Liver Growth Factor Induces Glia-Associated Neuroprotection in an In Vitro Model of Parkinson´s Disease

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. LGF Purification
2.2. Ethics Statement
2.3. Midbrain Primary Cultures of E14 Rat Embryos
2.3.1. Cultures Enriched in Neurons
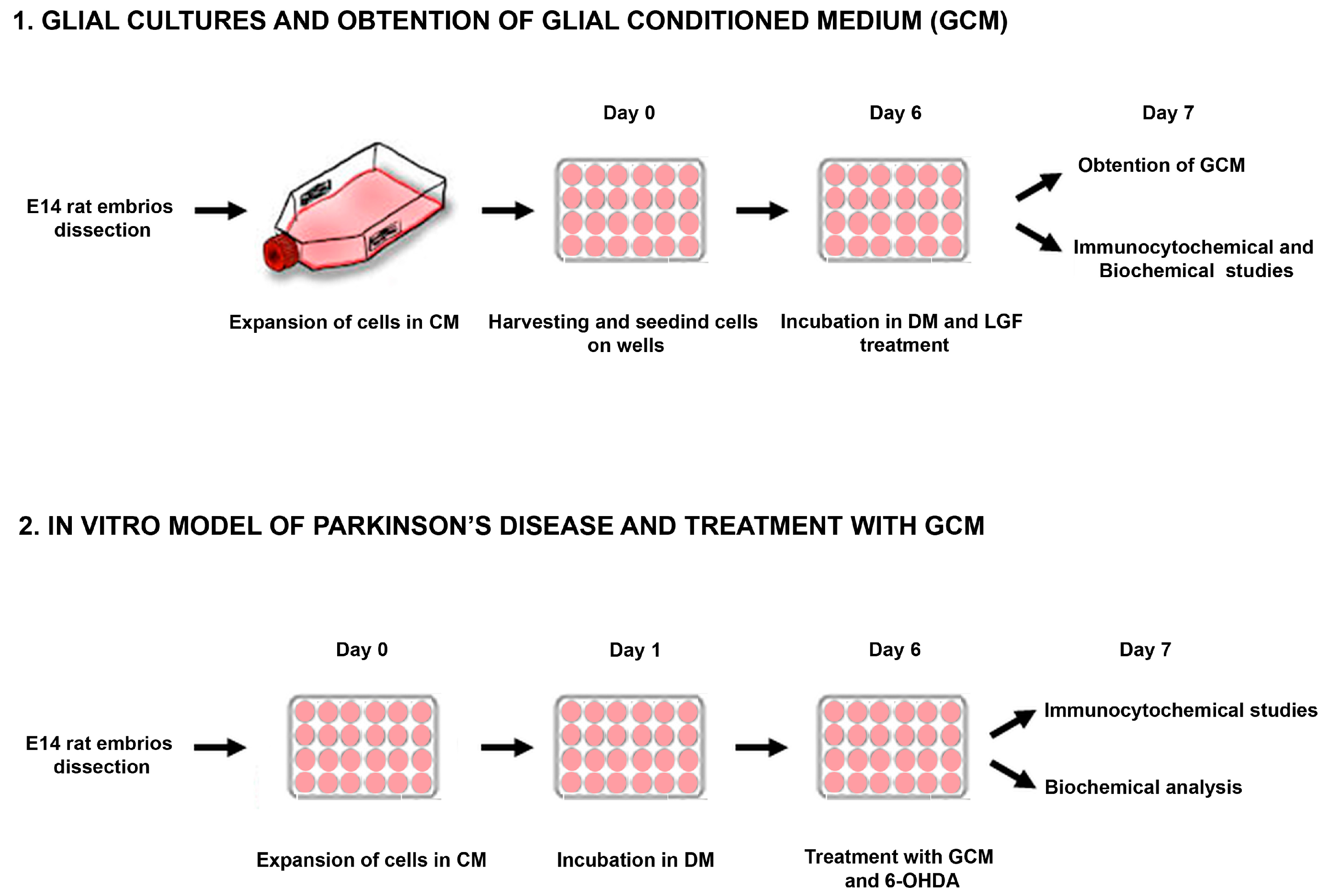
2.3.2. Glial Cultures
2.3.3. Treatments
2.4. Lactate Dehydrogenase Activity (LDH)
2.5. Immunocytochemical Characterization
2.5.1. Antibodies
2.5.2. Immunofluorescence
2.5.3. Immunoperoxidase
2.6. Biochemical Study
2.6.1. Collection and Homogenization of the Cells for Protein Extraction
2.6.2. Western Blot
2.7. Glutathione Determination
2.8. Statistical Analysis
3. Results
3.1. Characterization of Mesencephalic Glial Cultures
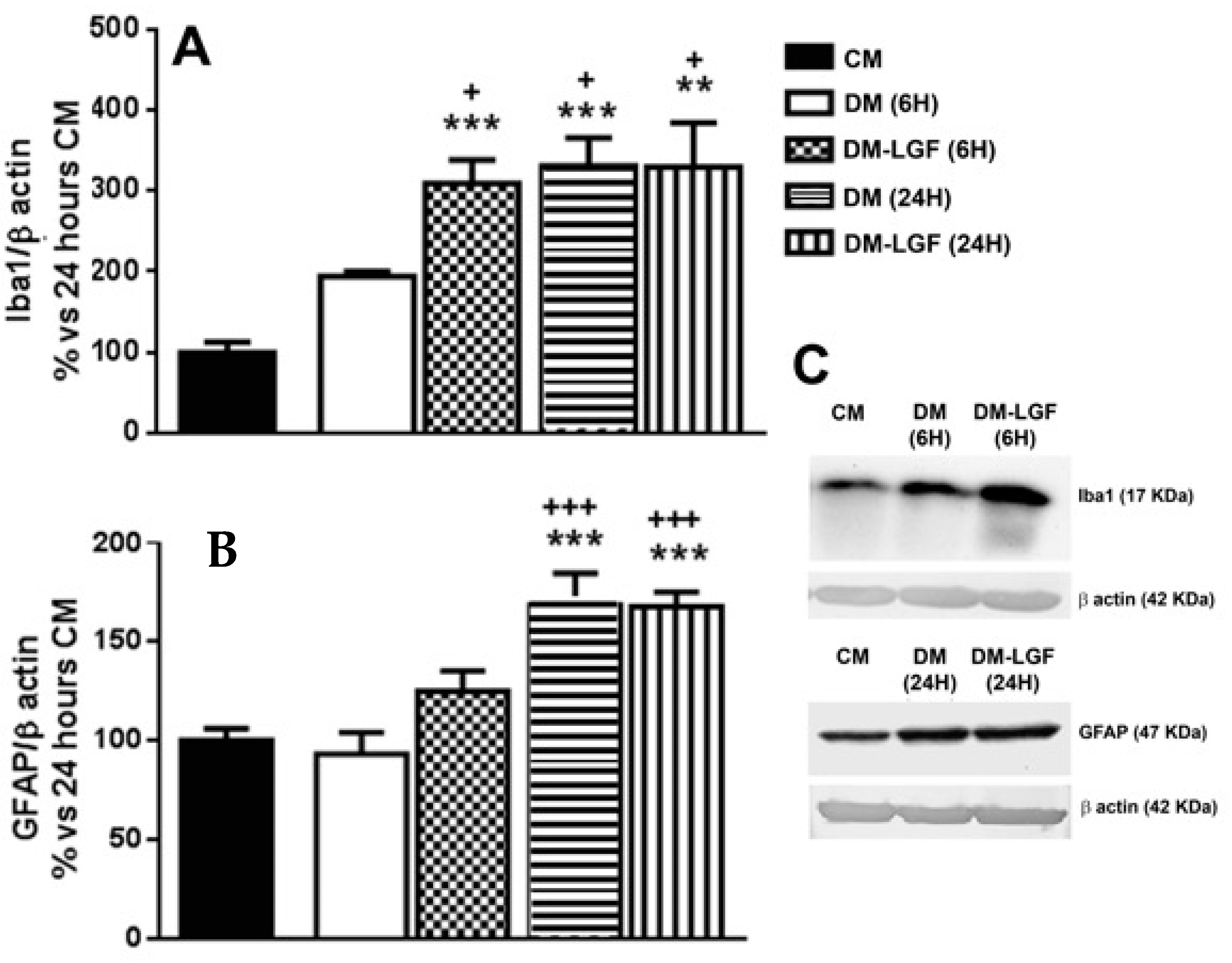
3.2. LGF Stimulates Microglia in Mesencephalic Glial Cultures
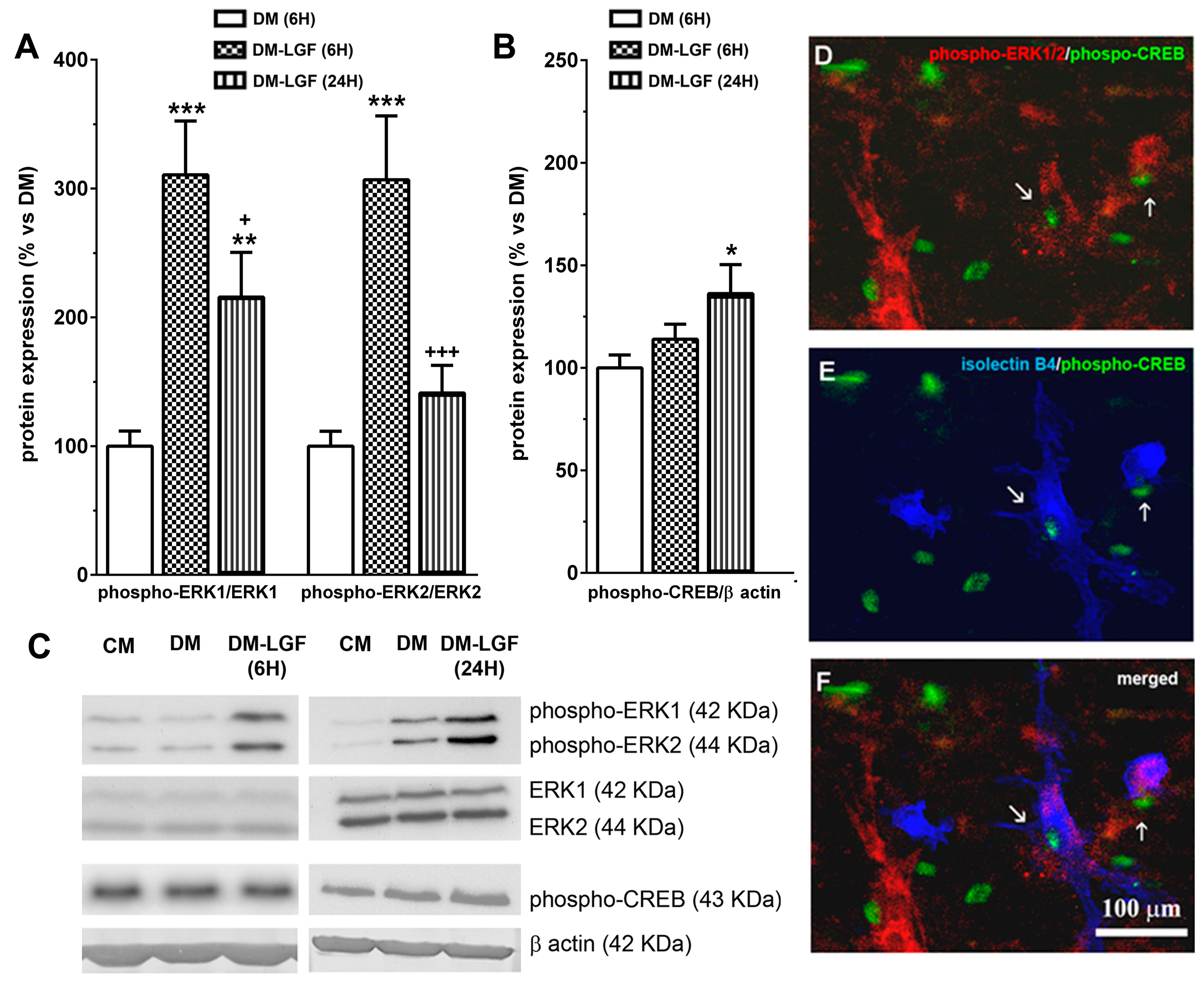
3.3. LGF Promotes the Activation of ERK1/2, CREB in Mesencephalic Glial Cultures
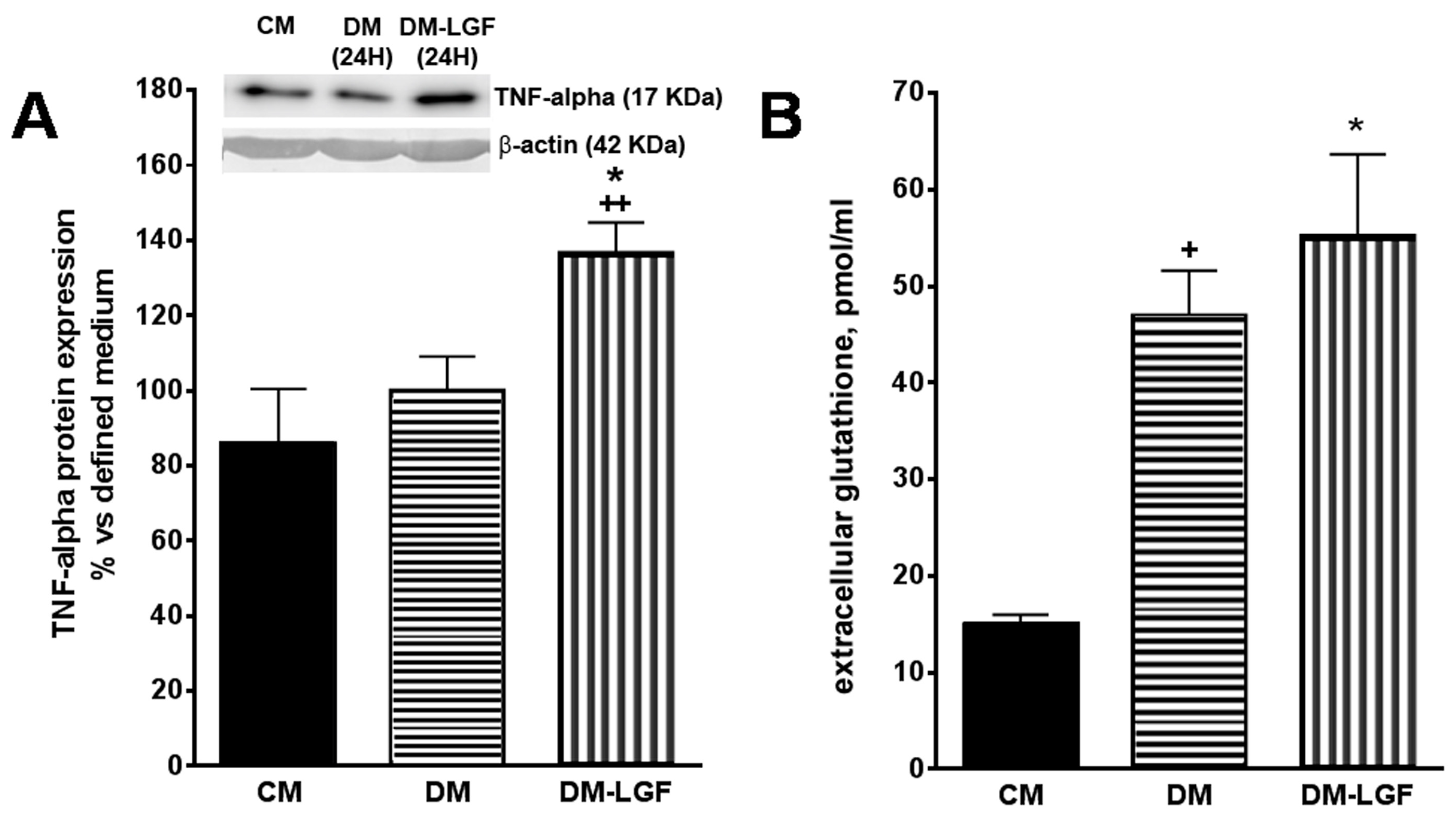
3.4. LGF Effects on TNF-Alpha Expression and Glutathione Concentration in Mesencephalic Glial Cultures
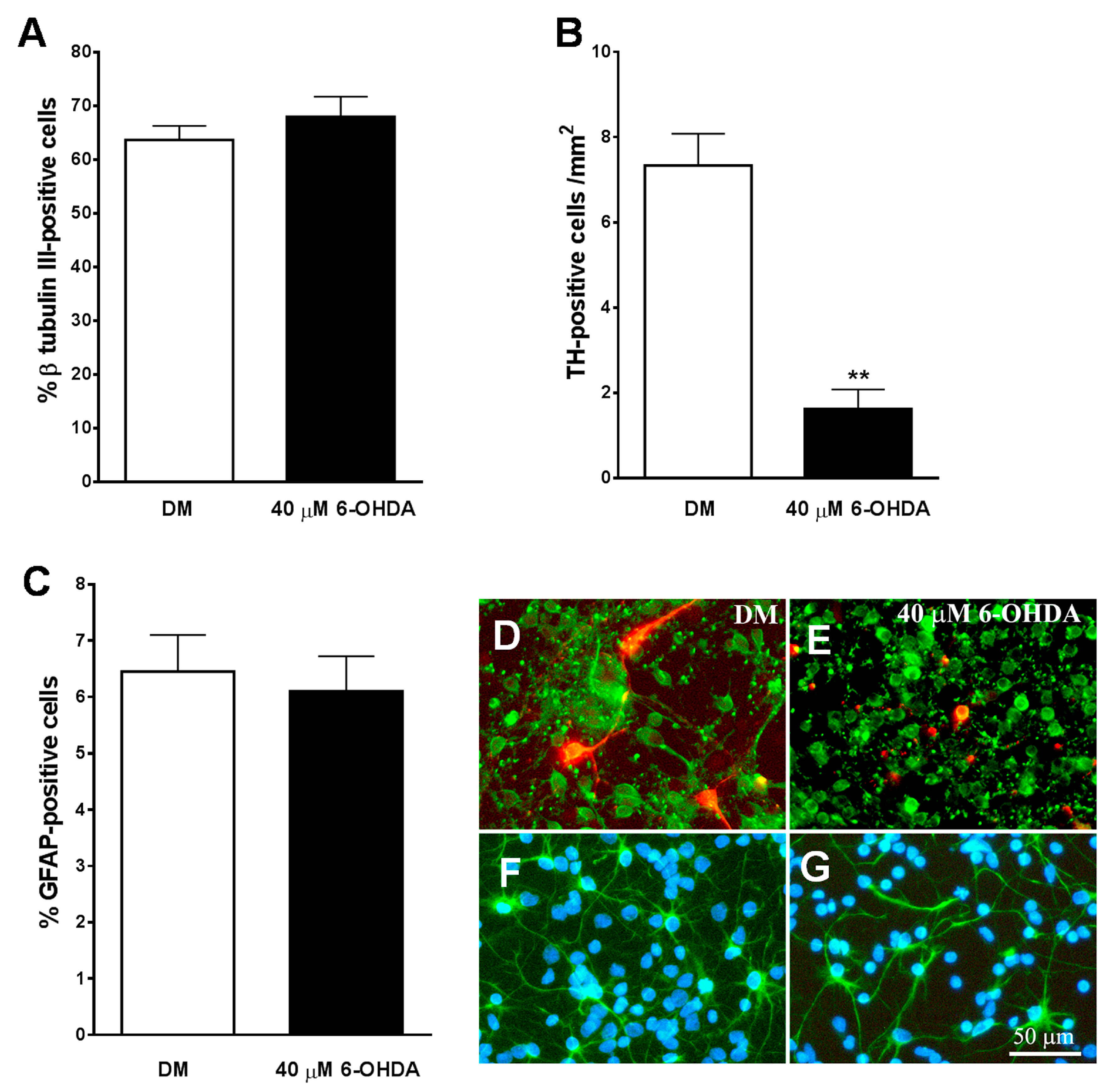
3.5. Development of an In Vitro Model of PD
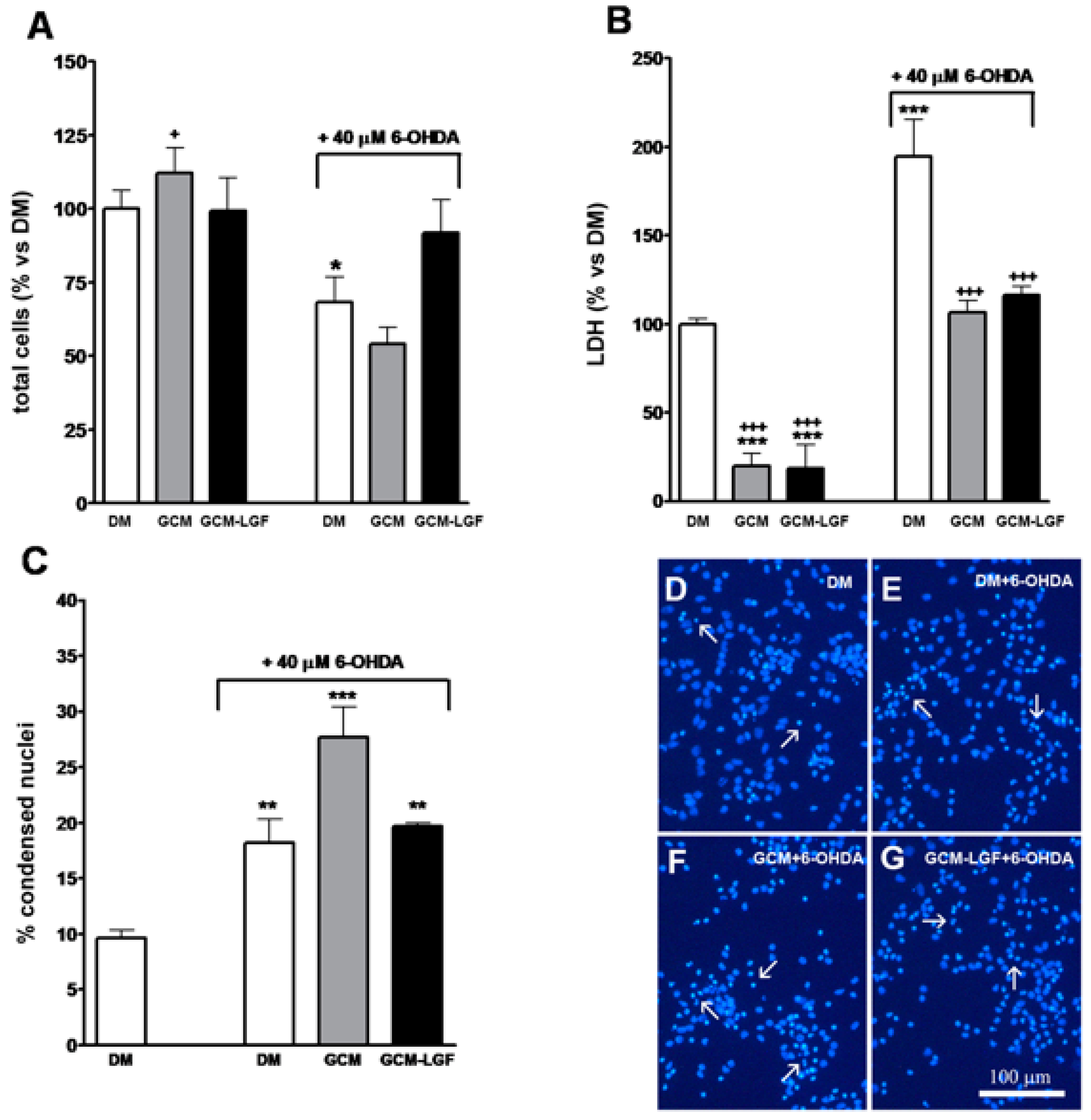
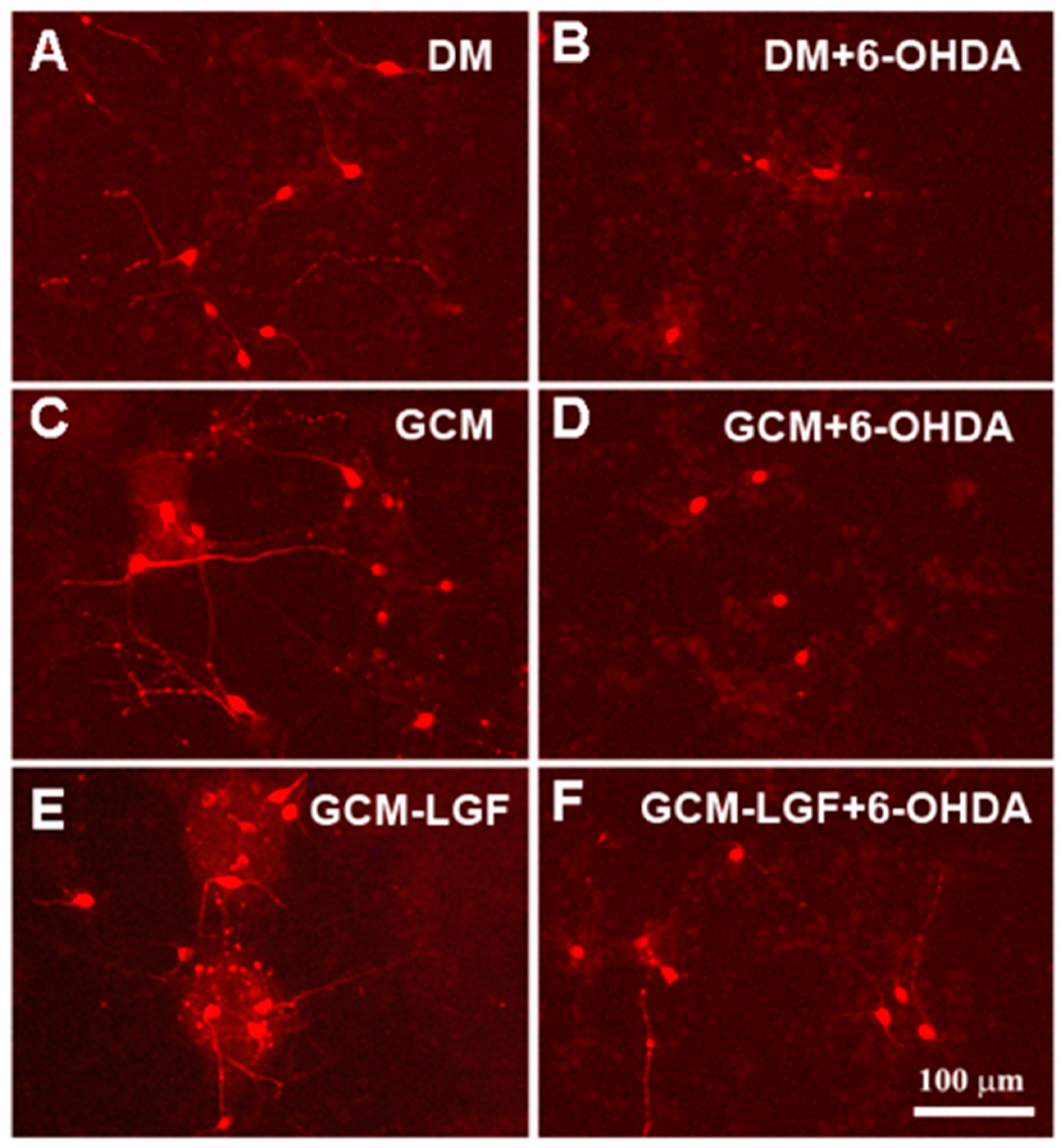
3.6. GCM Effects on Mesencephalic Neuronal Cultures Damaged by 6-OHDA
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Savitt, J.M.; Dawson, V.L.; Dawson, T.M. Diagnosis and treatment of Parkinson disease: Molecules to medicine. J. Clin. Investig. 2006, 116, 1744–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.C.; Ko, H.W.; Bok, E.; Park, E.S.; Huh, S.H.; Nam, J.H.; Jin, B.K. The role of neuroinflammation on the pathogenesis of Parkinson’s disease. BMB Rep. 2010, 43, 225–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hald, A.; Van Beek, J.; Lotharius, J. Inflammation in Parkinson’s disease: Causative or epiphenomenal? Subcell. Biochem. 2007, 42, 249–279. [Google Scholar] [PubMed]
- Schulz, J.B. Neuronal pathology in Parkinson’s disease. Cell Tissue Res. 2005, 320, 211. [Google Scholar] [CrossRef] [Green Version]
- Pezzoli, G.; Zini, M. Levodopa in Parkinson’s disease: From the past to the future. Expert Opin. Pharmacother. 2010, 11, 627–635. [Google Scholar] [CrossRef]
