
Abstract
A new series of tetrahydroacridine derivatives with the fluorobenzoyl moiety was synthesized and evaluated for cytotoxic activity against lung cancer cell lines A549 and colorectal cancer HT29. The cytotoxic activity of the compounds was compared on the somatic cell line—EAhy926. Compounds showed high cytotoxic activity on A549 cells (IC50 183.26–68.07 μM) and HT29 cells (IC50 68.41–19.70 μM), higher than controls—etoposide (IC50 451.47 μM) toward A549 and 5-fluorouracil (IC50 1626.85 μM) against HT29. Derivative 4 was the most cytotoxic to A549, whereas for the cell lines HT29 compound 6. Selected compounds showed similar cytotoxicity to the EAhy926 cell line (IC50 about 50 μM). In the hyaluronidase inhibition assay, all compounds exhibited anti-inflammatory activity, including 4 exhibiting the best inhibitory activity—IC50 of 52.27 μM when the IC50 heparin was 56.41 μM. Mathematical modeling was performed to determine LD50 after intraperitoneal, oral, intravenous and subcutaneous administration and to predict potential mutagenicity and carcinogenicity of the compounds analyzed. Obtained results showed that tested derivatives are slightly toxic compounds, and LD50 values (mg/kg) ranged from 680 to 1200 (oral rat model), the analyzed compounds have low mutagenic potential, and differences between derivatives are insignificant and very low probability of carcinogenicity. To confirm mathematical calculations, an in vivo test was carried out on a laboratory mouse model for two selected compounds. It allowed to qualify compounds: 6 to category 4 of the GHS scale, and 4 to category 3 of the GHS scale.
Similar content being viewed by others


Introduction
Cancer disease regards abnormal cell growth, which can further spread to other parts of the body and metastasize. Around 100 million people suffer from cancer worldwide. In 2018, there was about 18.1 million new cancer cases, which caused 9.6 million cancer deaths [1]. Cancer treatment costs 1.16 trillion USD per year. The most common types of cancer are lung, prostate, colorectal and stomach cancer in men, whereas breast, colorectal, lung and cervical cancer appear mostly in women [2]. People live longer and they have changed their lifestyle in the developing countries; therefore, new cases of cancer have started to appear more and more widely [3].
Lung cancer is the most common type of cancer in the world, causes 2.1 million new cases and 1.8 million deaths per year [1]. This makes it the most common cause of cancer deaths in men, and second, most often in women [2]. It is a malignant lung tumor, which symptoms regard coughing, weight loss, shortness of breath and chest pains [4]. Lung cancer mostly appear in people who smoke [5], but about 10–15% of cases occur in people who have never smoked [6]. The two main types are small-cell lung carcinoma (SCLC) and non-small-cell lung carcinoma (NSCLC) [7]. Treatment depends on the type of cancer, the stage and the general health of the person. Treatment involves surgery, chemotherapy and radiotherapy. NSCLC is more often treated surgically, while SCLC usually responds better to chemotherapy and radiotherapy [8].
Colon cancer is a malignant cancer, which symptoms may include blood in the stool, a change in bowel movements and weight loss [9]. In the world, colon cancer is the third most common type of cancer, with 1.8 million new cases and 880,000 deaths per year [2]. It occurs much more frequently in developed countries and more often in men than in women [10]. Most colon tumors are associated with bad lifestyle, inadequate diet, aging of the body, and only a small number of cases are caused by genetic disorders [11]. Colon cancer is commonly diagnosed by a sigmoidoscopy or colonoscopy [12]. Treatment of this type of cancer involves a combination of surgery, radiotherapy, chemotherapy and targeted therapy [9].
