
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infects host cells following binding with the cell surface ACE2 receptors, thereby leading to coronavirus disease 2019 (COVID-19). SARS-CoV-2 causes viral pneumonia with additional extrapulmonary manifestations and major complications, including acute myocardial injury, arrhythmia, and shock mainly in elderly patients. Furthermore, patients with existing cardiovascular comorbidities, such as hypertension and coronary heart disease, have a worse clinical outcome following contraction of the viral illness. A striking feature of COVID-19 pandemics is the high incidence of fatalities in advanced aged patients: this might be due to the prevalence of frailty and cardiovascular disease increase with age due to endothelial dysfunction and loss of endogenous cardioprotective mechanisms. Although experimental evidence on this topic is still at its infancy, the aim of this position paper is to hypothesize and discuss more suggestive cellular and molecular mechanisms whereby SARS-CoV-2 may lead to detrimental consequences to the cardiovascular system. We will focus on aging, cytokine storm, NLRP3/inflammasome, hypoxemia, and air pollution, which is an emerging cardiovascular risk factor associated with rapid urbanization and globalization. We will finally discuss the impact of clinically available CV drugs on the clinical course of COVID-19 patients. Understanding the role played by SARS-CoV2 on the CV system is indeed mandatory to get further insights into COVID-19 pathogenesis and to design a therapeutic strategy of cardio-protection for frail patients.
Similar content being viewed by others


Introduction
The novel coronavirus (CoV) that emerged in Wuhan, Hubei province, China, in December 2019, and known as severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2), rapidly spread throughout all Chinese provinces and, subsequently, propagated across the globe, with more than 700,000 cases and ≈ 30,000 deaths in 202 countries (https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/). The World Health Organization (WHO) has therefore declared coronavirus disease 2019 (COVID-19) as global pandemic on March 12, 2020. Europe is now the world’s COVID-19 epicenter, with Italy being the country with the highest number of reported cases and deaths. As of March 29, the Italian Institute of Health (ISS) reported 89,967 cases and 9200 deaths in Italy (https://www.epicentro.iss.it/coronavirus/), which overtook China as the country with most coronavirus-related casualties. The pandemic is now propagating across USA, where over 100,000 cases and 2000 deaths were reported as of March 29, 2020. Currently, SARS-CoV-2 presents an estimated R0 ranging between 2 and 3, which means that any infected individual may transmit the disease to other 2–3 people in a susceptible population (Madjid et al. 2020), such as older adults (D’Adamo et al. 2020) and those suffering from multiple comorbidities.
SARS-CoV-2, an enveloped, positive-strand RNA virus that belongs to the β-coronavirus (β-CoV) family, presents > 80% nucleotide identity with the SARS-like CoVZXC21 identified in bats and with human SARS-CoV, while nucleotide identity with MERS-CoV is reduced to ≈ 50% (Chan et al. 2020; Wan et al. 2020; Lu et al. 2020). SARS-CoV-2 mainly targets the respiratory tract of mammals, including humans, thereby inducing a wide spectrum of clinical features ranging from asymptomatic or paucisymptomatic forms to interstitial pneumonia or, upon progressive alveolar damage, severe acute respiratory failure, which may require advanced respiratory support and intensive care of two or more organ systems. In fact, COVID-19 may progress towards adult respiratory distress syndrome (ARDS) and/or multiple organ acute failure (e.g., kidney, liver, and heart), and even death (Huang et al. 2020; Zhou et al. 2020b; Wu et al. 2020a; Wang et al. 2020d). Although COVID-19 is a respiratory disease, one third of patients with more severe COVID-19 had conjunctivitis (Wu et al. 2020b) and some patients showed gastrointestinal symptoms (nausea, vomiting, or diarrhea) (Jin et al. 2020). Interestingly, epithelial cells of several body compartments, including oral (Xu et al. 2020a) and nasal mucosa (Hamming et al. 2004), respiratory tree and alveoli (Hamming et al. 2004), intestinal mucosa (Hamming et al. 2004) as well as human aqueous humor (Holappa et al. 2015), show high levels of a metallopeptidase named angiotensin-converting enzyme 2 (ACE2), which is the functional receptor for SARS-CoV2 (Lan et al. 2020). ACE2 is also expressed in coronary and intrarenal endothelial cells and in renal tubular epithelial cells (Donoghue et al. 2000). Since ACE2 catalyzes the cleavage of angiotensin II, a vasoconstrictor, into angiotensin (1–7) (Ang 1–7), a vasodilator, its extra-respiratory localization has supported hypotheses on the relationship of SARS-CoV-2 infection with the cardiovascular (CV) system. Indeed, adult patients affected by CVDs, arterial hypertension, diabetes, and/or chronic obstructive pulmonary disease (COPD) show worse clinical outcome following contraction of the viral illness with fatality rate attaining 10.5% (The Novel Coronavirus Pneumonia Emergency Response Epidemiology team and Novel Coronavirus Pneumonia Emergency Response Epidemiology Team 2020). In patients with COVID-19 pneumonia who developed ARDS, acute cardiac injury, shock, and arrhythmia represent the most common life-threatening complications along with acute kidney injury (Chen et al. 2020b; Huang et al. 2020; Wang et al. 2020a).
It has long been known that acute respiratory infections, including flu and pneumonias, may trigger severe CV disease (CVD) events and vice versa (Madjid et al. 2020). Although experimental evidence on this topic is still at its infancy, we will hypothesize and discuss more suggestive cellular and molecular mechanisms whereby SARS-CoV-2 may lead to detrimental consequences to the CV system. We will focus on aging, cytokine storm, NLRP3/inflammasome, hypoxemia, and air pollution, which is an emerging CV risk factor associated with rapid urbanization and globalization. We will finally discuss the impact of clinically available CV drugs on the clinical course of COVID-19 patients. Understanding the role played by SARS-CoV2 on the CV system is indeed mandatory to get further insights into COVID-19 pathogenesis and to design a therapeutic strategy of cardio-protection for frail patients (Zheng et al. 2020).
SARS-CoV-2 and the cardiovascular system: a suggestive relationship
First reports describing the clinical features of patients affected by COVID-19 hinted at a relationship between SARS-CoV-2-mediated infection and the CV system mainly due to the high inflammatory burden affecting myocardium and vessels (Table 1) (Zheng et al. 2020; Madjid et al. 2020). Although it is a suggestive causal relationship, more translational research should be carefully performed in order to avoid confounding factors and hasty conclusions. In order to support it, we will mention the clinical evidences as they emerged.
To best of our knowledge, the prevalence and risk of death of severe COVID-19 is higher in elderly patients with chronic comorbidities, such as arterial hypertension, type 2 diabetes, cardiac and cerebrovascular disorders, and COPD (Wang et al. 2020b). Of note, diagnosis of major cardiac complications (i.e., acute myocardial injury and lethal arrhythmias) recently emerged from analysis of initial representative populations of COVID-19 patients. The first report analyzed a cohort of 41 patients (median age = 49 years; 73% men), the majority of whom (n = 27, 66%) were exposed to Huanan seafood and live-animal market (Huang et al. 2020), the original epicenter of COVID-19 outbreak. Underlying comorbidities were reported in 32% of the patients, including diabetes (n = 8, 20%), CVD (n = 6, 15%), and hypertension (n = 6, 15%) (Huang et al. 2020). Moreover, increased blood levels of high-sensitivity cardiac troponin I (cTnI) were reported in 5 patients (12%) (Huang et al. 2020). This initial finding cautiously suggests the onset of acute cardiac ischemic injury in COVID-19 patients, yet measurements of cTnI levels should be always associated to electrocardiogram (ECG) or imaging findings of myocardial ischemia (Giannitsis and Katus 2013) to make a diagnosis. Intriguingly, rising levels of cTnI are also independent predictors of mortality in critically ill patients hospitalized with severe pneumonia without evidence of acute coronary syndrome (Lee et al. 2015). In this regard, we cannot exclude the onset of transient myopericarditis mimicking acute myocardial infarction in severe COVID-19 patients due to cytokine storm (Inciardi et al. 2020) as previously observed in severe ARDS patients (To et al. 2010). In fact, an increased serum level of pro-inflammatory cytokines, for instance interleukin-1β (IL-1β), interferon γ (IFNγ) and MCP1, was reported in both ICU-patients and non-ICU patients. This observation is consistent with the development of a cytokine storm syndrome, as previously reported in SARS (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) infections (De Wit et al. 2016).
A second report analyzed a cohort of 99 patients (median age = 55.5; 67% men), the half of which (n = 49, 49%) was also exposed to Huanan seafood market (Chen et al. Lancet 2020b). A large percentage of these subjects (n = 40, 40%, each) suffered of CVD and cerebrovascular disorders, which represent the most common chronic comorbidities in this cohort (Chen et al. 2020b). Cardiac injury was diagnosed by measuring changes in myocardial zymogram, which evaluates the activity of metalloproteinases. Myocardial zymogram results, however, are at risk of being over-interpreted since the assay is a TIMP (tissue inhibitor of metalloproteinases) free environment (Lindsey 2018). Since it is not considered a gold standard assay to diagnose acute cardiac injury in hospital setting, other established cardiac specific biomarkers should be tested. In fact, the authors have also reported high circulating levels of creatine kinase and lactate dehydrogenase in 13 (13%) and 75 (76%) patients, respectively (Chen et al. 2020b), which are biomarkers generally used to perform early diagnosis of acute myocardial infarction. Yet, since these enzymes are also present in other tissues than the myocardium, their diagnostic specificity is limited, and their diagnostic sensitivity also suffers from the presence of a sizeable baseline enzyme concentration in the circulation without any cardiac pathology (Bodor 2016). False-positive results, indeed, may occur in renal failure, muscle fatigue, vitamin D deficiency, pneumonia, asthma, malignancies, pulmonary embolism, hypoxia, and smokers. In particular, 2 of the dead patients presented a long history of tobacco smoke exposure (Chen et al. 2020b), which enhances the risk of respiratory and cardiovascular disease as well.