- Fernandez-Espejo, E. Pathogenesis of Parkinson’s disease: Prospects of neuroprotective and restorative therapies. Mol. Neurobiol. 2004, 29, 15–30. [Google Scholar] [CrossRef]
- Yasuda, T.; Mochizuki, H. Use of growth factors for the treatment of Parkinson’s disease. Expert Rev. Neurother. 2010, 10, 915–924. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Kordower, J.H. Are growth factors the answer? Parkinsonism Relat. Disord. 2009, 15 (Suppl. 3), S176–S180. [Google Scholar] [CrossRef]
- Díaz-Gil, J.J.; Escartin, P.; Garcia-Canero, R.; Trilla, C.; Veloso, J.J.; Sanchez, G.; Moreno-Caparros, A.; Enrique de Salamanca, C.; Lozano, R.; Gavilanes, J.G.; et al. Purification of a liver DNA-synthesis promoter from plasma of partially hepatectomized rats. Biochem. J. 1986, 235, 49–55. [Google Scholar] [CrossRef]
- Díaz-Gil, J.J.; Gavilanes, J.G.; Sanchez, G.; Garcia-Canero, R.; Garcia-Segura, J.M.; Santamaria, L.; Trilla, C.; Escartin, P. Identification of a liver growth factor as an albumin-bilirubin complex. Biochem. J. 1987, 243, 443–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Gil, J.J.; Sanchez, G.; Trilla, C.; Escartin, P. Identification of biliprotein as a liver growth factor. Hepatology 1988, 8, 484–486. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Gil, J.J.; Garcia-Monzon, C.; Rua, C.; Martin-Sanz, P.; Cereceda, R.M.; Miquilena-Colina, M.E.; Machin, C.; Fernandez-Martinez, A.; Garcia-Canero, R. The anti-fibrotic effect of liver growth factor is associated with decreased intrahepatic levels of matrix metalloproteinases 2 and 9 and transforming growth factor beta 1 in bile duct-ligated rats. Histol. Histopathol. 2008, 23, 583–591. [Google Scholar]
- Díaz-Gil, J.J.; Munoz, J.; Albillos, A.; Rua, C.; Machin, C.; Garcia-Canero, R.; Cereceda, R.M.; Guijarro, M.C.; Trilla, C.; Escartin, P. Improvement in liver fibrosis, functionality and hemodynamics in CCI4-cirrhotic rats after injection of the Liver Growth Factor. J. Hepatol. 1999, 30, 1065–1072. [Google Scholar] [CrossRef]