Acridines have been known as therapeutic drugs for many years and their use regard antibacterial, antiviral and anticancer mechanism of actions. Acridine and its derivatives intercalate into DNA [13] and intercalation process results in unwinding of DNA helix. These actions cause inhibition of replication and transcription, and this mechanism of actions is demanding in cancer treatment [14]. Nowadays one of acridine derivatives—amsacrine—is used as an antineoplastic drug to treat leukemia. Researcher’s team work on novel acridine derivatives, which further can be used in cancer therapy [7, 15,16,17,18,19,20]. We worked on novel tetrahydroacridine derivatives, which previously had been tested in in vivo biodistribution. Results had showed that compounds accumulated in large amount in the rat’s bowels and lungs [21]. Therefore, novel tetrahydroacridine derivatives were tested for cytotoxicity effect on lung and colon cancer cell lines.
Materials and methods
Synthesis of tetrahydroacridine derivatives
The synthesis of new tetrahydroacridine derivatives with fluorobenzo groups has been described previously by Czarnecka et al. [22]. This synthesis consisted of the combination of 9-chloro-1,2,3,4-tetrahydroacridine with 2-fluorobenzoic acid or 3-fluorobenzoic acid using diamine linker. Semi-finished products were used to obtain the final compounds. Reaction of 9-chloro-1,2,3,4-tetrahydroacridine with 2 equivalent of the corresponding a, x-diamine in the presence of phenol at 180 °C and catalytic amounts of the sodium iodide provided these intermediate products in good yield (80–90%). Subsequently, 2-chloro-4,6-dimethyl-1,3,5-triazine (CDMT) was used. The reaction mechanism included the activation of the carboxylic acid group (2-fluorobenzoic or 3-fluorobenzoic acid) by means of CDMT. The reaction may have been carried out in a suitable inert solvent such as dichloromethane, acetonitrile, dioxane or tetrahydrofuran at a temperature below 5 °C. The active ester was obtained, which was then reacted with the appropriate amine, added to the same pot. Compounds were obtained in good yield (about 80–90%), which were then purified by flash chromatography. In the last step, the obtained compounds were converted into the hydrochloride salts by dissolving in a small volume of methanol and adding HCl in ether. The HCl salts of the new compounds were synthesized to achieve higher solubility in polar solvents [22].
Cell culture
To assess the cytotoxicity of the new compounds, the A549 cell line (lung carcinoma from human) (European Collection of Cell Culture) was selected. Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (PAN-Biotech), which includes 10% Fetal Bovine Serum (Sigma Aldrich), 2 mM Glutamine (Sigma Aldrich), and 100 units/mL penicillin and 100 mg/ml streptomycin (Sigma Aldrich).
HT29 cell line (colorectal adenocarcinoma from human) (American Type Culture Collection) was chosen to evaluate the cytotoxicity of novel compounds. Cells were grown in McCoy’s Medium (Biological Industries) which includes 10% Fetal Bovine Serum (Sigma Aldrich), 2 mM Glutamine (Sigma Aldrich), and 100 units/mL penicillin and 100 mg/ml streptomycin (Sigma Aldrich).
To compare the cytotoxicity of the new compounds, the EAhy926 cell line (the human umbilical vein, somatic cell hybrid) (American Type Culture Collection) was selected. Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (PAN-Biotech) which includes 10% Fetal Bovine Serum (Sigma Aldrich), 2 mM Glutamine (Sigma Aldrich), and 100 units/mL penicillin and 100 mg/ml streptomycin (Biological Industries). Before the initiation of the assay, cells were plated and grown in an incubator at 37 °C with 5% CO2 to 80% confluence.
MTT cytotoxicity assay
Cell viability following exposure to synthetic compounds was estimated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) reduction assay. To perform the MTT test, cells were seeded into 96-well plates at density 104 cells per well and cultured for 24 h at 37 °C and 5% CO2. Next the medium was removed and cells were exposed to the 100 μl of the compound solutions over a range of concentrations (final DMSO concentration was below 0.2%) or nothing but culture medium (blank control), and pure DMSO was used as a positive control. Cells with compound solutions were incubated for 24 h. Medium was removed and 50 μl of the MTT solution was added to each well and incubated in the dark for additional 2 h at 37 °C. Later the MTT solution was carefully removed and 100 μl of DMSO was added. Plates were held for 10 min at room temperature. Before placing in a microplate reader (Synergy H1, BioTek, Winooski, VT, USA) 5 μl of Sorensen Buffer was added to each well. Plate was swayed and the absorbance was measured at a wavelength of 570 nm. The cell viability was expressed as a percentage of the control values (blank) [23, 24].