A third report analyzed a cohort of 138 patients (median age = 56; 54.3% men) with hypertension (n = 43, 31.2%), CVD (n = 20, 14.5%), diabetes (n = 14, 10.1%), and cancer (n = 10, 7.2%) as more common coexisting underlying comorbidities (Wang et al. 2020a), while a small fraction of subjects (n = 7, 5.1%) was suffering from cerebrovascular disease (Wang et al. 2020a). Of note, the admission rate to ICU (n = 36, 26.1%) was remarkably increased among patients of the same age displaying hypertension (n = 21, 58.3%), CVD (n = 9, 25%) and cerebrovascular disease (n = 6, 16.7%) as underlying comorbidities (Wang et al. 2020a). Finally, arrhythmias (n = 23, 16.7%) and acute cardiac injury (n = 10, 7.2%), defined as increased blood levels of hypersensitive cTnI, were the most common complications arising in ICU patients affected by ARDS (n = 27, 19.6%) and acute kidney injury (n = 5, 3.6%) (Wang et al. 2020a).
A fourth single-center study analyzed the epidemiological and clinical features of 140 hospitalized COVID-19 patients (median age = 57; 50.7% men), none of whom was exposed to Huanan market. Ninety patients (64.3%) suffered from at least 1 underlying comorbidity, the most common being hypertension (n = 42; 30%) followed by diabetes (n = 17; 12.1%). Of note, a lower fraction of severe COVID-19 patients suffered from preexisting arrhythmia (n = 5; 3.6%) and coronary heart disease (n = 7; 5%) (Zhang et al. 2020).
Finally, a single-center large study conducted on 1099 patients affected by COVID-19 from 552 hospitals in 30 Chinese provinces (median age = 47; 58.1% men) confirmed that 23.7% subjects had pre-existing comorbidities (Guan et al. 2020), such as hypertension (n = 165, 15%), diabetes (n = 81, 7.4%), coronary heart disease (n = 27, 2.5%), and cerebrovascular disease (n = 15, 1.4%) (Guan et al. 2020). In this study, acute cardiac injury was defined by the increase in circulating levels of creatine kinase and lactate dehydrogenase in 13.7% and 41.0% of the patients, respectively (Guan et al. 2020). No additional features of acute myocardial infarction were considered.
Among different clinical reports, a retrospective, multicenter cohort study, including 191 adult COVID-19 patients (median age = 56; 62% men), confirmed preexisting comorbidities in almost half of the individuals (n = 91; 48%), such as hypertension (n = 58; 30%), diabetes (n = 36; 19%), and coronary heart disease (n = 15; 8%) (Zhou et al. 2020a). Likewise, non-survivors displayed a higher rate of comorbidities as compared to survivors, including hypertension (48% vs. 23%), diabetes (31% vs. 14%), and coronary heart disease (24% vs. 1%) (Zhou et al. 2020a). In addition to sepsis (n = 112; 59%), respiratory failure (n = 103; 54%), ARDS (n = 59; 31%), and acute kidney injury (n = 28; 15%), the most common secondary outcomes in this cohort of patients were the onset of acute heart failure (n = 44; 23%) and acute cardiac injury (n = 33; 17%) (Zhou et al. 2020a). Of note, non-survivors presented higher rates of acute heart failure (52% vs. 12%) and acute cardiac injury (59% vs. 1%) as compared to survivors. Moreover, laboratory findings showed high levels of nonspecific biomarkers as lactate dehydrogenase, creatine kinase, and hypersensitive cTnI, which has an amino acid sequence similar to skeletal troponin C (Xu et al. 2013), and pro-inflammatory biomarkers, e.g., interleukin-6 (IL-6) and serum ferritin (Zhou et al. 2020a). Again, since measurement of circulating biomarkers is not enough to diagnose acute cardiac injury caused by SARS-CoV2 with certainty, further clinical investigations based on multimodal approach should be encouraged. Similarly, a retrospective multicenter study, conducted on a smaller cohort (n = 150) of COVID-19 patients, confirmed that risk of death was greater in the presence of CVD. Of note, isolated myocardial injury resulted to be one of the main causes of death in some patients (7%) in the presence of high levels of IL-6 and C-reactive protein.
The relationship between SARS-CoV-2 infection and CVD has been further corroborated by a recent descriptive analysis of all the 44,672 COVID-19 cases diagnosed in China as of February 11, 2020 (The Novel Coronavirus Pneumonia Emergency Response Epidemiology team and Novel Coronavirus Pneumonia Emergency Response Epidemiology Team 2020). This study confirmed that a fraction of patients was affected by CVD (14.2%) including arterial hypertension (12.8%). Although retrospective studies cannot be helpful to made a causal association, it further confirmed that the mortality rate was increased by CVD (+ 10.5%), diabetes (+ 7.3%), chronic respiratory disease (+ 6.3%), and arterial hypertension (+ 6%) (The Novel Coronavirus Pneumonia Emergency Response Epidemiology team and Novel Coronavirus Pneumonia Emergency Response Epidemiology Team 2020; Madjid et al. 2020).
Do these CVDs really make a difference in the worse prognosis of COVID-19 patients? Although Italy is the European country with the highest percentage of the population aged 80 or over and with the mortality rate due to COVID-19 among the highest in the world (Madjid et al. 2020), it is still unknown whether greater vulnerability to illness depends on higher percentage of older patients. Meanwhile, the ISS recently listed the most common comorbidities observed in deceased patients in Italy as of March 26, 2020 (Table 2). Only 1.6% of screened patients had no comorbidities, while majority of patients presented one (23.5%), two (26.6%), three, or more (48.6%) comorbidities. Finally, we can assume that clinical outcome of COVID-19 has progressively worsened in most Italian elderly patients due to the combined impact of multiple comorbidities and frailty.
Since cardiometabolic disease is most common in people over 50 and risk of developing it increases as people get older, it is conceivable that underlying CV and metabolic comorbidities, i.e., hypertension, ischemic heart disease, atrial fibrillation, stroke, and diabetes, contribute to unfavorable prognosis in older COVID-19 patients. In addition, 10.1% patients died because of acute myocardial injury (https://www.epicentro.iss.it/coronavirus/sars-cov-2-decessi-italia).
These first independent analyses suggest that, besides arising more frequently in individuals suffering from underlying CV comorbidities, SARS-CoV-2 may cause severe myocardial injury even in the absence of ARDS or multiple organ dysfunctions often leading to death. Besides local ischemia, fatal myocardial damage in COVID-19 patients may be due to acute myocarditis unresponsive to conventional therapy. Recently, the autopsy of a male COVID-19 patient revealed myocardial infiltration by interstitial mononuclear inflammatory cells (Xu et al. 2020b). Evidence for severe myocarditis associated to low cardiac output syndrome was even observed in some COVID-19 patients in both Italy (Inciardi et al. 2020) and China (Hu et al. 2020). These findings are consistent with the reported upregulation of IL-6 in the serum of COVID-19 patients, as chronic IL-6 production during viral infection has long been known to boost myocarditis (Fontes et al. 2015). However, further investigation about underlying mechanisms is required.
Although well-known epidemic viruses, i.e., SARS-CoV, MERS-CoV, and flu viruses (influenza A or B virus), may lead to arrhythmia, myocardial infarction, and acute heart failure through aberrant systemic release of pro-inflammatory cytokines, we cannot exclude that other factors activated by SARS-CoV2 exacerbate the cardiac injury characterizing COVID-19. Herein, we will analyze the potential role of angiotensin converting enzyme 2 (ACE2) pathway, NLRP3/inflammasome, hypoxemia, aging, and air pollution.
The role of renin-angiotensin system and ACE2 in SARS-CoV-2-induced cardiac injury
To the best of our knowledge, the critical initial step underlying SARS-CoV2 entry into target cells is the binding between the receptor binding domain (RBD) of the viral spike (S) protein and the ACE2 receptor, it follows S protein priming by Ser proteases TMPRSS2 and furin (Hoffmann et al. 2020; Lan et al. 2020). Since the binding affinity between S protein and ACE2 receptor is a major determinant of viral replication rates and disease severity (Li et al. 2005), the cleavage of spike protein plays a critical role.
COVID-19 is an airborne viral illness; therefore, the first ACE2-rich cells encountered by SARS-CoV2 delivered by particle droplets are those of the upper respiratory tract and in particular goblets and ciliated cells of the nasal epithelium (Sungnak et al. 2020). However, ACE2 is expressed and active in many other human tissues. Preliminary evidence indicated that ACE2 of cardiomyocytes and coronary pericytes could play a potential role in the myocardial vulnerability to SARS-CoV-2 infection and the onset of acute cardiac injury (Chen et al. 2020a). ACE2 is one of the major player of the renin-angiotensin system (RAS) and acts in balance with angiotensin-converting enzyme (ACE) (Tikellis et al. 2011). Contrary to ACE, which converts angiotensin I (Ang I) to angiotensin II (Ang II), ACE2 is mainly involved in the degradation of Ang II, thereby resulting in the formation of Ang 1–7 (Turner et al. 2004; Tikellis et al. 2011). While Ang II induces vasoconstrictive, pro-inflammatory and pro-oxidative effects by increasing the expression on endothelial von Willebrand factor and by activating the endothelial Ang II receptor type 1 (AT1) (Dushpanova et al. 2016), Ang 1–7 mediates anti-inflammatory, anti-oxidative and vasodilatory effects through binding Mas receptor (MasR), thus exerting a beneficial effect in several pathological conditions, such as hypertension, diabetes, and CVD (Tikellis et al. 2011 and reference therein). In order to maintain homeostasis, ACE2 opposes the actions of Ang II and negatively regulates RAS.