- Gonzalo-Gobernado, R.; Calatrava-Ferreras, L.; Perucho, J.; Reimers, D.; Casarejos, M.J.; Herranz, A.S.; Jimenez-Escrig, A.; Diaz-Gil, J.J.; Bazan, E. Liver growth factor as a tissue regenerating factor in neurodegenerative diseases. Recent Pat. CNS Drug Discov. 2014, 9, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Reimers, D.; Herranz, A.S.; Diaz-Gil, J.J.; Lobo, M.V.; Paino, C.L.; Alonso, R.; Asensio, M.J.; Gonzalo-Gobernado, R.; Bazan, E. Intrastriatal infusion of liver growth factor stimulates dopamine terminal sprouting and partially restores motor function in 6-hydroxydopamine-lesioned rats. J. Histochem. Cytochem. 2006, 54, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo-Gobernado, R.; Reimers, D.; Herranz, A.S.; Diaz-Gil, J.J.; Osuna, C.; Asensio, M.J.; Baena, S.; Rodriguez-Serrano, M.; Bazan, E. Mobilization of neural stem cells and generation of new neurons in 6-OHDA-lesioned rats by intracerebroventricular infusion of liver growth factor. J. Histochem. Cytochem. 2009, 57, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Reimers, D.; Osuna, C.; Gonzalo-Gobernado, R.; Herranz, A.S.; Diaz-Gil, J.J.; Jimenez-Escrig, A.; Asensio, M.J.; Miranda, C.; Rodriguez-Serrano, M.; Bazan, E. Liver growth factor promotes the survival of grafted neural stem cells in a rat model of Parkinson’s disease. Curr. Stem. Cell Res. Ther. 2012, 7, 15–25. [Google Scholar] [CrossRef]
- Gonzalo-Gobernado, R.; Calatrava-Ferreras, L.; Reimers, D.; Herranz, A.S.; Rodriguez-Serrano, M.; Miranda, C.; Jimenez-Escrig, A.; Diaz-Gil, J.J.; Bazan, E. Neuroprotective activity of peripherally administered liver growth factor in a rat model of Parkinson’s disease. PLoS ONE 2013, 8, e67771. [Google Scholar] [CrossRef] [Green Version]
- F, L.E.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Marchetti, B. Glia as a turning point in the therapeutic strategy of Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 349–372. [Google Scholar]
- Liberto, C.M.; Albrecht, P.J.; Herx, L.M.; Yong, V.W.; Levison, S.W. Pro-regenerative properties of cytokine-activated astrocytes. J. Neurochem. 2004, 89, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Glial reactions in Parkinson’s disease. Mov. Disord. 2008, 23, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P.; Schulz, J.B. Cellular pathology of Parkinson’s disease: Astrocytes, microglia and inflammation. Cell Tissue Res. 2004, 318, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, P.E.; Liberatore, G.T.; Wong, J.Y.; Porritt, M.J.; Frerichs, F.; Donnan, G.A.; Howells, D.W. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J. Neurosci. 1999, 19, 1708–1716. [Google Scholar] [CrossRef] [Green Version]