Hyaluronidase inhibition assay
All compounds were subjected in hyaluronidase inhibition test to determine their inhibitory activity toward enzyme. The hyaluronidase inhibition study was carried out by turbidimetric method modified to the 96-well plates and previously described by Michel et al. [25].
Analyzed compound solutions were prepared freshly right before the assay, and research began by adding 20 μl of the tested compound solution in monosodium phosphate buffer (pH 7.0) and 40 μl of hyaluronidase solution (22.55 U/ml, hyaluronidase from bovine testes Type I-S, Sigma Aldrich) to the wells of 96-well microtiter plates. The solutions were incubated in the dark for 10 min at the temperature 37 ºC and then 40 μl of hyaluronic acid solution (0.03%, Sigma Aldrich) in monosodium phosphate buffer (pH 5.35) was added to the wells. Next, the mixture was incubated in the dark for 45 min at 37 °C. At the end, 300 μl of bovine serum albumin (0.1%, Serva) in sodium acetate buffer (pH 3.75) was added to the wells and incubation was carried out at the room temperature for 10 min.
Using the microplate reader (BioTek, Winooski, VT, USA), the turbidity values were measured. The measurement was made at a length of 600 nM. The assay was run in three experiments in triplicate to calculate the half maximal inhibitory concentration—IC50 values. Heparin (WZF, Polfa Warsaw) was a positive control. The inhibitory activity of the tested compounds was calculated as inhibition percentage (% inhibition) of hyaluronidase according to the equation:
where AHA is the absorbance of solution without the enzyme (positive control), AHYAL the absorbance of solution without the tested compound (negative control), and AAN the absorbance of solution with the tested compound.
In silico toxicity calculations
Acute toxicity to rodents expressed in the dose of compound required to kill half the members of a tested population—LD50 values after intraperitoneal, oral, intravenous and subcutaneous administration—was calculated with the use of ACD/Percepta 14.0.0 software (ACD/Labs). The same software was also used for the prediction of the mutagenicity of the analyzed compounds and estimation of the probability of positive Ames test.
The carcinogenicity of the studied tetrahydroacridine derivatives was predicted with the use of Vega 1.1.4 software based on CAESAR 2.1.9 model.
In vivo acute oral toxicity: acute toxic class method
To determine the mathematical calculations, the effect on living organisms and qualify new tetrahydroacridine derivatives to a specific acute toxicity class, an in vivo test was performed on a laboratory mouse model according to OECD 423 "Acute Oral Toxicity—Acute Toxic Class Method" guidelines. The research was carried out based on the resolution of the Local Ethics Committee for Animal Experiments at the Medical University of Lodz No. 57/115 ŁB/2018.
The chemical substances were given by the oral route in one of four fixed doses (5, 50, 300, 2000 mg/kg) for a given stage of the study. The results will allow you to rank chemicals in commonly used classification systems. According to the diagram from the OECD 423 guidelines, a maximum of 6 test stages can be carried out. Three animals are involved in each stage, and the number of stages depends on the mortality of the animals, which is the final test parameter.
After the quarantine, handling, weighing and labeling, the substance was administered at a dose of 300 mg/kg per body weight (dose used in the absence of information on the toxicity of the test substance). If 2–3 animals are found after using the dose, substances should be given at a lower dose—50 mg/kg (the last dose of 5 mg/kg is used, if after 50 mg/kg, also 2–3 animals die). In the absence of death or death of only 1 animal, the substance is again administered to the stomach at the same dose—300 mg/kg, and in the case of subsequent deaths, a higher dose, 2000 mg/kg, is used. Absence or presence of the mortality of animals exposed at one stage determined the performance of the next stage of the study. After each dose, animals were observed for 14 days. The animals will then be killed by intraperitoneal administration of a lethal dose of sodium pentobarbital.