The physiological mechanisms that shift the balance towards a prevalence of the effects of ACE or ACE2 are not yet clear. ACE2 is a carboxypeptidase that removes single amino acids from the C-terminus of peptides, while ACE is a peptidyl dipeptidase that removes C-terminal dipeptides (Pagliaro and Penna 2005). In addition, ACE2 is considered to be an important enzyme outside of classical RAS, as it hydrolyzes apelin, ghrelin, des-Arg bradykinin, des-Arg, neurotensin, and dynorphin A1–13, as well as other peptide substrates (Pagliaro and Penna 2005). However, the precise interaction between tissue ACE, ACE2, their substrates, and by-products is not yet clear.
In humans, ACE2 gene is located in the X chromosome region (Xp22). It is well known that local ACE2 gene polymorphisms were associated with hypertension and CVD as well (Yagil et al. 1999; Donoghue et al. 2000; Komatsu et al. 2002; Crackower et al. 2002; Harmer et al. 2002; Warner et al. 2004; Fan et al. 2019) and with lower risk of fatal CV events in females (Vangjeli et al. 2011). This evidence is really important since women seem to be more protected against SARS-CoV2 infection, which preferentially targets elderly men. Although the mechanisms underlie gender-based tolerance are still unknown, the localization of ACE2 on the X chromosomes may be pivotal. It is conceivable that the second X chromosome could protect women from fatal polymorphisms that make the infection more aggressive in males, e.g., by favoring viral binding. Even hormonal changes may be responsible as estrogen-induced ACE2 expression is reduced in post-menopausal women (Hilliard et al. 2013), which would make them less amenable to SARS-CoV-2 infection compared to age-matched men and more prone to systemic toxic effects due to higher Ang II levels. Despite the scarcity of data in humans, this suggestion supports recent hypothesis supporting the view that worse outcome in older COVID-19 Italian patients is due to upregulation of Ang II proinflammatory pathways throughout the body in the presence of lower ACE2 levels (AlGhatrif et al. 2020). As mentioned above, ACE2 is largely expressed in lungs, kidney, heart, small intestine, colon & rectum and endothelium (Donoghue et al. 2000; Turner et al. 2004), which represent the main organs that are targeted and damaged by SARS-CoV-2. Herein, we first recall how ACE2 regulates respiratory and kidneys functions and we focus our attention on how ACE2 could contribute to acute myocardial injury in COVID-19 patients.
ACE2 in the lungs
RAS activity is known to be elevated in lung tissues. In fact, the endothelium of the pulmonary vessels is an important site of ACE and is therefore one of the main sources of systemic Ang II. ACE2 is also strongly expressed in the lung and upper airway. Pulmonary ACE2 may play a role in regulating the balance of circulating levels of Ang II/Ang 1–7. Of note, Ang II contributes to pulmonary vasoconstriction in response to hypoxia, an important pathophysiological mechanism useful to prevent shunt in patients with lung injury or pneumonia. However, the local increase in Ang II levels increases vascular permeability thus promoting the onset of pulmonary edema. In ARDS, RAS appears crucial in maintaining oxygenation (Lionetti et al. 2005). ACE2 knockout mice showed more severe symptoms than wild-type mice (Kiely et al. 1997; Imai et al. 2005), while overexpression appears protective. A correlation between an ACE polymorphism and ARDS severity has been described in humans (Matsuda et al. 2012). In acute lung injury, ACE, Ang II, and the AT1 receptor function as factors that promote lung injury (Oudit et al. 2009). Despite having normal lung structure and function, ACE2 knockout mice show a very serious pathology in case of ARDS/acute lung damage compared to wild-type mice since ACE2 plays an important anti-inflammatory role. ACE2 knockout mice are characterized by increased vascular permeability, pulmonary edema, and neutrophil accumulation. In some recent studies, ACE2 protects against pulmonary vasoconstriction, chronic pulmonary lesions, and fibrosis. Moreover, ACE2 inhibits neutrophil infiltration and lung inflammation by limiting IL-17 signaling and by reducing the activity of the STAT3 pathway in response to bacterial infection in rats (Sodhi et al. 2019). Therefore, ACE2 has been proposed as a new therapeutic target for acute lung injury, chronic lung disease, and bacterial pneumonia as well. Indeed, the efficacy of recombinant soluble human ACE2 protein on acute respiratory failure is currently being studied in humans in a phase II clinical trial (Kuba et al. 2013). In addition, some drugs have been developed to increase pulmonary ACE2 expression, such as anti-diabetic glucagon-like peptide 1 receptor (GLP-1R) agonist liraglutide, in order to improve lung function by normalizing expression of surfactant protein A and B (Romaní-Pérez et al. 2015), which is reduced in patients with severe viral pneumonia (Kerr and Paton 1999).
ACE2 in the kidney
ACE and ACE2 are highly expressed in the adult kidney even if in different areas. ACE2 is mainly expressed in the proximal tubule on the edge of the brush, while ACE seems to be evenly distributed on the apical and the basolateral membrane of the polarized cells of the tubules. Interestingly, both renal development and kidney function are normal in the ACE2 knockout mouse (Crackower et al. 2002). However, angiotensinogen, ACE, and AT1 receptor lack results in a series of alterations in kidney morphology. Therefore, ACE2 has a marginal role in regulating renal function, at least in the healthy state. However, the action of ACE2 is critical during RAS activation. In fact, Ang 1–7 exerts very limited renal effects in the healthy state but it plays profound beneficial effects in diabetic patients and in other states associated with renal damage. For instance, ACE2 deficiency in mouse models is associated to increased age-related glomerulosclerosis and enhanced Ang II-induced renal oxidative stress, leading to greater renal injury. In particular, downregulation of tubular ACE2 is associated with albuminuria or tubular injury in the diabetic kidney, where further inhibition of ACE2 results in augmented renal damage (Doan et al. 2001; Li et al. 2005; Oudit et al. 2006; Wong et al. 2007; Soler et al. 2007). Hence, ACE2 is proving to be a protective molecule against kidney disease especially in the context of a negative regulation of classical RAS axis. Deletion of ACE2 leads to spontaneous late-onset nephrotic glomerulosclerosis and accelerates diabetic kidney injury. Indeed, glomerular disease and albuminuria occur after pharmacological inhibition of ACE2 (Soler et al. 2013).
Note that in several forms of chronic kidney disease, and especially in diabetes, it has been found that tubular ACE2 expression is reduced. However, other studies showed that glomerular expression of ACE2 increases in acute kidney injury. It is likely that differential expression pattern of tubular and glomerular ACE2 may be an important determinant for determining the progression of kidney disease (Kuba et al. 2013).
ACE2 in the heart
In some cardiac resident cells, i.e., endothelial cells, pericytes and cardiomyocytes, ACE2 is the main enzyme involved in the local clearance of Ang II. Previous experimental evidences indicate that ACE2 is an essential regulator of cardiac function and is involved in the response of various cardiac cells to multiple injuries, especially those associated with overactivation of classical RAS pathway (Imai et al. 2005). In different experimental models, indeed, ACE2 deficiency induces progressive cardiac fibrosis with aging and/or pressure overload. Moreover, ACE2 deficiency accelerates post-infarct adverse ventricular remodeling and causes early cardiac hypertrophy in mice. These detrimental effects may be prevented or reversed following treatment with ACE inhibitors or AT1 receptor blockers (or sartans). On the other hand, Ang1–7 peptide treatment can hamper cardiac remodeling and improve myocardial performance and even survival in rodent heart failure models (Schindler et al. 2007). Furthermore, the use of Mas receptor selective ligands, such as AVE-0991, exerts cardioprotective effects similar to Ang1–7 in ischemia/reperfusion (I/R) injury, myocardial infarction (Zeng et al. 2012), or hypertension-induced cardiomyopathy models (Ferreira et al. 2007). These observations suggest that the altered balance between ACE and ACE2 in the CV system is a critical driver for progressive heart disease, which can be reverted by inhibiting classical ACE or upregulating ACE2 pathway. However, it is still unknown how myocardial ACE2 may worsen COVID-19 clinical scenario or contribute to vulnerability to SARS-CoV2 infection at least.
The role of cardiac ACE2 in SARS-CoV-2-induced myocardial injury: facts and hypotheses
Patients with CVDs are at higher risk for severe COVID-19 and show a worse prognosis in the presence of a systemic inflammatory response and immune system deregulation (Zheng et al. 2020; Madjid et al. 2020). So far, it is unclear whether cardiac ACE2 signaling may play a role in cardiac injury, yet the following clinical evidences will be helpful to support next focused investigations.
Firstly, since epithelial ACE2 is the gateway of SARS-CoV-2 entry into cells, the attractive hypothesis that SARS-CoV-2 may directly affect myocardial tissue through ACE2 of endothelial cells, pericytes or cardiomyocytes cannot be discounted a priori. In agreement with this hypothesis, preliminary reports provided evidence of symptoms of myocarditis in COVID-19 patients (Xu et al. 2020b; Ruan et al. 2020b; Hu et al. 2020; Inciardi et al. 2020), which is also consistent with the rise of hypersensitive cTnI levels even if reports lack histological detection of myocardial inflammation. While the myocardial localization of SARS-CoV-2 remains to be confirmed with certainty, we cannot ignore that other viruses, such as seasonal flu and parvovirus B-19, cause myocarditis (Fung et al. 2016). However, the detection of SARS-CoV viral RNA in 35% of cardiac biopsies performed in patients during the SARS outbreak in Toronto (2003) is suggestive to support the hypothesis of coronavirus cardiotropism. Furthermore, pulmonary infection with the human SARS-CoV led to ACE2-dependent myocarditis in mice (Oudit et al. 2009). It is, therefore, conceivable that SARS-CoV-2 also infects cardiomyocytes.
