Lung Tumor Cell-Derived Exosomes Promote M2 Macrophage Polarization

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. M1 and M2 Polarization
2.3. Isolation of Exosomes
2.4. NanoSight Analyses of Exosomes
2.5. Labeling Exosomes with PKH26
2.6. Co-Culture of Exosomes with THP-1 Cells
2.7. Flow Cytometry
2.8. ImageStream Flow Cytometry
2.9. qRT-PCR
2.10. Bioenergetics Assay
2.11. Statistical Analyses
3. Results
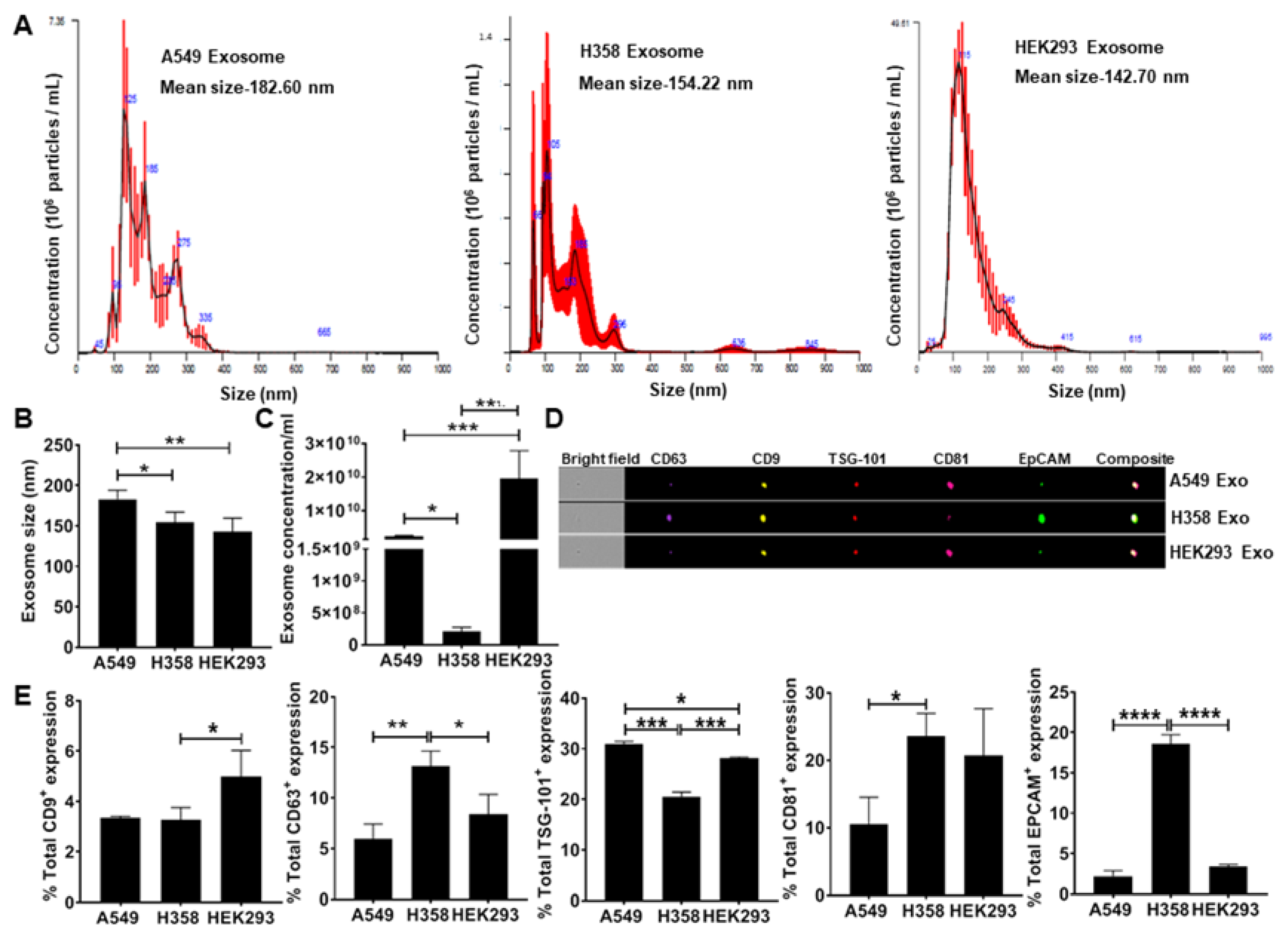