Statistical analysis
Values were represented as mean ± SD. One-way ANOVA with post-hoc analysis was made. Statistical analysis was performed in Statistica version 13.1 software.
Results and discussion
In vitro cytotoxicity of new compounds against cancer cell lines
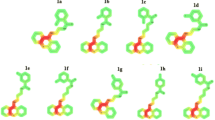
Tetrahydroacridine derivatives with the fluorobenzoic acid moiety (Figs. 1, 2) were tested in vitro by the MTT test against two human cancer cell lines—A549 and HT29; and one human non-neoplastic cell line EA.hy926. In cancer cells, p53 tumor suppressor protein plays a crucial role in cancer progression. Between both cancer cell lines, there is a difference between p53. A549 cells possess wild-type p53, whereas in HT-29, there is a mutant p53 (sequence codon change—273)—which results in increased invasion and cell scattering [26]. The IC50 value was determined by concentration–response analysis. The results of these tests are presented in Table 1. The positive control of the studies were chemotherapeutic—etoposide and 5-fluorouracil, which are commonly used clinical agents.

Compound no. 1 (n = 2), 2 (n = 3), 3 (n = 4), substitute: 2-fluorobenzoic acid

Compound no. 4 (n = 2), 5 (n = 3), 6 (n = 4), substitute: 3-fluorobenzoic acid
All tested tetrahydroacridine derivatives showed cytotoxic activity. IC50 values ranged from 183.26 μM to 68.07 μM relative to the lung cancer cell line and in the range of 68.41–19.70 μM relative to the colon cancer cell line. The strongest effect against A549 was demonstrated by compound 4 (Fig. 3), and against the HT29 cell by molecule 6 (Fig. 4). Therefore, they were chosen for research on human cells.

Cell viability of compound no. 4 against cell lines—A549. Experiments were done in triplicates. Statistical significance was assessed using one-way ANOVA with a post-hoc analysis was performed. ***p < 0.001, **p < 0.01, and *p < 0.05 were considered as significantly different in comparison to non-treated control

Cell viability of compound no. 6 against cell lines—HT29. Experiments were done in triplicates. Statistical significance was assessed using one-way ANOVA with a post-hoc analysis was performed. ***p < 0.001, **p < 0.01, and *p < 0.05 were considered as significantly different in comparison to non-treated control
Control compounds showed significantly lower cytotoxic activity than the tested derivatives—IC50 etoposide 451.47 μM and 654.03 μM, 5-fluorouracil IC50 1626.85 μM and > 1800 μM. For the A549 cell line, IC50 values were higher than for HT29 cells, indicating that colon adenocarcinoma cells are more sensitive to tetrahydroacridine derivatives than lung adenocarcinoma cells. The compounds were divided into two groups, depending on the position of the fluoride. Against lung and colorectal carcinoma, the structure–activity relationship showed that a longer carbon linkage and fluoride substitution in meta, and preferably in a pair, positions significantly increased cytotoxic activity.
The cytotoxicity effects on EA.hy926 cells were comparable for both compositions and were approximately 50 μM. There was a significant difference between cancer and non-cancer cell line IC50 results. These results suggest that the compounds were more selective for colon cancer cells than normal cells. Compound 6 (Fig. 5) was slightly more toxic than compound 4. Etoposide showed a higher cytotoxic activity on EA.hy926 cells (IC50 155.19 μM) than A549 cells (IC50 451.47 μM) that showed toxic effects on endothelial cells. While 5-fluorouracil exhibited similar cytotoxicity to HT29 and EA.hy926 cell lines.