Secondly, the loss of ACE2 is associated to increased susceptibility to heart failure, while augmented ACE2 levels prevent and reverse the heart failure phenotype, emerging as a key protective pathway in the heart pathology (Patel et al. 2016). Accordingly, CVD is associated with decreased baseline levels of ACE2 expression (Chamsi-Pasha et al. 2014). During its replication, SARS-CoV is able to progressively downregulate ACE2 protein expression (Dijkman et al. 2012). It has, therefore, been hypothesized that the loss of ACE2 function may also occur during SARS-CoV-2 infection (Garami 2020; Henry et al. 2020). In particular, the binding of SARS-CoV-2 to ACE2 might reduce the residual ACE2 activity, thereby altering the ACE/ACE2 balance and generating a RAS dysregulation with higher ACE and/or lower ACE2, a combination of phenomena that can result in co-occurrence of lung, heart and renal acute failure (Garami 2020; Henry et al. 2020). The predominance of the ACE/Ang II/AT1 axis function compared to the ACE2/Ang1–7/Mas counterpart could be responsible of the typical vasoconstrictive, pro-inflammatory and pro-oxidative effects of Ang II occurring in these conditions, that could ultimately evolve to acute heart failure and ARDS (Zhang and Baker 2017; Henry et al. 2020). Accordingly, the ACE2 production is dramatically impaired during ARDS, resulting in dominant ACE activity (Wösten-Van Asperen et al. 2011). Considering that peripheral Ang II levels were found to be significantly higher in COVID-19 patients with respect to healthy subjects and that positively correlated with the viral load and lung injury (Liu et al. 2020b), the hypothesis of the RAS pathway dysregulation in both lung and cardiac injury during SARS-CoV-2 infection should be investigated (Garami 2020; Henry et al. 2020).
Third, the SARS-CoV-2-dependent ACE2 downregulation could trigger monocyte/macrophage activation and induce severe lung inflammation, contributing to the pathogenesis of acute lung injury up to the onset of respiratory failure (Sodhi et al. 2018; Garami 2020). The hypoxemia consequent to severe interstitial pneumonia can stimulate renin secretion and renin gene expression (Kramer et al. 1998), thereby leading to a vicious circle (Garami 2020) that could enhance cardiac damage in COVID-19 patients. Notably, high Ang II levels were associated with hypertension, heart failure (Packer and McMurray 2017), and diabetes (Hao et al. 2013), which both represent the most common underlying comorbidities in patients more vulnerable to SARS-CoV-2 infection. Of course, the relationship between virus load, onset and magnitude of COVID-19, and ACE2 downregulation-dependent RAS dysregulation in lungs and heart requires a lot more compelling evidence. Moreover, additional studies are mandatory to investigate whether the acute myocardial injury due to SARS-CoV-2 infection might be related to ACE2 expression rather than activity.
Is there a role for ACE inhibitors or ARBs in the development of SARS-CoV-2 infection?
Inhibiting the classical RAS pathway with ACE inhibitors or angiotensin receptor blocker (ARB) represents a first-line pharmacological strategy to treat arterial hypertension (Paz Ocaranza et al. 2020), which is usually associated with reduced lung function (Schnabel et al. 2011b). Intriguingly, ACE2 expression in humans is enhanced by these treatments (Thomson 2020), whereas the ARBs, losartan and olmesartan, were found to increase by threefold cardiac ACE2 expression in a rat model of acute myocardial infarction (AMI) (Gurwitz 2020). This observation led some investigators to speculate about a mechanistic link between the preexisting use of these drugs and predisposition to SARS-CoV-2 infection (Thomson 2020; Kuster et al. 2020; Esler and Esler 2020). As described in the “ACE2 in the heart” section, however, ACE2 confers protection against both lung and cardiac injury, so that it has also been proposed to use ACE inhibitors or ARB as therapeutics for treating COVID-19 patients prior to ARDS development (Gurwitz 2020). Although ACE inhibitors provide imperfect renoprotection in advanced type 2 diabetes and CV risk remains elevated (Zoja et al. 2011), preliminary evidence from a limited cohort of patients (n = 187) demonstrated that there was no difference in fatality rate between COVID-19 patients pretreated or not with ACE inhibitors or ARB (Guo et al. 2020a). Moreover, inhibition of classical RAS pathway proved to exert a beneficial outcome in patients affected by viral pneumonia, by likely reducing the respiratory inflammatory burden and cytokine storm induced by virus infection because of increased ACE2 expression (Henry et al. 2018). The effect of these drugs on ACE2 levels in the lungs is still unknown and larger clinical studies are required to assess the impact of ACE inhibitors and ARB on severity of SARS-CoV-2 infection. A recent joint statement of the American Heart Association, American College of Cardiology and Heart Failure Society of American recommended not interrupting these medications in COVID-19 patients affected by chronic arterial hypertension, unless otherwise required by changes in patients’ conditions (Madjid et al. 2020). Indeed, these life-saving drugs are difficult to replace, since other common anti-hypertensive drugs such as beta-blockers, cause bronchoconstriction aggravating lung function in general population (Schnabel et al. 2011a) and common calcium-channel blockers may cause cardiac conduction defects, impair myocardial contractility and cause peripheral edema.
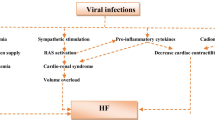
The role of virus infection-dependent cytokine storm in SARS-CoV-2-induced acute myocardial injury
While the involvement of ACE2 in SARS-CoV-2-induced acute myocardial injury remains controversial, there is no doubt that the cytokine storm associated to virus infection is highly detrimental to the heart. Acute respiratory infections, such as those characterizing interstitial pneumonia by SARS-CoV and MERS-CoV, have long been known to result in major acute cardiac complications, which may lead to a 60% increase in short-term fatality rate (Corrales-Medina et al. 2013). Cardiac complications associated to viral pneumonia include malignant arrhythmias, AMI and heart failure, which likely result from the increased myocardial workload during infections that accelerate myocardial injury or infarction (Bonow et al. 2020). In addition, SARS-CoV-2 has been shown to trigger an exaggerated systemic inflammatory response that, besides acute lung injury and ARDS, may ultimately lead to multiple CV complications (Zheng et al. 2020), including AMI, unstable angina, tachycardia, heart failure, and stroke (Gorla et al. 2018). Interestingly, SARS-CoV-2 promotes an aberrant release of pro-inflammatory cytokines and chemokines by alveolar epithelial cells, vascular endothelial cells, activated monocytes and lymphocytes, which express ACE2 on their cells surface. Furthermore, SARS-CoV-2 could also induce lymphocyte death through pyroptosis, which would explain why COVID-19 patients undergo lymphopenia, thereby further contributing to exacerbate organ injury. The “cytokine hypothesis” for cardiac damage postulates that the deleterious effects of chronic cytokine release on myocardium and vascular endothelium may facilitate the onset of AMI or heart failure. For instance, pro-inflammatory cytokines, i.e., interleukin-1 (IL-1), IL-6, and TNFα, exert a negative inotropic effect on cardiac contractility. Likewise, the sustained activation of inflammatory signaling, e.g., through TNFα and IL-1β, may induce massive cardiomyocyte apoptosis and lead to adverse left ventricular remodeling, which favors the onset of acute heart failure. Additionally, the cytokine storm stimulates monocytes/macrophages to release matrix metalloproteinases (MMPs), which boost atherosclerotic plaque growth and rupture, favor the secretion of pro-coagulant factors and induce hemodynamic changes, thereby increasing the probability for AMI to occur (Mann 2015; Newby 2016; Toldo and Abbate 2018). Indeed, while short-term IL-6 release exerts a cardioprotective effect, chronic activation of IL-6 signaling leads to autoimmune myocarditis (Fontes et al. 2015). In this scenario, underlying CVDs are predicted to worsen the outcome of patients with viral infections on the heart, as already established for pneumonia and seasonal flu.
Several observations support the contribution of deregulated inflammatory signaling in SARS-CoV-2-induced myocardial injury (Zheng et al. 2020). For instance, Huang et al. observed that plasma levels of inflammatory mediators, including IL-1β, IFNγ, IP10 (CXCL10), and MCP1 (CCL2), were upregulated in COVID-19 patients; interestingly, this study also showed that cytokine levels were remarkably higher in critically ill patients. In particular, the serum levels of granulocyte-colony stimulating factor (G-CSF), IP10, MCP1, MIP1A (CCL3), and TNFα were higher in ICU-patients than those that did not require ICU admission, thereby suggesting that the cytokine storm is a clinical predictor of mortality (Huang et al. 2020). As IFNγ is the signature cytokine of the T-helper-1 (TH1) response (Rose 2011), these findings suggest that the balance between the pro-inflammatory TH1 response and the anti-inflammatory T-helper-2 (TH2) response is tipped in favor of the former (Zheng et al. 2020). In line with these observations, Zhu et al. reported that hypersensitive cTnT and inflammatory biomarkers (e.g., serum ferritin and IL-6) were significantly higher in non-survivors COVID-19 patients (Zhou et al. 2020a). The high inflammatory burden in severe COVID-19 has been further confirmed by another independent study, which reported higher levels of serum ferritin, IL-6 and C-reactive protein in patients who did not survive, likely because of fulminant myocarditis (Ruan et al. 2020b). Of note, over-production of inflammatory cytokines and chemokines was common in SARS-CoV (Wong et al. 2004) and MERS-CoV (Mahallawi et al. 2018) patients, where acute lung injury was diagnosed.
Further investigations are required to elucidate the potential role of the cytokine storm in the pathogenesis of CV complications including cardiomyopathy, acute myocardial injury and myocarditis following SARS-CoV-2 infection and to understand the potential mechanistic relationships between CVD and COVID-19 outcomes. As discussed in the next paragraph, the NLRP3/inflammasome might play a triggering role in the cytokine storm induced by SARS-CoV-2.
NLRP3/Inflammasome: a common feature in viral infections, CVD and COVID-19
Both innate and acquired immune systems represent a crucial defense against pathogens. Indeed, it is conceivable that viral antigens firstly enroll innate immunity by activating pattern recognition receptors (PRRs) to suppress viral replication and modulate the adaptive immune responses through the expression of type I interferon and pro-inflammatory cytokines (Bowie and Unterholzner 2008). During the last decades, the NLRP3/inflammasome pathway emerged as an important mediator of virus-induced inflammation.