3.1. Secretion of EpCAM+ Lung Tumor Cell-Derived Exosomes is p53 Dependent
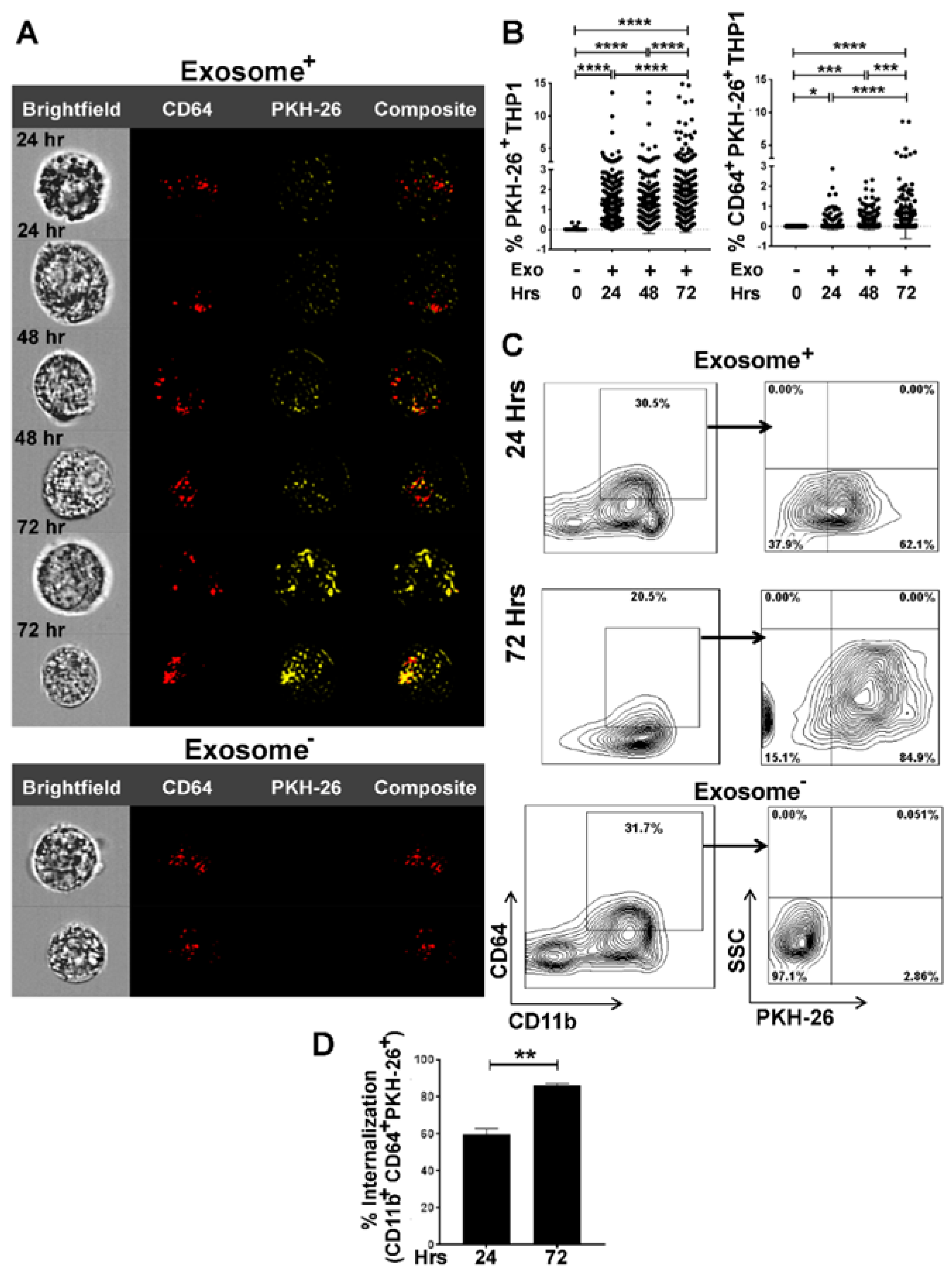
3.2. Internalization of Tumor Cell-Derived Exosomes by Macrophages is Time-Dependent
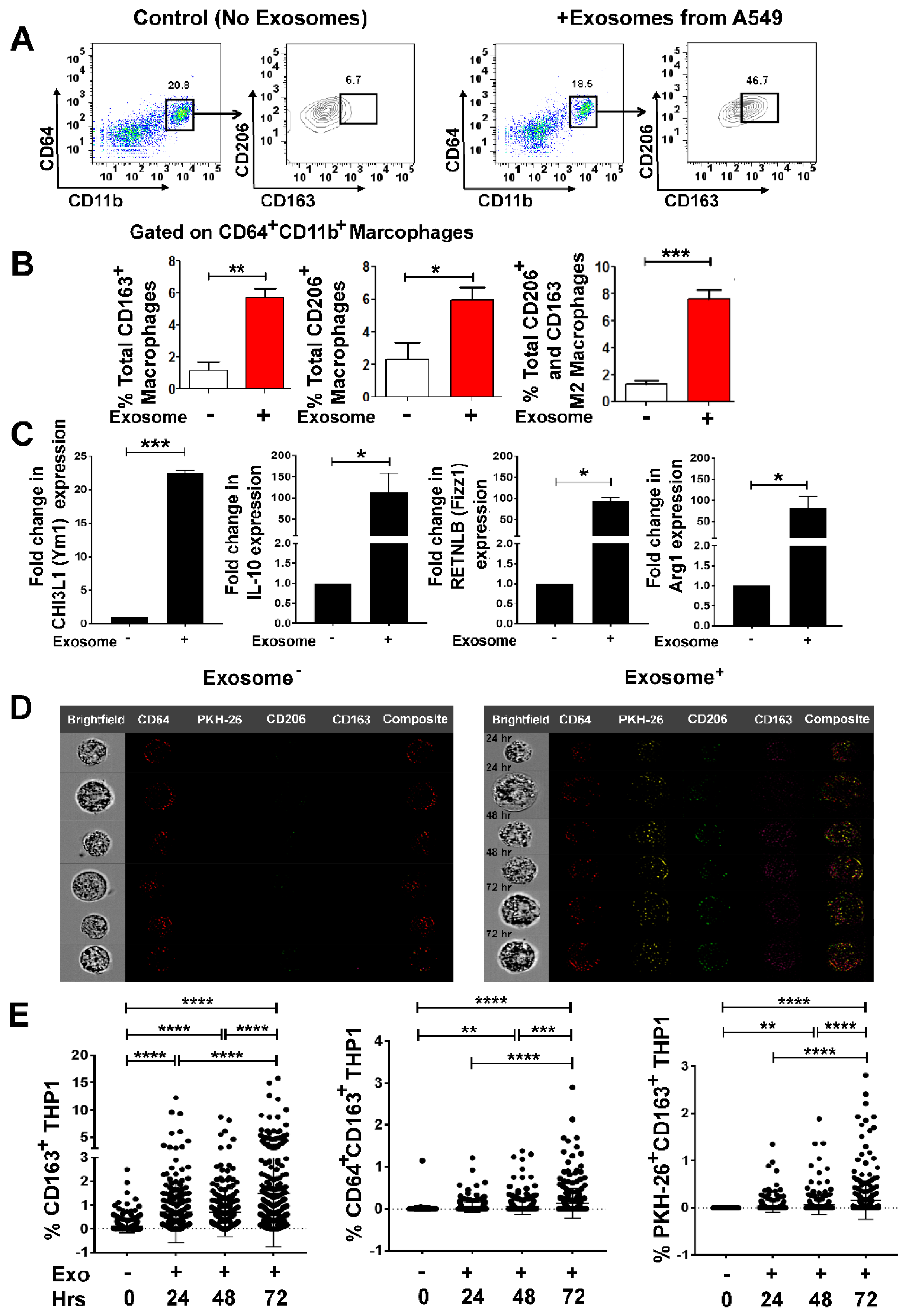
3.3. Lung Tumor Cell-Derived Exosomes Promote M2 Macrophage Polarization in a Time-Dependent Manner
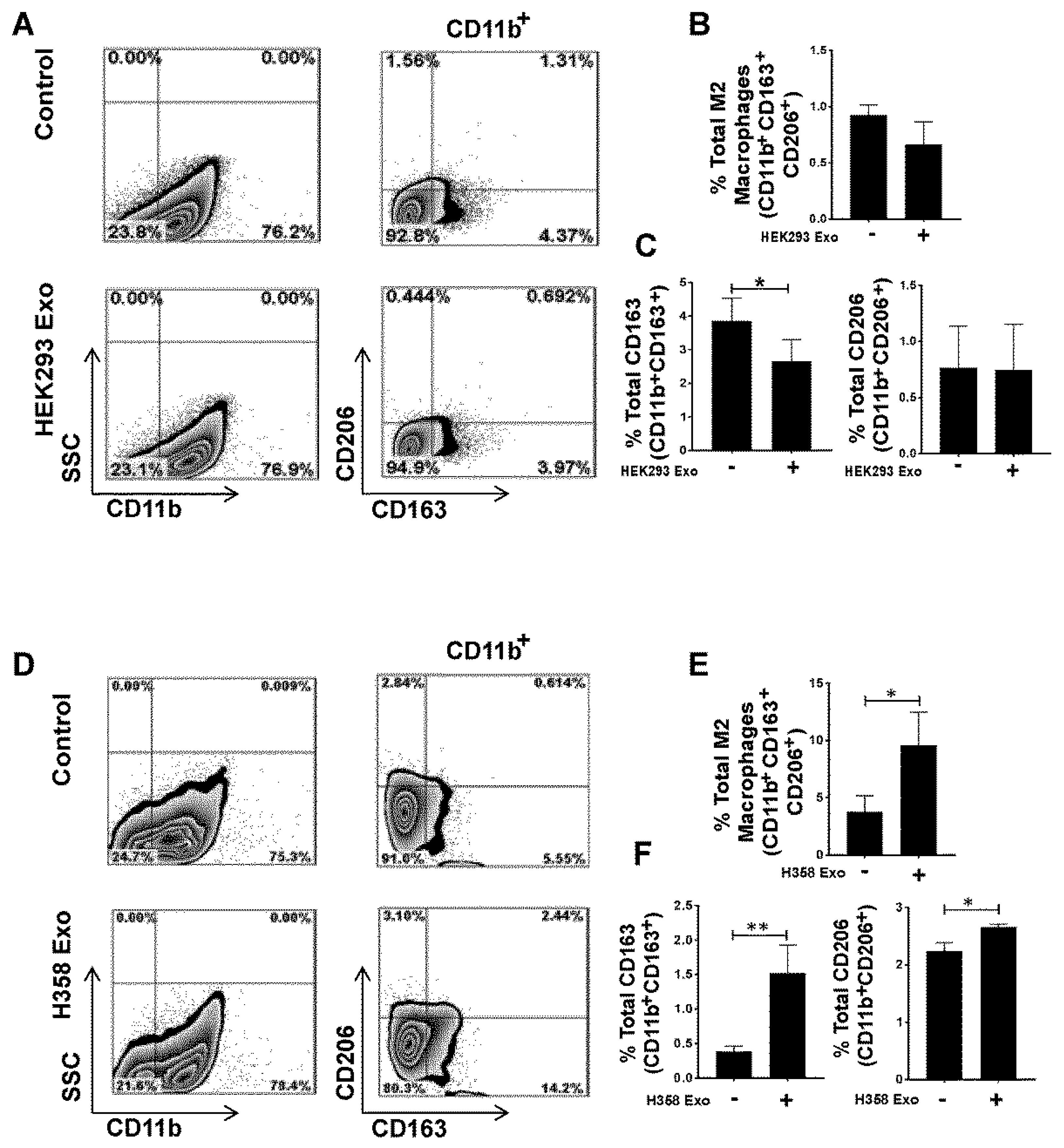
3.4. Non-Tumor Cell-Derived Exosomes are Unable to Polarize M0 to M2 While M2 Macrophage Polarization by Lung Tumor Cell-Derived Exosomes May Not Be p53 Dependent