Cell viability of compound no. 6 against cell lines—EA.hy9263. Experiments were done in triplicates. Statistical significance was assessed using one-way ANOVA with a post-hoc analysis was performed. ***p < 0.001, **p < 0.01, and *p < 0.05 were considered as significantly different in comparison to non-treated control
In vitro inflammation study on HYAL
Hyaluronidase is an enzyme that undergoes depolymerization of hyaluronan (part of the extracellular matrix) and thus the integrity of the tissue is attenuated during the inflammatory process. This is the reason for the interest of many scientists studying new drugs with additional anti-inflammatory properties [27,28,29,30].
In the in vitro inflammation study on HYAL, inhibitory activity of new compounds was determined by spectrophotometric method. All compounds tested showed significant inhibitory activity. The IC50 values ranged from 52.27 to 272.31 μM, which with positive control (heparin) are shown in Table 2.
The inhibitory activity among compounds was influenced by the fluorine position and the number of methylene groups. For 2-fluorobenzoic acid derivatives, the higher number of methylene groups accelerated the inhibition, while for 3-fluorobenzoic acid the reverse situation was observed. Compound 4 had the best inhibitory activity (IC50 52.27 µM), comparable to the positive control (IC50 56.41 µM). It can be concluded that tetrahydroacridine derivatives have some anti-inflammatory activity.
In silico assessment of toxicity
The estimation of toxicity of recently synthesized pharmacological active compounds is one of the most important part of the investigation of new drugs. At this early stage of research, low-cost and fast methods based on in silico mathematical calculations are very useful. In this study, a well-known software was used to predict the acute toxicity, mutagenicity and carcinogenicity of the new synthesized compounds (Table 3).
Percepta software was used to estimate the acute toxicity to rodents on six models: mice intraperitoneal, mice oral, mice intravenous, mice subcutaneous, rat intraperitoneal and rat oral. The obtained results expressed in LD50 values clearly show that the tested tetrahydroacridine derivatives are slightly toxic compounds and LD50 [mg/kg] values ranged from 680 to 1200 (rat oral model). It can also be observed that the acute toxicity of the studied compounds increases with the rise of the atoms of carbon in aliphatic chain.
The same software was also used for the prediction of mutagenicity and estimation of the probability of positive Ames test. The obtained results indicate that the analyzed compounds possess low mutagenic potential (probability ranged 0.66–0.74) and the differences between the derivatives are insignificant.
Vega software was used to predict the carcinogenicity of the studied tetrahydroacridines and a very low probability of carcinogenic activity (0.07–0.33) was obtained. It should be also noticed that the position of fluorine in fluorobenzoic moiety has no influence on the carcinogenic potential as well as mutagenic and acute toxicity of the studied compounds.
Acute oral toxicity: acute toxic class method
Two best-performing compounds, i.e. no. 4 and 6, were tested.
For substance no. 6, 300 mg/kg was given to 2 groups. After first dose of 300 mg/kg, no mouse died, and after second dose of 300 mg/kg, 1 mouse died. Then, dose 2000 mg/kg was given. In this step, all 3 mice died. Therefore, dose 2000 mg/kg was not repeated for the second time and compound can be classified in category 4 of GHS scale, with LD50 cut-off 500 mg/kg. This result was comparable with in silico assessment of toxicity—compound 6 had LD50 value of 540 mg/kg.
After first dose of substance No. 4 of 300 mg/kg, 2 mice died. Therefore, this dose was not repeated and lower dose, 50 mg/kg, was given to the mice. In this step, 1 mouse died. Dose 50 mg/kg was repeated and none mouse died. Therefore, compound can be classified in category 3 of GHS scale, with LD50 cut-off 300 mg/kg. In the in vivo test, compound 4 turned out to be much more toxic than in silico assessment (LD50 value of 1300 mg/kg).
As a result of the tests, it can be concluded that substance no. 6 can be classified as category 4, while substance no. 4 can be classified as category 3 (Fig. 6).