- Batchelor, P.E.; Porritt, M.J.; Martinello, P.; Parish, C.L.; Liberatore, G.T.; Donnan, G.A.; Howells, D.W. Macrophages and Microglia Produce Local Trophic Gradients That Stimulate Axonal Sprouting Toward but Not beyond the Wound Edge. Mol. Cell Neurosci. 2002, 21, 436–453. [Google Scholar] [CrossRef]
- Polazzi, E.; Contestabile, A. Reciprocal interactions between microglia and neurons: From survival to neuropathology. Rev. Neurosci. 2002, 13, 221–242. [Google Scholar] [CrossRef]
- Streit, W.J. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 2002, 40, 133–139. [Google Scholar] [CrossRef]
- Suzuki, T.; Hide, I.; Ido, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J. Neurosci. 2004, 24, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Remy, S.; Naveilhan, P.; Paille, V.; Brachet, P.; Neveu, I. Lipopolysaccharide and TNFalpha regulate the expression of GDNF, neurturin and their receptors. Neuroreport 2003, 14, 1529–1534. [Google Scholar] [CrossRef]
- Schulte-Herbruggen, O.; Nassenstein, C.; Lommatzsch, M.; Quarcoo, D.; Renz, H.; Braun, A. Tumor necrosis factor-alpha and interleukin-6 regulate secretion of brain-derived neurotrophic factor in human monocytes. J. Neuroimmunol. 2005, 160, 204–209. [Google Scholar] [CrossRef]
- Cho, S.G.; Yi, S.Y.; Yoo, Y.S. IFNgamma and TNFalpha synergistically induce neurite outgrowth on PC12 cells. Neurosci. Lett. 2005, 378, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Wang, C.X.; Song, D.K.; Wang, P.; Shuaib, A.; Hao, C. Interferon gamma induces neurite outgrowth by up-regulation of p35 neuron-specific cyclin-dependent kinase 5 activator via activation of ERK1/2 pathway. J. Biol. Chem. 2005, 280, 12896–12901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, G.; Goldshmit, Y.; Turnley, A.M. Interferon-gamma but not TNF alpha promotes neuronal differentiation and neurite outgrowth of murine adult neural stem cells. Exp. Neurol. 2004, 187, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, V.; Mohand-Said, S.; Hanoteau, N.; Fuchs, C.; Pfizenmaier, K.; Eisel, U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: Opposite roles of TNF receptor 1 and TNF receptor 2. J. Neurosci. 2002, 22, RC216. [Google Scholar] [CrossRef] [PubMed]