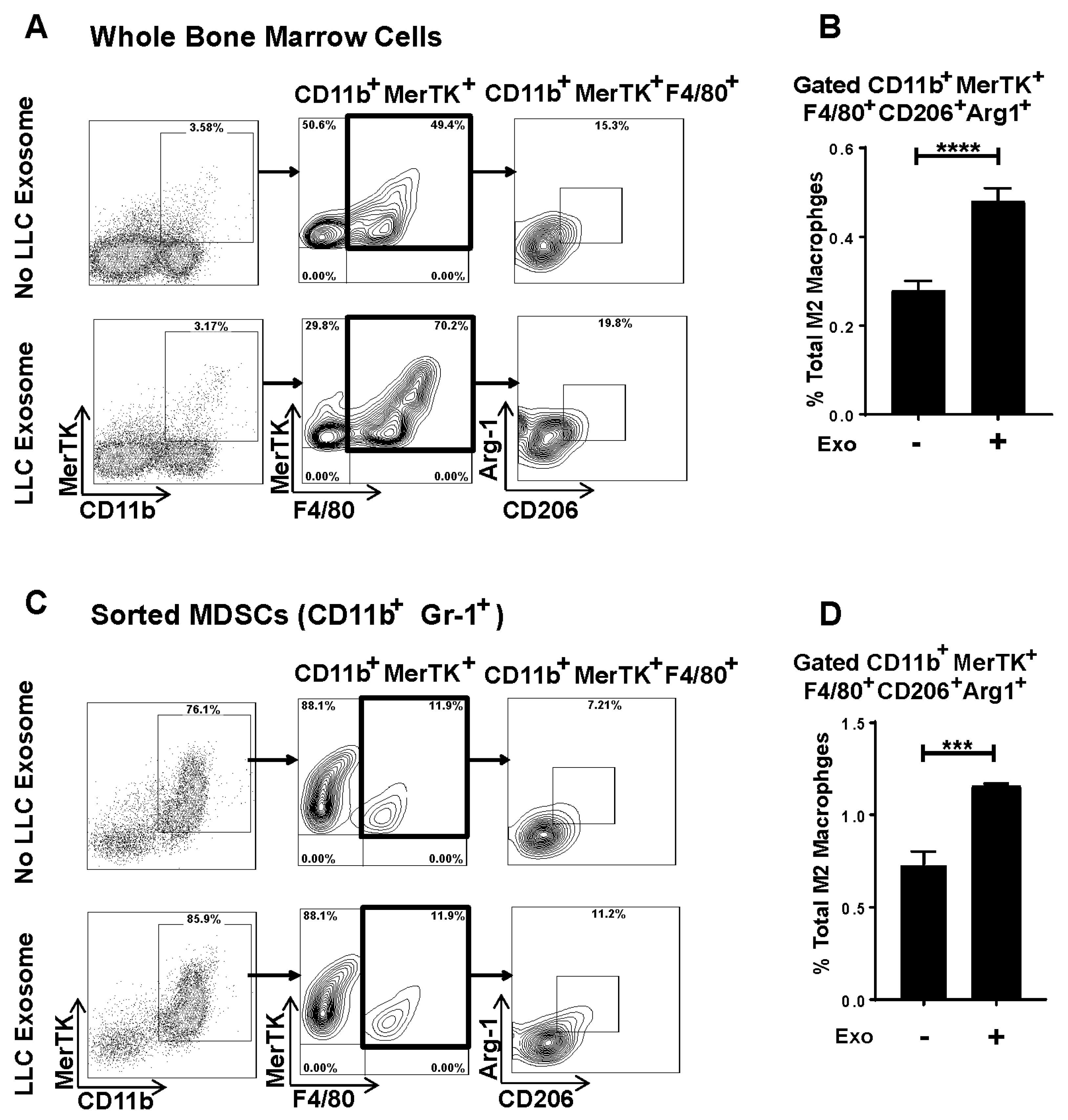
3.5. Murine Lung Tumor Cell-Derived Exosomes Differentiate Bone Marrow (BM) Cells and MDSCs to M2 Macrophages
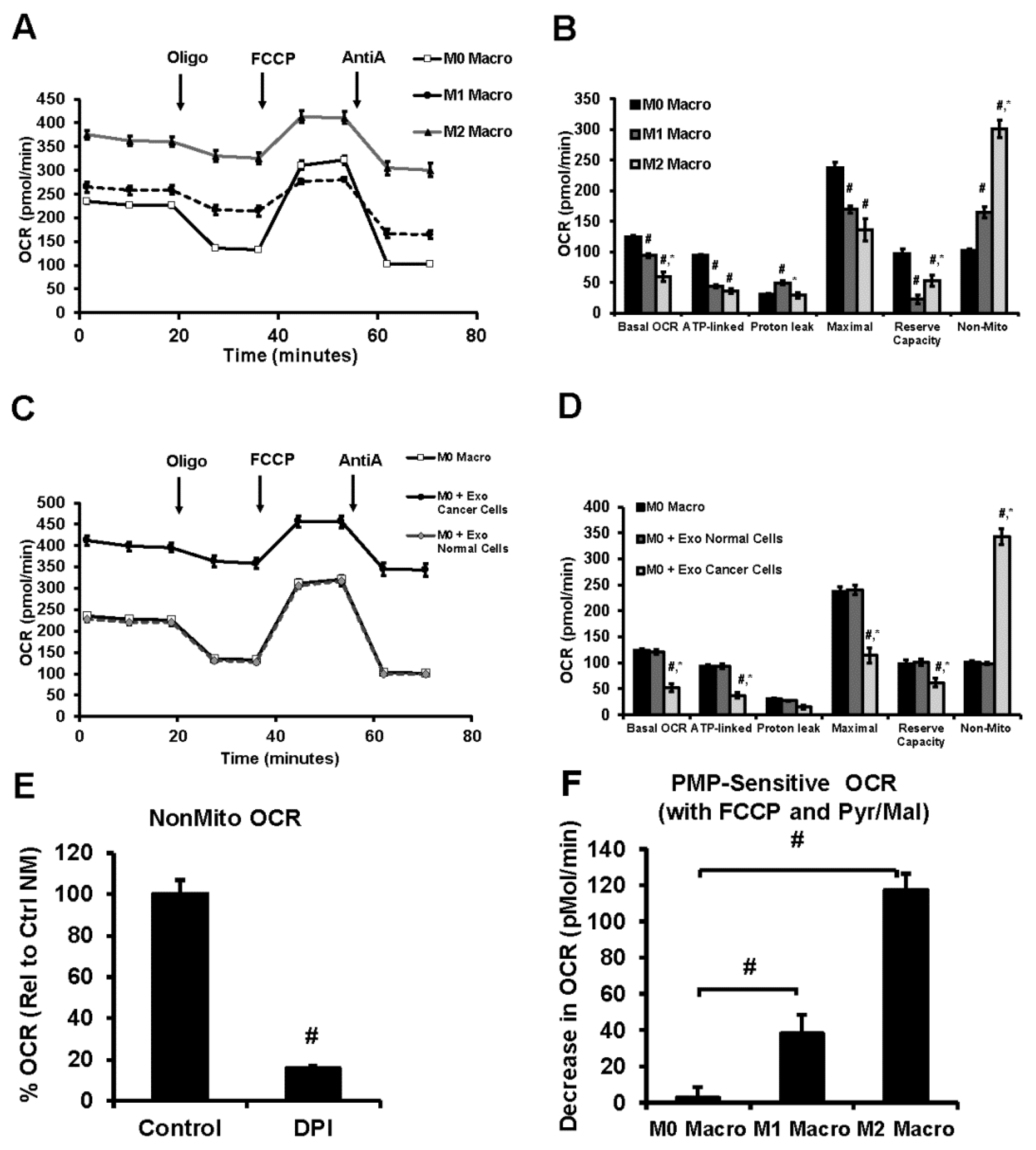
3.6. Lung Tumor Cell-Derived Exosomes Modulate Cellular Bioenergetics of Macrophages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TME | Tumor microenvironment |
| MDSCs | Myeloid derived suppressor cells |
| LWC | Lewis lung carcinoma |
| NSCLC | Non-small-cell lung carcinoma |
| TAMs | Tumor associated macrophages |
| BMDMs | Bone marrow-derived macrophages |
| BM | Bone marrow |
| RPS 6 | Ribosomal protein S6 |
| OCR | Oxygen consumption rates |
| EpCAM | Epithelial cell adhesion molecule |
| TSG-101 | Tumor susceptibility gene 101 |
| PMA | Phorbol 12-myristate-13-acetate |
| CHI3L1 | Chitinase-3-like protein 1 |
| RETNLB | Resistin-like beta |
| Arg-1 | Arginase-1 |
| MerTK | Proto-oncogene tyrosine-protein kinase MER |
| DPI | Diphenyleneiodonium chloride |
| PMP | Plasma Membrane Permeabilizer |
| HIF-1a | Hypoxia-inducible factor-1α |
| NADPH oxidase | Nicotinamide adenine dinucleotide phosphate oxidase |
| LPS | Lipopolysaccharides |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Cancer Facts and Figures. 2017. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-Figures/cancer-facts-Figures-2017.html (accessed on 1 April 2020).