Classification scheme of acute oral toxicity
Conclusion
The aim of the study was to determine the cytotoxic activity of six new tetrahydroacridine derivatives with a fluorobenzoic moiety on human cell lines A549 (lung adenocarcinoma), HT-29 (colon adenocarcinoma) and EA.hy926 (hybrid umbilical vein). The results confirmed cytotoxicity activities of novel compounds. Moreover, all compounds presented higher cytotoxicity activity than the reference drugs—etoposide and 5-fluorouracil. HT29 cells were more sensitive to the novel compounds than AA549 cells and the normal cell line. It can be concluded that novel compounds might be used in the treatment of colorectal cancer, with less harm to the vein cells and stronger toxicity toward cancer cells. Among all derivatives, compounds 4 and 6 showed the highest activity. Both belong to the derivatives with position of fluorine in meta position, which could indicate higher activity of compounds with substitution in meta than in ortho position. The anti-inflammatory activity was determined in the hyaluronidase inhibition assay. Compound 4 showed the highest inhibitory activity, what is more, slightly higher than a positive control. Compounds confirmed their inhibitory activity and in result, anti-inflammatory properties. Comparing results from the studies, compound 4 turned out to be the best compound to further studies on lung cancer, whereas compound 6 might be considered in the research against colorectal cancer. Therefore, these compounds were tested on animals. The results of tests carried out on the mouse model in accordance with OECD 423 allowed to qualify the substance no. 6 to category 4 of the GHS scale and substance no. 4 to category 3 of the GHS scale. The mathematical calculations showed that the tested tetrahydroacridine derivatives are slightly toxic compounds and the LD50 values [mg/kg] ranged from 680 to 1200 (oral rat model), the analyzed compounds have low mutagenic potential (the probability ranged from 0.66 to 0.74), and differences between derivatives are insignificant and very low probability of carcinogenicity (0.07–0.33).
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. https://doi.org/10.3322/caac.21492.
Stewart B, Wild C. World Cancer Report. Lyon: International Agency for Research on Cancer; 2014.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. https://doi.org/10.3322/caac.20107.
Horn L, Lovly C, Johnson D. Chapter107: neoplasms of the lung. In: Kasper DLHS, Jameson JL, Fauci AS, Longo DL, Loscalzo J, editors. Harrison's principles of internal medicine. 19th ed. New York: McGraw-Hill; 2015.
Alberg A, Brock M, Samet J. Chapter 52: epidemiology of lung cancer. Murray & Nadel's textbook of respiratory medicine. 6th ed. Philadelphia: Saunders Elsevier; 2016.
Thun MJ, Hannan LM, Adams-Campbell LL, Boffetta P, Buring JE, Feskanich D, et al. Lung cancer occurrence in never-smokers: an analysis of 13 cohorts and 22 cancer registry studies. PLoS Med. 2008;5(9):e185-e. https://doi.org/10.1371/journal.pmed.0050185.
Olszewska P, Mikiciuk-Olasik E, Błaszczak-Świątkiewicz K, Szymański J, Szymański P. Novel tetrahydroacridine derivatives inhibit human lung adenocarcinoma cell growth by inducing G1 phase cell cycle arrest and apoptosis. Biomed Pharmacother. 2014;68(8):959–67. https://doi.org/10.1016/j.biopha.2014.10.018.
Chapman S, Robinson G, Stradling J, West S, Wrightson J. Chapter 31. Oxford handbook of respiratory medicine. 3rd ed. Oxford: Oxford University Press; 2014.
Ballinger AB, Anggiansah C. Colorectal cancer. BMJ (Clinical research ed). 2007;335(7622):715–8. https://doi.org/10.1136/bmj.39321.527384.BE.
Kuipers EJ, Grady WM, Lieberman D, Seufferlein T, Sung JJ, Boelens PG, et al. Colorectal cancer. Nat Rev Dis Primers. 2015;1:15065. https://doi.org/10.1038/nrdp.2015.65.
