)2] 4-dpds. Crystals 2020, 10, 419. https://doi.org/10.3390/cryst10050419
Wang C-C, Huang Z-L, Tseng Y-Y, Sheu G-B, Lu S-I, Lee G-H, Sheu H-S. Synthesis, Structural Characterization and Hirshfeld Surface Analysis of a 2D Coordination Polymer, [Co(4-dpds)(bdc)(H2O)2] 4-dpds. Crystals. 2020; 10(5):419. https://doi.org/10.3390/cryst10050419
Chicago/Turabian StyleWang, Chih-Chieh, Zi-Ling Huang, Yueh-Yi Tseng, Gia-Bin Sheu, Shih-I Lu, Gene-Hsiang Lee, and Hwo-Shuenn Sheu. 2020. "Synthesis, Structural Characterization and Hirshfeld Surface Analysis of a 2D Coordination Polymer, [Co(4-dpds)(bdc)(H2O)2] 4-dpds" Crystals 10, no. 5: 419. https://doi.org/10.3390/cryst10050419