- World Health Organization. The Global Burden of Disease: 2004 Update; World Health Organization: Geneva, Switzerland, 2004. [Google Scholar]
- Woo, E.Y.; Chu, C.S.; Goletz, T.J.; Schlienger, K.; Yeh, H.; Coukos, G.; Rubin, S.C.; Kaiser, L.R.; June, C.H. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001, 61, 4766–4772. [Google Scholar]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [Green Version]
- Olkhanud, P.B.; Damdinsuren, B.; Bodogai, M.; Gress, R.E.; Sen, R.; Wejksza, K.; Malchinkhuu, E.; Wersto, R.P.; Biragyn, A. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4+ T cells to T-regulatory cells. Cancer Res. 2011, 71, 3505–3515. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Schafer, C.; Hough, K.; Tousif, S.; Duncan, S.R.; Kearney, J.; Ponnazhagan, S.; Hsu, H.-C.; Deshane, J.S. Myeloid-Derived Suppressor Cells Impair B Cell Responses in Lung Cancer through IL-7 and STAT5. J. Immunol. 2018, 201, 278–295. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.-Z.; Condeelis, J.S. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Toh, B.; Toh, B.; Abastado, J.P.; Abastado, J.P. Myeloid cells: Prime drivers of tumor progression. Oncoimmunology 2012, 1, 1360–1367. [Google Scholar] [CrossRef] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunology 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Marleau, A.M.; Chen, C.-S.; A Joyce, J.; Tullis, R.H. Exosome removal as a therapeutic adjuvant in cancer. J. Transl. Med. 2012, 10, 134. [Google Scholar] [CrossRef] [Green Version]
- Muller, L.; Muller-Haegele, S.; Mitsuhashi, M.; Gooding, W.; Okada, H.; Whiteside, T.L. Exosomes isolated from plasma of glioma patients enrolled in a vaccination trial reflect antitumor immune activity and might predict survival. OncoImmunology 2015, 4, e1008347. [Google Scholar] [CrossRef] [Green Version]
- Ismail, N.; Wang, Y.; A Dakhlallah, D.; Moldovan, L.; Agarwal, K.; Batte, K.; Shah, P.; Wisler, J.; Eubank, T.D.; Tridandapani, S.; et al. Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood 2013, 121, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.W.; Yu, T.J.; Zhang, J.; Li, Y.; Chen, H.L.; Yang, G.F.; Yu, W.; Liu, Y.Z.; Liu, X.X.; Duan, C.F.; et al. CYP4A in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene 2017, 36, 5045–5057. [Google Scholar] [CrossRef] [Green Version]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Xu, D.; Yu, X.; Ding, T.; Rao, H.; Zhan, Y.; Zheng, L.; Li, L. Association of Intra-tumoral Infiltrating Macrophages and Regulatory T Cells Is an Independent Prognostic Factor in Gastric Cancer after Radical Resection. Ann. Surg. Oncol. 2011, 18, 2585–2593. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Rodriguez, J.-C.; Traves, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martin-Sanz, P.; Cascante, M.; Boscá, L. Substrate Fate in Activated Macrophages: A Comparison between Innate, Classic, and Alternative Activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; A Wagner, R.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Soo, C.Y.; Song, Y.; Zheng, Y.; Campbell, E.C.; Riches, A.C.; Gunn-Moore, F.; Powis, S.J. Nanoparticle tracking analysis monitors microvesicle and exosome secretion from immune cells. Immunology 2012, 136, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Hough, K.; Chanda, D.; Duncan, S.R.; Thannickal, V.J.; Deshane, J.S. Exosomes in immunoregulation of chronic lung diseases. Allergy 2016, 72, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Boil. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nature 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, B.G.; Shiva, S.; Ballinger, S.; Zhang, J.; Darley-Usmar, V. Bioenergetics and translational metabolism: Implications for genetics, physiology and precision medicine. Boil. Chem. 2019, 401, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Hough, K.; Trevor, J.L.; Strenkowski, J.G.; Wang, Y.; Chacko, B.; Tousif, S.; Chanda, D.; Steele, C.; Antony, V.B.; Dokland, T.; et al. Exosomal transfer of mitochondria from airway myeloid-derived regulatory cells to T cells. Redox Boil. 2018, 18, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Würdinger, T.; Van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O.; Sena-Esteves, M.; et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nature 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Dickman, C.T.; Lawson, J.; Jabalee, J.; MacLellan, S.A.; LePard, N.E.; Bennewith, K.L.; Garnis, C. Selective extracellular vesicle exclusion of miR-142-3p by oral cancer cells promotes both internal and extracellular malignant phenotypes. Oncotarget 2017, 8, 15252–15266. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. Exosomes and tumor-mediated immune suppression. J. Clin. Investig. 2016, 126, 1216–1223. [Google Scholar] [CrossRef] [Green Version]