Hughes K, Bellis MA, Hardcastle KA, Sethi D, Butchart A, Mikton C, et al. The effect of multiple adverse childhood experiences on health: a systematic review and meta-analysis. Lancet Glob Health. 2017;2(8):e356–e366366. https://doi.org/10.1016/S2468-2667(17)30118-4.
Niedermaier T, Weigl K, Hoffmeister M, Brenner H. Fecal immunochemical tests in combination with blood tests for colorectal cancer and advanced adenoma detection-systematic review. United Eur Gastroenterol J. 2018;6(1):13–211. https://doi.org/10.1177/2050640617737004.
Demeunynck M. Antitumour acridines. Expert Opin Ther Pat. 2004;14(1):55–70. https://doi.org/10.1517/13543776.14.1.55.
Ferguson LR, Denny WA. Genotoxicity of non-covalent interactions: DNA intercalators. Mutat Res. 2007;623(1):14–23. https://doi.org/10.1016/j.mrfmmm.2007.03.014.
Vispé S, Vandenberghe I, Robin M, Annereau JP, Créancier L, Pique V, et al. Novel tetra-acridine derivatives as dual inhibitors of topoisomerase II and the human proteasome. Biochem Pharmacol. 2007;73(12):1863–72. https://doi.org/10.1016/j.bcp.2007.02.016.
Szymański P, Olszewska P, Mikiciuk-Olasik E, Różalski A, Maszewska A, Markiewicz Ł, et al. Novel tetrahydroacridine and cyclopentaquinoline derivatives with fluorobenzoic acid moiety induce cell cycle arrest and apoptosis in lung cancer cells by activation of DNA damage signaling. Tumour Biol. 2017;39(3):1–13. https://doi.org/10.1177/1010428317695011.
Oppegard LM, Ougolkov AV, Luchini DN, Schoon RA, Goodell JR, Kaur H, et al. Novel acridine-based compounds that exhibit an anti-pancreatic cancer activity are catalytic inhibitors of human topoisomerase II. Eur J Pharmacol. 2009;602(2):223–9. https://doi.org/10.1016/j.ejphar.2008.11.044.
Sondhi SM, Singh J, Rani R, Gupta PP, Agrawal SK, Saxena AK. Synthesis, anti-inflammatory and anticancer activity evaluation of some novel acridine derivatives. Eur J Med Chem. 2010;45(2):555–63. https://doi.org/10.1016/j.ejmech.2009.10.042.
de Almeida SMV, Lafayette EA, Gomes da Silva LPB, Amorim CAC, de Oliveira TB, Gois Ruiz ALT, et al. Synthesis, DNA binding, and antiproliferative activity of novel acridine-thiosemicarbazone derivatives. Int J Mol Sci. 2015;16(6):13023–42. https://doi.org/10.3390/ijms160613023.
Kožurková M, Sabolová D, Kristian P. A review on acridinylthioureas and its derivatives: biological and cytotoxic activity. J Appl Toxicol. 2017;37(10):1132–9. https://doi.org/10.1002/jat.3464.
Szymański P, Lázničková A, Lázniček M, Bajda M, Malawska B, Markowicz M, et al. 2,3-dihydro-1H-cyclopenta[b]quinoline derivatives as acetylcholinesterase inhibitors—synthesis, radiolabeling and biodistribution. Int J Mol Sci. 2012;13(8):10067–90. https://doi.org/10.3390/ijms130810067.
Czarnecka K, Szymański P, Girek M, Mikiciuk-Olasik E, Skibiński R, Kabziński J, et al. Tetrahydroacridine derivatives with fluorobenzoic acid moiety as multifunctional agents for Alzheimer’s disease treatment. Bioorg Chem. 2017;72(Supplement C):315–22. https://doi.org/10.1016/j.bioorg.2017.05.003.