- Kuno, R.; Yoshida, Y.; Nitta, A.; Nabeshima, T.; Wang, J.; Sonobe, Y.; Kawanokuchi, J.; Takeuchi, H.; Mizuno, T.; Suzumura, A. The role of TNF-alpha and its receptors in the production of NGF and GDNF by astrocytes. Brain Res. 2006, 1116, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Gil, J.J.; Majano, P.L.; Lopez-Cabrera, M.; Sanchez-Lopez, V.; Rua, C.; Machin, C.; Trilla, C.; Garcia-Canero, R.; Moreno-Otero, R. The mitogenic activity of the liver growth factor is mediated by tumor necrosis factor alpha in rat liver. J. Hepatol. 2003, 38, 598–604. [Google Scholar] [CrossRef]
- Singh, J.; Bowers, L.D. Quantitative fractionation of serum bilirubin species by reversed-phase high-performance liquid chromatography. J. Chromatogr. 1986, 380, 321–330. [Google Scholar] [CrossRef]
- Diaz Gil, J.J.; Rua, C.; Machin, C.; Cereceda, R.M.; Garcia-Canero, R.; de Foronda, M.; Perez de Diego, J.; Trilla, C.; Escartin, P. Hepatic growth induced by injection of the liver growth factor into normal rats. Growth Regul. 1994, 4, 113–122. [Google Scholar]
- O’Malley, E.K.; Black, I.B.; Dreyfus, C.F. Local support cells promote survival of substantia nigra dopaminergic neurons in culture. Exp. Neurol. 1991, 112, 40–48. [Google Scholar] [CrossRef]
- Mena, M.A.; Pardo, B.; Paino, C.L.; De Yebenes, J.G. Levodopa toxicity in foetal rat midbrain neurones in culture: Modulation by ascorbic acid. Neuroreport 1993, 4, 438–440. [Google Scholar] [CrossRef]
- Mena, M.A.; Casarejos, M.J.; Carazo, A.; Paino, C.L.; Garcia de Yebenes, J. Glia conditioned medium protects fetal rat midbrain neurones in culture from L-DOPA toxicity. Neuroreport 1996, 7, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Ungerstedt, U. Stereotaxic mapping of the monoamine pathways in the rat brain. Acta Physiol. Scand. Suppl. 1971, 367, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Mena, M.A.; Casarejos, M.J.; Garcia de Yebenes, J. The effect of glia-conditioned medium on dopamine neurons in culture. Modulation of apoptosis, tyrosine hydroxylase expression and 1-methyl-4-phenylpyridinium toxicity. J. Neural Transm. (Vienna) 1999, 106, 1105–1123. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martin, E.; Canals, S.; Casarejos, M.J.; de Bernardo, S.; Handler, A.; Mena, M.A. L-DOPA and glia-conditioned medium have additive effects on tyrosine hydroxylase expression in human catecholamine-rich neuroblastoma NB69 cells. J. Neurochem. 2001, 78, 535–545. [Google Scholar] [CrossRef]
- Rodriguez-Martin, E.; Casarejos, M.J.; Bazan, E.; Canals, S.; Herranz, A.S.; Mena, M.A. Nitric oxide induces differentiation in the NB69 human catecholamine-rich cell line. Neuropharmacology 2000, 39, 2090–2100. [Google Scholar] [CrossRef]
- Susin, L. Capacidad antioxidante del Liver Growth factor: Estudios in Vitro e in Vivo en Ratas Espontáneamente Hipertensas. Ph.D. Thesis, Department of Physiology, School of Medicine, Universidad Autónoma de Madrid, Madrid, Spain, 2008. [Google Scholar]
- Polazzi, E.; Altamira, L.E.; Eleuteri, S.; Barbaro, R.; Casadio, C.; Contestabile, A.; Monti, B. Neuroprotection of microglial conditioned medium on 6-hydroxydopamine-induced neuronal death: Role of transforming growth factor beta-2. J. Neurochem. 2009, 110, 545–556. [Google Scholar] [CrossRef]
- Ralay Ranaivo, H.; Wainwright, M.S. Albumin activates astrocytes and microglia through mitogen-activated protein kinase pathways. Brain Res. 2010, 1313, 222–231. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.T.; Chang, Y.C.; Tu, Y.F.; Huang, C.C. CREB activation mediates VEGF-A’s protection of neurons and cerebral vascular endothelial cells. J. Neurochem. 2010, 113, 79–91. [Google Scholar] [CrossRef]
- Mastroeni, D.; Grover, A.; Leonard, B.; Joyce, J.N.; Coleman, P.D.; Kozik, B.; Bellinger, D.L.; Rogers, J. Microglial responses to dopamine in a cell culture model of Parkinson’s disease. Neurobiol. Aging 2009, 30, 1805–1817. [Google Scholar] [CrossRef] [Green Version]
- Saha, R.N.; Liu, X.; Pahan, K. Up-regulation of BDNF in astrocytes by TNF-alpha: A case for the neuroprotective role of cytokine. J. Neuroimmune Pharmacol. 2006, 1, 212–222. [Google Scholar] [CrossRef] [Green Version]
- Lai, A.Y.; Todd, K.G. Differential regulation of trophic and proinflammatory microglial effectors is dependent on severity of neuronal injury. Glia 2008, 56, 259–270. [Google Scholar] [CrossRef]