- Sevko, A.; Umansky, V. Myeloid-Derived Suppressor Cells Interact with Tumors in Terms of Myelopoiesis, Tumorigenesis and Immunosuppression: Thick as Thieves. J. Cancer 2012, 4, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.; Lima, L.G.; Chai, E.P.Z.; Müller, A.; Lobb, R.J.; Krumeich, S.; Wen, S.W.; Wiegmans, A.P.; Moller, A. Breast Cancer-Derived Exosomes Alter Macrophage Polarization via gp130/STAT3 Signaling. Front. Immunol. 2018, 9, 871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mears, R.; Craven, R.A.; Hanrahan, S.; Totty, N.; Upton, C.; Young, S.L.; Patel, P.; Selby, P.J.; E Banks, R. Proteomic analysis of melanoma-derived exosomes by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry. Proteomics 2004, 4, 4019–4031. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.; To, J.; O’Brien, B.A.; Donnelly, S. The choice of phorbol 12-myristate 13-acetate differentiation protocol influences the response of THP-1 macrophages to a pro-inflammatory stimulus. J. Immunol. Methods 2016, 430, 64–70. [Google Scholar] [CrossRef]
- Stossi, F.; Erdogan, Z.M.; Katzenellenbogen, B.S. Macrophage-elicited loss of estrogen receptor-α in breast cancer cells via involvement of MAPK and c-Jun at the ESR1 genomic locus. Oncogene 2011, 31, 1825–1834. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Chacko, B.K.; A Kramer, P.; Ravi, S.; Johnson, M.S.; Hardy, R.W.; Ballinger, S.W.; Darley-Usmar, V. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab. Investig. 2013, 93, 690–700. [Google Scholar] [CrossRef]
- Ravi, S.; Chacko, B.; Sawada, H.; Kramer, P.A.; Johnson, M.S.; Benavides, G.A.; O’Donnell, V.B.; Marques, M.; Darley-Usmar, V. Metabolic Plasticity in Resting and Thrombin Activated Platelets. PLOS ONE 2015, 10, e0123597. [Google Scholar] [CrossRef] [Green Version]
- Kramer, P.A.; Chacko, B.K.; George, D.J.; Zhi, D.; Wei, C.-C.; Dell’Italia, L.J.; Melby, S.J.; George, J.F.; Darley-Usmar, V. Decreased Bioenergetic Health Index in monocytes isolated from the pericardial fluid and blood of post-operative cardiac surgery patients. Biosci. Rep. 2015, 35, e00237. [Google Scholar] [CrossRef]
- Yu, X.; Harris, S.L.; Levine, A.J. The Regulation of Exosome Secretion: A Novel Function of the p53 Protein. Cancer Res. 2006, 66, 4795–4801. [Google Scholar] [CrossRef] [Green Version]
- Soldano, S.; Trombetta, A.C.; Contini, P.; Tomatis, V.; Ruaro, B.; Brizzolara, R.; Montagna, P.; Sulli, A.; Paolino, S.; Pizzorni, C.; et al. Increase in circulating cells coexpressing M1 and M2 macrophage surface markers in patients with systemic sclerosis. Ann. Rheum. Dis. 2018, 77, 1842–1845. [Google Scholar] [CrossRef] [PubMed]
- Genin, M.; Clément, F.; Fattaccioli, A.; Raes, M.; Michiels, C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 2015, 15, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Levin, M.; Kaplan, D. Bioelectric modulation of macrophage polarization. Sci. Rep. 2016, 6, 21044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunology 2016, 44, 303–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Muller, L.; Mitsuhashi, M.; Simms, P.; Gooding, W.E.; Whiteside, T.L. Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci. Rep. 2016, 6, 20254. [Google Scholar] [CrossRef] [Green Version]
- Hough, K.; Wilson, L.S.; Trevor, J.L.; Strenkowski, J.G.; Maina, N.; Kim, Y.-I.; Spell, M.L.; Wang, Y.; Chanda, D.; Dager, J.R.; et al. Unique Lipid Signatures of Extracellular Vesicles from the Airways of Asthmatics. Sci. Rep. 2018, 8, 10340. [Google Scholar] [CrossRef]
- Lu, J.; Li, J.; Liu, S.; Wang, T.; Ianni, A.; Bober, E.; Braun, T.; Xiang, R.; Yue, S. Exosomal tetraspanins mediate cancer metastasis by altering host microenvironment. Oncotarget 2017, 8, 62803–62815. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. p53, the Cellular Gatekeeper for Growth and Division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Zhang, J.; Chen, S.; Lu, M.; Luo, X.; Yao, S.; Liu, S.; Qin, Y.; Chen, H. Tumor-associated macrophages provide a suitable microenvironment for non-small lung cancer invasion and progression. Lung Cancer 2011, 74, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L. Session 2: Inflammation and Cancer. Toxicol. Pathol. 2004, 32, 732. [Google Scholar] [CrossRef]
- Mantovani, A. Inflammation by remote control. Nature 2005, 435, 752–753. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ng, D.S.W.; Mah, W.-C.; Almeida, F.F.; A Rahmat, S.; Rao, V.K.; Leow, S.C.; Laudisi, F.; Peh, M.T.; Goh, A.M.; et al. A unique role for p53 in the regulation of M2 macrophage polarization. Cell Death Differ. 2014, 22, 1081–1093. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lü, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Duan, C.; Pang, H.; Chen, Q.; Han, B.; Zha, C.; Dinislam, M.; Wu, P.; Li, Z.; Zhao, S.; et al. DNA damage repair alterations modulate M2 polarization of microglia to remodel the tumor microenvironment via the p53-mediated MDK expression in glioma. EBioMedicine 2019, 41, 185–199. [Google Scholar] [CrossRef] [Green Version]
- Bardi, G.T.; Smith, M.A.; Hood, J. Melanoma exosomes promote mixed M1 and M2 macrophage polarization. Cytokine 2018, 105, 63–72. [Google Scholar] [CrossRef]