The mechanisms of NLRP3/inflammasome activation
NLRP3 is a PRR belonging to the Nod (nucleotide oligomerization domain)-like receptors (NLRs) able to recognize both damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) (Schroder and Tschopp 2010), such as specific virus molecules, stimulating downstream pathways that contribute to the systemic immune response (Farag et al. 2020). NLRP3 inflammasome is a multi-protein platform made up of a sensor protein (NLRP3), the adaptor protein ASC, and a cysteine protease precursor procaspase-1 (Schroder and Tschopp 2010; Zhou et al. 2011). Activated NLRP3 induces the inflammasome assembly interacting with ASC that, in turn, triggers the conversion of the zymogen procaspase-1 into the active caspase-1 (Franchi and Núñez 2012). Lastly, caspase-1, an established inflammatory caspase (Martinon and Tschopp 2007), converts the inactive pro-IL-1β and pro-IL-18 into active pro-inflammatory cytokines, such as IL-1β and IL-18 (Epstein and Dinarello 1984). Finally, IL-1β stimulates the release of TNFα and IL-6, whereas IL-18 increases circulating levels of IFN-γ, IL-13, IL-4, IL-8, thereby contributing to the immune inflammatory responses. Both IL-1β and IL-18 represent key factors of the host defense (Davis et al. 2011).
The role of the NLRP3/inflammasome in COVID-19
Viral infection has been always associated with inflammation (Farag et al. 2020) and viroporins seem to play a triggering role rather than viral PAMPs. Viroporins are a new class of virus proteins composed of 15–20 amino acids that homo-oligomerize forming hydrophilic pores across the organelle membranes, thereby increasing the permeability to monovalent and divalent cations (i.e., Na+, K+, and Ca2+) (Wang et al. 2011; Nieva et al. 2012). These structures appear to play a crucial role in the virus infection/life cycle, genome replication, and assembly (Farag et al. 2020), paving the way to consider the possibility to target viroporins with antiviral strategies (Scott and Griffin 2015). An increase in intracellular Ca2+ concentration, as well as a reduction in cytosolic K+, are regarded among the most suitable triggering signals for NLRP3/inflammasome activation (Muñoz-Planillo et al. 2013; Horng 2014; Okada et al. 2014; Zhu et al. 2017).
Of note, viroporins mediate NLRP3/inflammasome activation and excessive production of IL-1β (Guo et al. 2015; Farag et al. 2020). To date, NLRP3/inflammasome activation following a viral infection has been reported for influenza A virus (IAV), hepatitis C virus (HCV), encephalomyocarditis virus (EMCV), and SARS-CoV (Liu et al. 2004; Ichinohe et al. 2010; Farag et al. 2017). The reported increase in pro-inflammatory cytokines levels, such as IL-18 and IL-1β, not only in the plasma, but also in lungs and lymphoid tissues of patients affected by SARS, MERS, and, more recently, by COVID-19, is potentially consistent with NLRP3/inflammasome activation (Chen et al. 2019; Huang et al. 2020). It is well known that all coronaviruses encode for viroproteins E and 3a, which are responsible at least for replication, virulence, and NLRP3/inflammasome activation (i.e., SARS-CoV) (Castaño-Rodriguez et al. 2018; Chen et al. 2019; Farag et al. 2020). SARS-CoV proteins E and 3a IC differ for intracellular distribution and biophysical properties. Protein E locates in the endoplasmic reticulum (ER)-Golgi intermediate compartment, its functionality is strictly dependent on the lipid environment composition (Nieto-Torres et al. 2015), and induces Ca2+ efflux during SARS-CoV-2 infection, which activates the inflammasome complex, leading to the acute proinflammatory response associated with virus pathogenicity. On the other hand, the detailed targeting of protein 3a remains still elusive (Castaño-Rodriguez et al. 2018; Chen et al. 2019; Farag et al. 2020).
In addition, in vitro studies revealed that also the SARS-CoV open reading frame 3a (ORF3a) accessory protein, which is a strong inducer of the NLRP3/inflammasome (Siu et al. 2019), may serve as a viroprotein (Chen et al. 2011). Therefore, it is plausible that SARS-CoV-2 may use the same mechanism of action to induce the NLRP3-dependent storm of cytokines and the resulting severe systemic inflammation that often causes fatal outcome. It is not surprising that inhibition of the NLRP3/inflammasome has recently been put forward as a promising strategy to treat or prevent acute cardiac injury, such as I/R injury (Liu et al. 2018; Toldo and Abbate 2018; Pasqua et al. 2018). Likewise, targeting NLRP3/inflammasome may represent an attractive therapeutic strategy in the treatment of virus-associated diseases, such as CVD (Liu et al. 2018; Pasqua et al. 2018). For instance, it has been demonstrated that NLRP3/inflammasome activation by hypertension induces a “sterile inflammation” that results in a chronic low-grade inflammation characterized by an increased activity of NF-κB (Zhu et al. 2017; Bullón et al. 2017). Sustained NF-κB signaling, in turn, leads to increased tissue and/or circulating levels of proinflammatory mediators, such as IL-1β and IL-18, that induces the activation of immune cells and their consequent detrimental effects on both respiratory tract and heart (Dalekos et al. 1997; Rabkin 2009; Krishnan et al. 2014). Based upon these evidences, it is reasonable to refer to NLRP3/Inflammasome as a common pathogenic mechanism included in a vicious cycle that triggers and supports both COVID-19 and its most common CV comorbidities, such as arterial hypertension. In this context, targeting NLRP3/inflammasome might serve as a therapeutic tool in counteracting the severe systemic inflammation following SARS-CoV-2 infection, while at same time also mitigating the underlying CVD and reducing the mortality rate of hospitalized patients. In this regard, promising data were provided by a drug recently acquired by Novartis, namely IFM-2427, which is already under clinical trial (Mullard 2019).
COVID-19 and the hypoxia challenge to the cardiovascular system
In agreement with other pulmonary diseases, COVID-19 poses a remarkable challenge to health because of hurdled blood oxygenation and oxygen delivery to tissues. When the oxygen supply to tissues becomes insufficient to sustain normal function, a situation called hypoxia ensues. Whereas some organs/tissues can survive short in response to non-severe hypoxia periods, for other organs/tissues hypoxia represents an intolerable hit that may lead to lethal outcomes. As myocardium and cerebral tissues are the most endangered, heart and brain represent the primary targets of acute respiratory failure.
There are several criteria that recommend the transfer of patients to ICU. One of these is arterial blood oxygen saturation < 90% and PaO2 < 60 mmHg (Smith and Nielsen 1999). As the COVID-19 pathology mainly concerns lung dysfunction, most of these patients may face severe hypoxia for several days, and most treatments aim at correcting hypoxia by oxygen therapy and mechanical ventilation. There are several concerns related to the effects of prolonged hypoxia on the CV system.
First, the hypoxia-driven hyperventilation response, mediated by the carotid bodies and aimed at restoring normal oxygenation, necessarily implies excessive washout of carbon dioxide, which shifts the acid-base balance towards alkalosis. Such response has been found in healthy subjects even at moderate altitude for periods of time ranging from few days (Samaja et al. 1997) to several months (Porcelli et al. 2017). Although often partially compensated by metabolic acidosis, alkalosis of either origin represents a common finding in critically ill patients (Mæhle et al. 2014; Kreü et al. 2017). Although not yet documented, alkalosis is expected to damage brain and pulmonary tissue as it enhances oxidant-induced apoptosis in several cell lineages (Auer 2004; Ando et al. 2007).
Second, chronic hypoxia weakens the myocardial resistance against stress (Milano et al. 2010). Acute hypoxia-induced pulmonary vasoconstriction at last induces histological changes in the right ventricular (RV) myocardium (Netzer et al. 2017), contributing to increase RV systolic pressure and hypertrophy, common features of pulmonary hypertension (McLaughlin and McGoon 2006). In addition to the effects elicited by acute hypoxia, chronic hypoxia leads to pulmonary remodeling, which include increased pulmonary vessel density, increased frequency of small (< 50 μm wall thickness) vessels and increased thickness of pulmonary lamina (Nydegger et al. 2018, 2019), all changes imputable to excessive stimulation of bone marrow-derived progenitor cells (Favre et al. 2017). Chronic hypoxia is well known to contribute significantly to pulmonary hypertension (Ramchandran et al. 2012) and is associated with many pulmonary hypertension-related diseases, including chronic obstructive pulmonary disease, cystic fibrosis, diffuse interstitial fibrosis, bronchopulmonary dysplasia, infiltrative lung tumors, congenital hearts defects, and ischemic events.
Third, at odds with what happens in healthy subjects exposed to altitude hypoxia, which is well known to stimulate erythropoiesis (Piperno et al. 2011; Goetze et al. 2013), systemic hypoxia represses erythropoiesis in ARDS patients and these often develop severe anemia (< 8 g/dl), which correlates with a poor prognosis, and may require blood transfusion (Silver 2005; Jenq et al. 2018). This clinical scenario is common in ICU patients (Nguyen et al. 2003) and it may be attributable to anemia of inflammation, whose etiology is scarcely known. The physiological response to hypoxia, with compensatory increase in hemoglobin, requires an increase in iron transport and use. In fact, it demands stabilization of ferroportin, the transmembrane protein that enables iron release from the cells with increased intestinal absorption. The degradation of ferroportin is mediated by hepcidin (Hep) that decreases in healthy subjects thereby enabling the compensatory response to hypoxia (Gassmann and Muckenthaler 2015). However, Hep levels increase in ARDS patients (Galesloot et al. 2011), despite their hypoxic condition. The consequent loss of Hep regulation may limit the efficacy of iron-replacement therapies (Lasocki et al. 2010). This mechanism may be relevant in ARDS and COVID-19 patients because iron release into the circulation and deregulated uptake by tissues that use iron may saturate transferrin, increase the non-transferrin-bound aliquot of iron, with possibly triggering of unspecific oxidative stress injury.