Akbarzadeh A, Samiei M, Joo SW, Anzaby M, Hanifehpour Y, Nasrabadi HT, et al. Synthesis, characterization and in vitro studies of doxorubicin-loaded magnetic nanoparticles grafted to smart copolymers on A549 lung cancer cell line. J Nanobiotechnology. 2012;10:46. https://doi.org/10.1186/1477-3155-10-46.
Girek M, Kłosiński K, Grobelski B, Pizzimenti S, Cucci MA, Daga M, Barrera G, Pasieka Z, Czarnecka K, Szymański P. Novel tetrahydroacridine derivatives with iodobenzoic moieties induce G0/G1 cell cycle arrest and apoptosis in A549 non-small lung cancer and HT-29 colorectal cancer cells. Mol Cell Biochem. 2019;460:123–50. https://doi.org/10.1007/s11010-019-03576-x.
Michel P, Owczarek A, Matczak M, Kosno M, Szymański P, Mikiciuk-Olasik E, et al. Metabolite profiling of eastern teaberry (Gaultheria procumbens L.) lipophilic leaf extracts with hyaluronidase and lipoxygenase inhibitory activity. Molecules. 2017;22(3):412. https://doi.org/10.3390/molecules22030412.
Muller PAJ, Trinidad AG, Timpson P, Morton JP, Zanivan S, van den Berghe PVE, Nixon C, Karim SA, Caswell PT, Noll JE, Coffill CR, Lane DP, Sansom OJ, Neilsen PM, Norman JC, Vousden KH. Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion. Oncogene. 2013;32:1252–65. https://doi.org/10.1038/onc.2012.148.
de Santana Souza MT, Almeida JRGS, de Souza Araujo AA, Duarte MC, Gelain DP, Moreira JCF, et al. Structure-activity relationship of terpenes with anti-inflammatory profile—a systematic review. Basic Clin Pharmacol Toxicol. 2014;115(3):244–56. https://doi.org/10.1111/bcpt.12221.
Nishida Y, Sugahara S, Wada K, Toyohisa D, Tanaka T, Ono M, et al. Inhibitory effects of the ethyl acetate extract from bulbs of Scilla scilloides on lipoxygenase and hyaluronidase activities. Pharm Biol. 2014;52(10):1351–7. https://doi.org/10.3109/13880209.2014.891140.
Zeng H, Ma J, Yang R, Jing Y, Qu L. Molecular interactions of flavonoids to hyaluronidase: insights from spectroscopic and molecular modeling studies. J Fluoresc. 2015;25(4):941–59. https://doi.org/10.1007/s10895-015-1576-3.
Salvamani S, Gunasekaran B, Shukor MY, Shaharuddin NA, Sabullah MK, Ahmad SA. Anti-HMG-CoA reductase, antioxidant, and anti-inflammatory activities of Amaranthus viridis leaf extract as a potential treatment for hypercholesterolemia. Evid Based Complement Altern Med. 2016;10:8090841. https://doi.org/10.1155/2016/8090841.
Acknowledgements
Financial support by a grant from the Medical University of Lodz No. 502-02/3-015-01/502-34-091 and No. 502-03/1-153-02/502-14-325 is gratefully acknowledged. The in vitro and in vivo tests were carried out at the Animal House of the Faculty of Pharmacy, Medical University of Lodz.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential conflict of interest.
Ethical standards
The Animal tests were performed based on OECD 423 "Acute Oral Toxicity—Acute Toxic Class Method" and "3R rules", i.e., replacements, restrictions and refinement with the approval of the resolution of the Local Ethics Committee for Animal Experiments at the Medical University of Lodz No. 57/115 ŁB/2018.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kłosiński, K., Girek, M., Czarnecka, K. et al. Biological assessment of new tetrahydroacridine derivatives with fluorobenzoic moiety in vitro on A549 and HT-29 cell lines and in vivo on animal model. Human Cell 33, 859–867 (2020). https://doi.org/10.1007/s13577-020-00376-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-020-00376-0