- Cortez-Retamozo, V.; Etzrodt, M.; Newton, A.; Rauch, P.J.; Chudnovskiy, A.; Berger, C.; Ryan, R.; Iwamoto, Y.; Marinelli, B.; Gorbatov, R.; et al. Origins of tumor-associated macrophages and neutrophils. Proc. Natl. Acad. Sci. USA 2012, 109, 2491–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, C.; Wang, Y.; Hough, K.; Sawant, A.; Grant, S.C.; Thannickal, V.J.; Zmijewski, J.; Ponnazhagan, S.; Deshane, J.S. Indoleamine 2,3-dioxygenase regulates anti-tumor immunity in lung cancer by metabolic reprogramming of immune cells in the tumor microenvironment. Oncotarget 2016, 7, 75407–75424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawant, A.; Schafer, C.; Jin, T.H.; Zmijewski, J.; Tse, H.M.; Roth, J.; Sun, Z.; Siegal, G.P.; Thannickal, V.J.; Grant, S.C.; et al. Enhancement of Antitumor Immunity in Lung Cancer by Targeting Myeloid-Derived Suppressor Cell Pathways. Cancer Res. 2013, 73, 6609–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Tedesco, S.; De Majo, F.; Kim, J.; Trenti, A.; Trevisi, L.; Fadini, G.P.; Bolego, C.; Zandstra, P.W.; Cignarella, A.; Vitiello, L. Convenience versus Biological Significance: Are PMA-Differentiated THP-1 Cells a Reliable Substitute for Blood-Derived Macrophages When Studying in Vitro Polarization? Front. Pharmacol. 2018, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Kurynina, A.V.; Erokhina, M.V.; Makarevich, O.A.; Sysoeva, V.Y.; Lepekha, L.N.; Kuznetsov, S.A.; Onishchenko, G.E. Plasticity of Human THP–1 Cell Phagocytic Activity during Macrophagic Differentiation. Biochemistry 2018, 83, 200–214. [Google Scholar] [CrossRef]
- Russell, D.G.; Huang, L.; VanderVen, B. Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol. 2019, 19, 291–304. [Google Scholar] [CrossRef]
- Blouin, C.C.; Pagé, E.L.; Soucy, G.M.; Richard, D.E. Hypoxic gene activation by lipopolysaccharide in macrophages: Implication of hypoxia-inducible factor 1α. Blood 2004, 103, 1124–1130. [Google Scholar] [CrossRef]
- Bossche, J.V.D.; Baardman, J.; Otto, N.A.; Van Der Velden, S.; Neele, A.E.; Berg, S.M.V.D.; Luque-Martin, R.; Chen, H.-J.; Boshuizen, M.C.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Liu, J.; Ma, Q.; Cao, L.; Fattah, R.J.; Yu, Z.; Bugge, T.H.; Finkel, T.; Leppla, S.H. Solid tumor therapy by selectively targeting stromal endothelial cells. Proc. Natl. Acad. Sci. USA 2016, 113, E4079–E4087. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Choksi, S.; Qu, J.; Jang, J.; Choe, M.; Banfi, B.; Engelhardt, J.F.; Liu, Z. NADPH Oxidases Are Essential for Macrophage Differentiation. J. Boil. Chem. 2016, 291, 20030–20041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helfinger, V.; Palfi, K.; Weigert, A.; Schröder, K. The NADPH Oxidase Nox4 Controls Macrophage Polarization in an NFκB-Dependent Manner. Oxidative Med. Cell. Longev. 2019, 2019, 3264858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene 2019, 38, 5158–5173. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.S.; Roy, A.; Rajpoot, S.; Liu, D.; Savai, R.; Banerjee, S.; Kawada, M.; Faisal, S.M.; Saluja, R.; Saqib, U.; et al. Tumor-derived exosomes in the regulation of macrophage polarization. Inflamm. Res. 2020, 69, 435–451. [Google Scholar] [CrossRef]
- Garaude, J.; Acín-Pérez, R.; Martínez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villán, E.; Hervas-Stubbs, S.; Pelegrín, P.; Sander, L.E.; Enríquez, J.A.; et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016, 17, 1037–1045. [Google Scholar] [CrossRef] [Green Version]
- Lapuente-Brun, E.; Loshuertos, R.M.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodríguez-Hernández, Á.; et al. Supercomplex Assembly Determines Electron Flux in the Mitochondrial Electron Transport Chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence |
|---|---|
| IL-10 Forward | AAGCCTGACCACGCTTTCTA |
| IL-10 Reverse | CCCAAGCCCAGAGACAAGAT |
| Arg1 Forward | GCCCTTTGCTGACATCCCTA |
| Arg1 Reverse | CGCTTGCTTTTCCCACAGAC |
| CHI3L1 (Ym1) Forward | AGGTCACCATTGACAGCAGC |
| CHI3L1 (Ym1) Reverse | ATCCTCCTGACCTCGGAACA |
| RETNLB (Fizz1) Forward | TCAAAAGCCAAGGCAGACCG |
| RETNLB (Fizz1) Reverse | AACATCCCACGAACCACAGC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pritchard, A.; Tousif, S.; Wang, Y.; Hough, K.; Khan, S.; Strenkowski, J.; Chacko, B.K.; Darley-Usmar, V.M.; Deshane, J.S. Lung Tumor Cell-Derived Exosomes Promote M2 Macrophage Polarization. Cells 2020, 9, 1303. https://doi.org/10.3390/cells9051303
Pritchard A, Tousif S, Wang Y, Hough K, Khan S, Strenkowski J, Chacko BK, Darley-Usmar VM, Deshane JS. Lung Tumor Cell-Derived Exosomes Promote M2 Macrophage Polarization. Cells. 2020; 9(5):1303. https://doi.org/10.3390/cells9051303
Chicago/Turabian StylePritchard, Alexandra, Sultan Tousif, Yong Wang, Kenneth Hough, Saad Khan, John Strenkowski, Balu K. Chacko, Victor M. Darley-Usmar, and Jessy S. Deshane. 2020. "Lung Tumor Cell-Derived Exosomes Promote M2 Macrophage Polarization" Cells 9, no. 5: 1303. https://doi.org/10.3390/cells9051303