Fourth, although the correction of hypoxia with oxygen therapy remains the major life-saving concern in ICU patients, excess oxygen delivery or hyperoxia may cause considerable harm (Ottolenghi et al. 2020). In facts, hyperoxia caused by breathing mixtures with > 40% oxygen markedly elevates reactive oxygen species (ROS) release in the circulation and contributes to tissue injury by oxidative stress (Nagatomo et al. 2012). Hyperoxia-induced injury may occur even in patients undergoing laparoscopic surgery breathing mixtures containing 40% to 80% oxygen (Ottolenghi et al. 2019). Hyperoxia-driven oxidative stress is expected to alter the expression of genes that regulate pathways underlying different responses to oxidative stress (Terraneo and Samaja 2017). Intuitively, the direction of the alterations driven by hyperoxia is opposite to those elicited by hypoxia. However, there is evidence that this is not the case in many instances, for example the regulation of gene transcription by the hypoxia-inducible factors (HIFs). HIFs are well known to respond to hypoxia by acting as transcriptional factors for an array of genes that participate in various cell processes as angiogenesis, metabolism, cell proliferation, control of ROS-induced damage, and many others (Giordano 2005), including apoptosis and autophagy (Ziello et al. 2007; Gui et al. 2016). But recent studies show that HIF-1α and HIF-2α are indeed up-regulated in the response to hyperoxia as well, at least in brain (Benderro et al. 2012; Terraneo et al. 2017), some types of cancer (Terraneo et al. 2010, 2014), and hepatocytes (Wikenheiser et al. 2009; Marconi et al. 2014), but not in heart (Gyongyosi et al. 2018). Although the injury led by hyperoxia has not yet been studied as that led by hypoxia, it is established that hyperoxia reduces the expression of angiogenic vascular endothelial growth factor (VEGF) signaling and capillary density (Benderro et al. 2012), reduces the anti-apoptotic defense by increasing the expression of the pro-apoptotic proteins Bax and Bad and decreasing the activation of anti-apoptotic proteins Bcl-2 and Bcl-xl (Brutus et al. 2009), increases neuronal apoptosis concomitantly with decreased protective p-Akt and erythropoietin signaling (Terraneo et al. 2017), accumulates oxidized lipids and proteins (Tatarkova et al. 2011), down-regulates neurotrophins that provide tropic support to developing neurons (Felderhoff-Mueser et al. 2004), and stimulates ferroptosis, a newly discovered iron-dependent form of cell death thereby contributing to brain damage (Li et al. 2017).
Impact of COVID-19 in elderly population
A striking feature of COVID-19 pandemics is the high incidence of fatalities in elderly patients (Guan et al. 2020; Wang et al. 2020a; Zhou et al. 2020a). A recent analysis by the Italian Institute of Health revealed that the overall-case fatality rate increased with aging in both China and Italy; however, the fatality rate in Italy was remarkable higher as compared to China (7.2% vs. 2.3%) because of the higher death tolls in Italy among subjects aged > 70 years (12.8% vs. 8.0%) and among those older than 79 (20.2% vs. 14.8%). This increase in COVID-19-related mortality rate in Italy may be ascribed to the demographic characteristics of this Country, where 23% of the population was aged > 65 years in 2019 (Onder et al. 2020). Of note, aging represents one of the most detrimental risk factors for the CV system (Obas and Vasan 2018). This addresses the issue of frailty in older population as a critical parameter to be accounted for in the control of COVID-19 pandemics.
Age-associated frailty
Frailty, which was labeled as “the most problematic expression of population ageing” (Clegg et al. 2013), is often confounded with disability or co-morbidity. But frailty is assuming separate distinguished features, which highlight the presence of a well-defined phenotype that characterizes elders. Many elders have multi-organ dysfunction, with frailty representing a “unifying notion in the care of elderly patients that directs attention away from organ-specific diagnoses” (Clegg et al. 2013). The concept of frailty is now universally defined as an individual’s state of increased vulnerability and susceptibility to adverse health outcomes or death when exposed to a stressor, in this case the pulmonary diseases arising from SARS-CoV-2 infection in the presence of an age-dependent excess of ROS and reactive nitrogen species (RNS).
Frailty may be viewed as the result of the disruption of the organism’s homeostatic balance that leads to heterogeneous clinical manifestations when the individual is exposed to endogenous or exogenous stressors. Only a minor but non negligible number of people > 65 years old is considered frail: the prevalence of physical frailty ranges from 4 to 17% (mean 9.9%), with a higher prevalence in persons > 80 years old, among patients with multiple comorbidities (Collard et al. 2012).
Frailty and CVD in the elder
In the absence of pandemics, the prevalence of frailty and CVD increase with age, making their coexistence common. Epidemiological studies suggest that frailty may represent an important prognostic marker for patients with several types of CVDs, including coronary artery disease, heart failure, as well as older people undergoing surgical and transcatheter interventions (Afilalo et al. 2014). Despite unclear biological underpinnings of frailty and unexplored interaction with heart failure, it appears that frailty and CVD share common pathophysiological pathways (Bellumkonda et al. 2017). Among CVDs, AMI is a major cause of death, morbidity and loss of quality of life in industrialized countries, accounting for more than 7 million deaths per year worldwide (Steg et al. 2012). Aging is classically regarded to as the greatest risk factor for AMI. Indeed, endothelial cell senescence due to increased ROS production and dysfunction of nuclear factor-erythroid-2-related factor 2 (Nrf2)-mediated antioxidant response pathway may compromise endothelial nitric oxide (NO) bioavailability, exacerbate inflammatory response and pathogenesis of age-related vascular diseases (i.e., AMI and stroke) in older patients (Fulop et al. 2018; Ungvari et al. 2019). An excess of ROS and RNS is central in many pathways leading to cardiac hypertrophy and failure (Tocchetti et al. 2015). Accordingly, the incidence of AMI and the case-fatality mortality rate are markedly higher in elderly patients, who are also more prone to develop post-AMI heart failure (Afilalo et al. 2014). When compared to younger subjects, aged patients are less amenable to receive percutaneous coronary intervention, while they are less sensitive to fibrinolytics (Carro et al. 2011). Therefore, the impaired resilience of the cardiovascular cells to oxidative stressors in the presence of deficiency of relevant anti-oxidant mechanisms (i.e., Nrf2-related pathway) may lead to SARS-CoV-2 detrimental consequences to the aged heart and vasculature. Moreover, the correct fine-tuning of the nitroso-redox balance is fundamental for the function of the main components of the excitation-contraction coupling machinery (Tocchetti et al. 2015).
Cardiovascular therapy in young and frail patients
Despite the predominance of older patients with CVD among the population, randomized clinical trials have generally focused into young subjects without co-morbidities, thereby generating data that cannot be extended to elderly patients. This feature has been related to the higher case fatality rate in older vs. younger subjects hospitalized with AMI. Individuals aged ≥ 75 years old do not benefit from the same pharmacological interventions established for their younger counterparts after AMI (Fleg et al. 2011). The lower sensitivity to CV drugs in the elderly depends on age-associated physiological changes in pharmacodynamics and pharmacokinetics, together with intrinsic deficits in the signaling pathways to protection.
The reasons for the higher incidence/severity of AMI in elderly subjects are multifactorial and not easy to reconcile with a single triggering event. Endothelial dysfunction has been regarded as an early mechanism among those leading to the mismatch between oxygen supply and demand that finally culminates in AMI (Moccia et al. 2018). In addition, aged endothelial cells lose their capability to proliferate and to replace neighboring damaged cells, thereby aggravating the impact of coronary endothelium dysfunction (Moccia et al. 2018). Consequently, aged subjects are more susceptible to develop coronary artery disease since the aging endothelial cells of coronary microcirculation are unable to express cytoprotective (pro-survival, antioxidant, macromolecular damage repair, and anti-inflammatory) genes due to deregulation of relevant transcription factors pathways, such as Nrf2 signaling. Oxidative damage is quite remarkable in microvascular endothelial cells affected by NAD+ depletion and Sirtuin 1 dysregulation (Kiss et al. 2019), dampening their angiogenic activity and thus leading to microvascular rarefaction, which is an established hallmark of aging (Valcarcel-Ares et al. 2012). Likewise, Nrf2 signaling is disrupted during aging also in cardiac myocytes (Silva-Palacios et al. 2016) and fibroblasts (Nacarelli et al. 2018), thereby contributing to enhance cardiac vulnerability to oxidative stress. It thus appears that the impairment of Nrf2 transcriptional activity is key to increase CV risk during aging and could, therefore, worsen CV damage in older COVID-19 patients. This scenario could be even worsened when oxygen therapy is required, as hyperoxia-induced ROS release is postulated to exert a stronger cytotoxic effect on aged vasculature due to its lower resilience to oxidative stress. In agreement with these hypotheses, a recent study demonstrated that genetic depletion of Nrf2 worsened age-related impairment of endothelial NO bioavailability and of endothelial monolayer integrity. In addition, genetic silencing of Nrf2 resulted in a dramatic increase in the secretion of proinflammatory cytokines (Fulop et al. 2018), also known as senescence-associated secretory phenotype (SASP). These cytokines include IL-1β and TNFα (Ungvari et al. 2018), which are recently shown to mediate COVID-19-induced cytokine storm. Enhancing the pro-inflammatory phenotype might represent another mechanism whereby aging-dependent Nrf2 dysfunction contributes to exacerbate the detrimental consequences of SARS-CoV-2 on the CV system.