- Liang, J.; Takeuchi, H.; Jin, S.; Noda, M.; Li, H.; Doi, Y.; Kawanokuchi, J.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Glutamate induces neurotrophic factor production from microglia via protein kinase C pathway. Brain Res. 2010, 1322, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Shein, N.A.; Grigoriadis, N.; Horowitz, M.; Umschwief, G.; Alexandrovich, A.G.; Simeonidou, C.; Grigoriadis, S.; Touloumi, O.; Shohami, E. Microglial involvement in neuroprotection following experimental traumatic brain injury in heat-acclimated mice. Brain Res. 2008, 1244, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.D.; McPherson, C.A.; Harry, G.J. Heterogeneity of microglia and TNF signaling as determinants for neuronal death or survival. Neurotoxicology 2009, 30, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Taoufik, E.; Petit, E.; Divoux, D.; Tseveleki, V.; Mengozzi, M.; Roberts, M.L.; Valable, S.; Ghezzi, P.; Quackenbush, J.; Brines, M.; et al. TNF receptor I sensitizes neurons to erythropoietin- and VEGF-mediated neuroprotection after ischemic and excitotoxic injury. Proc. Natl. Acad. Sci. USA 2008, 105, 6185–6190. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, K.R.; Boato, F.; Diestel, A.; Hechler, D.; Kruglov, A.; Berger, F.; Hendrix, S. Hypothermia-induced neurite outgrowth is mediated by tumor necrosis factor-alpha. Brain Pathol. 2010, 20, 771–779. [Google Scholar] [CrossRef]
- Lian, H.; Zheng, H. Signaling pathways regulating neuron-glia interaction and their implications in Alzheimer’s disease. J. Neurochem. 2016, 136, 475–491. [Google Scholar] [CrossRef]
- Sagare, A.P.; Winkler, E.A.; Bell, R.D.; Deane, R.; Zlokovic, B.V. From the liver to the blood-brain barrier: An interconnected system regulating brain amyloid-beta levels. J. Neurosci. Res. 2011, 89, 967–968. [Google Scholar] [CrossRef]
- Thornalley, P.J. Cell activation by glycated proteins. AGE receptors, receptor recognition factors and functional classification of AGEs. Cell Mol. Biol. (Noisy-le-grand) 1998, 44, 1013–1023. [Google Scholar]
- Park, I.H.; Yeon, S.I.; Youn, J.H.; Choi, J.E.; Sasaki, N.; Choi, I.H.; Shin, J.S. Expression of a novel secreted splice variant of the receptor for advanced glycation end products (RAGE) in human brain astrocytes and peripheral blood mononuclear cells. Mol. Immunol. 2004, 40, 1203–1211. [Google Scholar] [CrossRef]