Moreover, aging is associated to a significant perturbation of the Ca2+ signaling toolkit in cardiomyocytes, which leads to a dramatic impairment of heart muscle contraction (Hamilton and Terentyev 2019). The higher mortality for AMI in these individuals has further been linked to the age-related decline in the endogenous protective mechanism against myocardial I/R injury. Indeed, pre- and post-conditioning are powerful interventions aimed to reduce the infarct size by eliciting endogenous cardioprotection in response to one or several brief cycles of sub-lethal myocardial I/R that precede or follow, respectively, a sustained potentially lethal ischemic insult. These conditioning protocols recruit a complex cascade of intracellular signaling pathways, mainly RISK and SAFE, which converge on mitochondria and limit cell death (Pagliaro et al. 2011). Although pharmacological conditioning recapitulates the endogenous mechanisms of cardioprotection induced by both pre- and post-conditioning and might be exploited in the therapy of AMI, these conditioning protocols are barely effective in aged hearts, thereby exacerbating I/R injury. The loss of cardioprotection in elderly people is not irreversible and might be either rescued or preserved by alternative strategies, such exercise (Boengler et al. 2009), which also promotes angiogenesis and increases vascular density in aging mouse models of AMI (Bei et al. 2015).
Lessons gained from CVD in the treatment of COVID-19 patients
Presently, there is no experimental evidence-based difference in the clinical management of young vs. elderly, presumably frail, subjects, at least for CVD; conversely, the sharp decrease in the endogenous mechanisms of cardioprotection and angiogenic repair during aging suggests that older subjects should undergo a distinct treatment as compared to younger individuals. Older patients are left with few, if any, effective therapeutic methods to alleviate symptoms, restore distal perfusion and preserve tissue viability, calling for innovative approaches to restore the efficiency of cardioprotective mechanisms with aging, for example by pursuing alternative strategies to foster pre-conditioning, post-conditioning, and endogenous protection (Boengler et al. 2009). As a matter of facts, actual strategies and treatments for COVID-19 patients are essentially the same for young and elder patients. But if CV patients respond differently depending on their age or frailty, why shouldn’t this occur in COVID-19 patients? One could, however, suggest that restoring endothelial function, e.g. at microvascular level, or improving vascular health during aging could represent an effective strategy to treat COVID-19 in critical patients. For instance, activation of Nrf2 by drugs (e.g., propofol during mechanical ventilation or oltipraz) (Zhou et al. 2020c; Ruan et al. 2020a) or by epigenetic compounds (e.g., nicotinamide mononucleotide, resveratrol, histone deacetylase inhibitors) (Csiszár et al. 2015; Pooladanda et al. 2019; Kiss et al. 2019) could rescue, at least partially, stress resilience of both endothelial and cardiac cells in aging individuals affected by COVID-19. Otherwise, regular physical exercise and dietary intake of functional foods (Lionetti et al. 2019) has also been shown to activate Nrf2 in the myocardium, as well as across a variety of tissues, and could be advised to frail older people to reduce the risk to develop major CV complications in the case of SARS-CoV-2 infection (McPhee et al. 2016; Done and Traustadóttir 2016).
Correlation between PM exposure, CVD and COVID-19: just a conjecture?
Preliminary evidence has been provided in favor of a relationship between the number of days of exceeding the limits for particulate matter (PM) 10 (i.e., fine material with an airborne diameter < 10 μm) recorded in the control units of some cities, and the number of hospitalizations from COVID-19 in Northern Italy (Setti et al. 2020), the area of this country which suffered the highest number of cases and fatalities. The PM is worldwide recognized as an important prompt for CVD, as the inhalation of PM is certainly a trigger for multiple CV disorders, such as AMI, heart failure, and stroke, in urban communities (Liang et al. 2020). Inhalation of smaller PM, such as PM 2.5 (i.e., fine material with an airborne diameter < 2.5 μm), leads to pulmonary inflammation and cardiotoxicity via secondary systemic effects (Mills et al. 2009) as it can easily reach the alveolar space by irritating the alveolar wall up to onset of lung dysfunction (Xing et al. 2016). Cohen and colleagues have reported that more than four million deaths are attributable to PM 2.5, with nearly 60% of these due to CVDs (Cohen et al. 2017). This link is emphasized in the countries where air pollution concentration is continuing to rise (Mills et al. 2009). The exposure to PM is still associated with the incidence of coronary events in Europe even though the limit value was set at 50 μg/m3 daily (Cesaroni et al. 2014). The scientific literature reports that air pollution and PM has been associated with a plethora of CV events such as angina, AMI, arrhythmia and heart failure (Dockery et al. 1993; Samet et al. 2000; Peters et al. 2001; Mann et al. 2002; Brook et al. 2003; Bhatnagar 2006; Mills et al. 2009). Hence, any comorbidity with other pathologies as obesity and hypertension seems to impact the CVD-associated mortality in adults who are daily exposed to higher levels of PM (Mazidi and Speakman 2018). A time-stratified case-crossover study design evaluated, in the Milan area (Lombardy region, Italy) during the winter period 2016–2017, the independent short-term effect of flu outbreaks, air pollution and temperature (Murtas and Russo 2019). They observed a statistically significant relationship between PM 10 levels and flu for CVD mortality and between flu and temperature for deaths by natural causes. In Wuhan region (China), where air quality index is extremely low and the average daily PM concentration is 115.60 μg/m3, similar association between air pollution (mainly NO2) and CVD mortality during 4-year period (2006–2009) has been observed, albeit no correlation was found directly among PM 10 and CVD (Liu et al. 2015). A positive relationship between PM 2.5 and influenza-like illness at the flu season, reported for the period January 2008–December 2014, was then observed via time-series analysis in Beijing (Feng et al. 2016).
Both Lombardy and Wuhan areas are hitherto clusters of the worldwide pandemic of COVID-19 with higher number of people deceased in contrast with the rest of the nations (Italy and China) where the virus also spread but with less aggressiveness (as to March 27). Chen at al. provided evidence that avian influenza H5N1 may be subjected to intercontinental transportation via Saharian dust (Chen et al. 2010). Thus, one wonders if air pollution has an impact upon COVID-19 spread (Ciencewicki and Jaspers 2007). A position paper of the Italian Society of Environmental Medicine proposed that PM could represent a factor that favors long-term persistence of viruses in the atmosphere (http://www.simaonlus.it/wpsima/wp-content/uploads/2020/03/COVID_19_position-paper_ENG.pdf). Indeed, only for the Lombardy region, the number of infected persons before quarantine and lockdown and the mean number of PM 10 exceedances/monitoring stations results in a R2 correlation of 0.974.
The study concluded that there is a tangible risk that PM may serve as carrier for the viruses in the Northern regions of Italy, whereas further investigation and more solid data are necessary to disentangle this hypothesis. To strengthen this hypothesis, the exceedance in PM during the period February 10–29 2020 and the number of COVID-19 cases reported as to March 26 from the Italian National Institute of Health were visually correlated. It is clear from this analysis that, in the selected provinces in Lombardy, Piedmont, and Emilia-Romagna, where the majority of COVID19 cases occurred so far, the PM 10 exceedances increment was higher. While these preliminary data hint at a correlation between air pollution, which represents an established risk factor, and COVID-19, further in-depth retrospective cross-sectional investigations are required, keeping in mind the quarantine have drastically reduced the PM concentration in the aforementioned regions (https://www.eea.europa.eu/highlights/air-pollution-goes-down-as).
In search for an effective treatment: a proposal for repurposing of anti-hypertensive drugs to treat COVID-19
The COVID-19 outbreak poses a complex challenge to the biomedical community: to design an immediate and effective therapy to reduce the high fatality rate among patients, especially those older and/or with comorbidities. At present, no specific antiviral drugs provided satisfactory outcomes in the treatment of the disease, whereas a suitable large-scale vaccine is still far from being available. In this regard, it is worth noticing that even SARS-CoV, a pandemic emerged about 20 years ago (2002–2003), still awaits specific antivirals and approved vaccines.
Conventional safety protocols, including quarantine of affected individuals, tracing of patients’ contacts, social distancing, self-monitoring, use of masks and travel restriction, were adopted to mitigate the spread of COVID-19. Nevertheless, this approach will not be sufficient to constrain disease propagation when the basic reproduction number (R0) is not lower than 1.5 and/or the percentage of identified contacts is not higher than 80% (Mitjà and Clotet 2020). Under this scenario, a growing number of patients throughout the globe require an effective treatment.
Repurposing of clinically available antivirals to treat COVID-19
As already described in the case of Ebola and SARS-CoV outbreaks (Kruse 2020), repurposing of clinically approved drugs has been invocated to effectively treat patients who are at greater risk of death. For instance, the raising hope that lopinavir and ritonavir, which are employed as human immunodeficiency virus (HIV) protease inhibitors, could display clinical efficacy against COVID-19, was recently dashed by the failure of a clinical trial in China (Cao et al. 2020). As reviewed in Li and De Clercq (Li and De Clercq 2020), clinical trials were already launched in China and USA to evaluate the therapeutic efficacy of multiple Food and Drug Administration (FDA)-approved antiviral treatments, including the nucleoside analogues, favipiravir and ribavirin, and the experimental nucleoside analogue, remdesivir (NCT04280705) (Holshue et al. 2020). The list of antiviral agents that were proposed in the fight against COVID-19 include oseltamivir, penciclovir/acyclovir, ganciclovir, sarilumab, galidesivir, nitazoxanide, rapamycin, recombinant interferons, and chlorpromazine (Li and De Clercq 2020; Guo et al. 2020b).
Of note, a randomized controlled clinical trial of sarilumab (NCT04315298), a monoclonal antibody against the IL-6 receptor, is currently assessing whether the modification of the inflammatory response induced by this treatment could provide benefit in reducing lung inflammation and improving lung function in COVID-19 patients.