- Wang, Z.; Li, D.D.; Liang, Y.Y.; Wang, D.S.; Cai, N.S. Activation of astrocytes by advanced glycation end products: Cytokines induction and nitric oxide release. Acta Pharmacol. Sin. 2002, 23, 974–980. [Google Scholar] [PubMed]
- Martin, H.L.; Teismann, P. Glutathione—A review on its role and significance in Parkinson’s disease. FASEB J. 2009, 23, 3263–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, J.K.; Gardaneh, M.; Iwasiow, R.; Lanthier, P.; Gangaraju, S.; Ribecco-Lutkiewicz, M.; Tremblay, R.; Kiuchi, K.; Sikorska, M. Astrocyte-secreted GDNF and glutathione antioxidant system protect neurons against 6OHDA cytotoxicity. Neurobiol. Dis. 2009, 33, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gil, J.J.; Gavilanes, J.G.; Garcia-Canero, R.; Garcia-Segura, J.M.; Santamaria, L.; Trilla, C.; Martin, A.M.; Guerra, M.A.; Garcia-Escandon, F.; Iniguez, J.A.; et al. Liver growth factor purified from human plasma is an albumin-bilirubin complex. Mol. Biol. Med. 1989, 6, 197–207. [Google Scholar]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalo-Gobernado, R.; Reimers, D.; Casarejos, M.J.; Calatrava Ferreras, L.; Vallejo-Muñoz, M.; Jiménez-Escrig, A.; Diaz-Gil, J.J.; Ulzurrun de Asanza, G.M.; Bazán, E. Liver Growth Factor Induces Glia-Associated Neuroprotection in an In Vitro Model of Parkinson´s Disease. Brain Sci. 2020, 10, 315. https://doi.org/10.3390/brainsci10050315
Gonzalo-Gobernado R, Reimers D, Casarejos MJ, Calatrava Ferreras L, Vallejo-Muñoz M, Jiménez-Escrig A, Diaz-Gil JJ, Ulzurrun de Asanza GM, Bazán E. Liver Growth Factor Induces Glia-Associated Neuroprotection in an In Vitro Model of Parkinson´s Disease. Brain Sciences. 2020; 10(5):315. https://doi.org/10.3390/brainsci10050315
Chicago/Turabian StyleGonzalo-Gobernado, Rafael, Diana Reimers, María José Casarejos, Lucía Calatrava Ferreras, Manuela Vallejo-Muñoz, Adriano Jiménez-Escrig, Juan José Diaz-Gil, Gonzalo M. Ulzurrun de Asanza, and Eulalia Bazán. 2020. "Liver Growth Factor Induces Glia-Associated Neuroprotection in an In Vitro Model of Parkinson´s Disease" Brain Sciences 10, no. 5: 315. https://doi.org/10.3390/brainsci10050315