Blocking SARS-CoV-2 entry into the cytoplasm as a target for vaccines and plasma-derived therapy
An alternative, but not mutually exclusive, strategy consists in the identification of a small molecule drug that could be repurposed to inhibit the interaction between virus and target cells (Gunaratne et al. 2018b; Kruse 2020). As mentioned in Section 3, SARS-CoV-2 entry requires priming of S protein by furin and TMPRSS2 (Hoffmann et al. 2020; Walls et al. 2020; Coutard et al. 2020). A promising report revealed that camostat mesylate, an FDA-approved drug which blocks TMPRSS2 activity, effectively prevented SARS-CoV-2 spread in vitro (Hoffmann et al. 2020). In addition, once fully surface-exposed, S glycoprotein represents a source of accessible epitopes for the production of neutralizing antibodies and thus a target for ongoing vaccine design efforts (Walls et al. 2020). In this line, the use of antibody-rich plasma donated by patients who fully recovered from COVID-19 is under evaluation to determine if it could shorten the length, or lessen the severity, of the illness.
Blocking SARS-CoV-2 entry by targeting the endosomal compartment with chloroquine and Ca2+ antagonists
This finding opens the way to the repurposing of clinically available drugs interfering with the endocytic pathway mediating SARS-CoV-2 entry into the target cell, e.g., through inhibition of pH-mediated endocytosis, bulk alkalinization of endolysosomal (EL) vesicles, or blockade of EL proteases. For instance, FDA-approved cardiotonic steroids (ouabain and bufalin), known for their selective blockage on the Na+/K+-ATPase, can significantly impair MERS-CoV infection through the endocytic pathway (Burkard et al. 2015). Remarkably, the lysosomotropic compound, chloroquine, a cheap and widely available anti-malarial drug, was shown to block SARS-CoV-2 infection in vitro at a clinically relevant concentration (Wang et al. 2020c) and, according to preliminary reports, proved to exert beneficial exerts in COVID-19 patients (Colson et al. 2020; Mitjà and Clotet 2020; Gao et al. 2020; Inciardi et al. 2020). Chloroquine could indeed accumulate within the acidic vesicles and interfere with the EL vesicle trafficking (Golden et al. 2015). Of note, chloroquine, which is also employed in autoimmune disorders, has the potential to counteract the cytokine storm associated with COVID-19 severity (Liu et al. 2020a). Obviously, the therapeutic efficacy of chloroquine in COVID-19 patients’ needs to be supported by randomized clinical trials conducted on large cohorts of patients since the margin between the effective therapeutic dose and toxic dose is narrow and chloroquine poisoning has been associated with serious CV disorders (Frisk-Holmberg et al. 1983). Nevertheless, these preliminary evidences endorse the view that, the usage of a single compound potentially able to block viral-host cell interaction and attenuate the inflammatory response, could provide an effective weapon against the disease.
In this view, a hint at an alternative therapy against COVID-19 is suggested by the CV comorbidities affecting these patients. Ca2+ antagonists have long been used as first line anti-hypertensive drugs due to their ability to block L-type voltage-gated Ca2+ channels in vascular smooth muscle cells (Godfraind 2017; Oparil et al. 2018). Ca2+ antagonists include dihydropyridines, e.g., nifedipine, nicardipine, and nimodipine, as well as phenylalkylamines, e.g., verapamil, and the benzothiazepine, diltiazem (Godfraind 2017; Rosenfeldt et al. 2018). Nifedipine, verapamil, and diltiazem, often used in combination with ACE inhibitors or sartans, reduce arterial hypertension with quite tolerable side effects (Godfraind 2017; Rosenfeldt et al. 2018). Furthermore, nicardipine is strongly recommended as antihypertensive agent in stroke (Rosenfeldt et al. 2018), whereas verapamil is largely employed to treat both hypertension and ventricular arrhythmia (Godfraind 2017). It has long been known that Ca2+ antagonists may block endolysosomal (EL) two-pore channels 1–2 (TPC1–2) (Genazzani et al. 1997; Moccia et al. 2003), which play a crucial role in EL trafficking (Patel 2015), at the same concentrations as those reported to block L-type voltage-gated Ca2+ channels. TPCs are gated by multiple agonists, including nicotinic acid adenine dinucleotide phosphate (NAADP), sphingosine, and the EL phospholipid, phosphatidylinositol 3,5-bisphosphate (Galione 2019). TPCs-mediated Ca2+ release recruits EL-located Ca2+-sensitive decoders, including calmodulin, synaptotagmin VII, and Arf6 (Faris et al. 2018), to regulate vesicular fusion and trafficking (Galione 2019). A screening campaign aiming at repurposing FDA-approved drugs as novel antiviral compounds recently revealed that classical Ca2+ antagonists blocked Ebola virus and MERS-CoV infections in vitro (Sakurai et al. 2015; Gunaratne et al. 2018a, b). Ebola virus entry into host cells occurs by macropinocytosis followed by relocation to the endosomal compartment. Thereafter, the virus binds to the endosomal membrane protein NPC1 to fuse with acidic late endosomes/lysosomes and release the capside into the cytoplasm (Falzarano and Feldmann 2015). A recent series of investigations demonstrated that inhibiting TPCs-mediated EL Ca2+ release with nifedipine, verapamil, nicardipine, and diltiazem prevented viral genome entry in the cytoplasm (Sakurai et al. 2015; Penny et al. 2019). Subsequently, the same strategy was worked out to find clinically available tools to treat MERS, which was first described in Saudi Arabia in 2012 (Zaki et al. 2012) and subsequently reported to affect more than 1500 subjects (Zumla et al. 2015). As described above for SARS-CoV-2, the viral S protein mediates MERS-CoV entry upon binding to the host cell receptor, dipeptidyl peptidase 4, followed by proteolytic priming via TMPRSS2 (Raj et al. 2013; Park et al. 2016). Following clathrin-mediated endocytosis, MERS-CoV is redirected through the EL system where the S protein is proteolytically cleaved by lysosomal proteases, which results in vesicular fusion and capside release into the cytoplasm (Millet and Whittaker 2014; Burkard et al. 2014). Notably, MERS-CoV pseudovirus translocation was also attenuated by blocking EL TPCs with verapamil, nicardipine, and nimodipine (Gunaratne et al. 2018b). TPCs function as homodimers, in which each subunit comprises two homologous six-transmembrane domain motifs. Therefore, they represent the molecular ancestors of the α (CaV) subunits of voltage-gated Na+ and Ca2+ channels, with which they share the structural motifs within the pore regions (Patel 2015). In the same line, docking analysis and Ca2+ fluxes measurements confirmed that Ca2+ antagonists avidly bind to the TPC pore (Rahman et al. 2014).
As a further support to the proposal to probe Ca2+ antagonists to treat COVID-19, it is worth recalling that chloroquine inhibits SARS-CoV-2 virus entry by promoting the alkalinization of EL vesicles (Wang et al. 2020c; Liu et al. 2020a), which is a common strategy to perturb EL membrane fusion. Notably, elevating EL pH would also result in a significant reduction in EL Ca2+ concentration, usually around 500 μM, as EL Ca2+ refilling is mainly driven by the H+ gradient (Morgan et al. 2011; Faris et al. 2018). For instance, the lysomotropic weak base NH4Cl abrogates EL Ca2+ storage (Morgan et al. 2011) as well as MERS-CoV (Gunaratne et al. 2018b) and SARS-CoV (Yang et al. 2004; Vincent et al. 2005) entry in the cytoplasm. Furthermore, lysosomotropic agents also affected ACE2 localization “in vitro”; the viral receptor remains trapped within perinuclear vacuoles upon pretreatment with chloroquine, NH4Cl or Bafilomycin A1 (Wang et al. 2008).
Besides supporting viral entry into the host cells, preliminary evidence suggested that TPCs contribute to deliver the EL Ca2+ signal responsible for NLRP3 inflammasome activation (Tseng et al. 2017). It thus appears that blocking TPCs-mediated EL Ca2+ release, with either chloroquine or Ca2+ antagonists, might represent a potential perspective to treat COVID-19 patients by interfering with both SARS-CoV-2 infection and the subsequent inflammatory response. While this hypothesis remains to be probed by a devoted experimental pipeline, a clinical trial has been launched in China to evaluate the therapeutic outcome of tetrandrine on SARS-CoV-2-infected subjects (ClinicalTrials.gov Identifier: NCT04308317). Tetrandrine is an alkaloid isolated from the root of Stephania tetrandra, a vine that grows in China and Taiwan, and is widely employed in China to treat autoimmune diseases, CV disorders, hypertension, and cancer. Tetrandrine targets multiple intracellular signaling pathways, including L-type voltage-gated Ca2+ channels, which could explain its beneficial effects on the CV system (Yao and Jiang 2002). Of note, tetrandrine was recently shown to inhibit TPCs-mediated EL Ca2+ release and to attenuate Ebola (Sakurai et al. 2015) and MERS-CoV-2 (Gunaratne et al. 2018b) entry into the host cells. These findings, however, may suggest the hypothesis that repurposing of Ca2+ antagonists could prove effective in treating SARS-CoV-2 infection while, at the same time, attenuating CV and respiratory dysfunction co-occurring in older COVID-19 patients.
Conclusions
The COVID-19 pandemic has rapidly spread throughout the globe by preferentially affecting older individuals with underlying CV comorbidities. Besides ARDS and acute kidney injury, cardiovascular complications also represent a common consequence of the disease and, at the same time, a cause for adverse outcome in COVID-19 patients. Although we cannot ignore emerging events of drug-induced sudden cardiac death among treated COVID-19 patients (Chorin et al. 2020), clinical reports well support careful evaluation of the pathogenic mechanisms that promote the onset of severe COVID-19 due to CVD following SARS-CoV-2 infection. These include, for example, classical RAS pathway and ACE2, NLRP3/inflammasome, and hypoxemia (Fig. 1). Furthermore, the prevalence and death toll of COVID-19 increases with aging as well as frailty, and seems correlated with prolonged exposure to air pollution, both of which represent life-threatening CV risk factors. In our opinion, understanding the cellular and molecular mechanisms that exacerbate the impact of COVID-19 in patients with existing CV pathologies and lead to major cardiac complications is mandatory to design a truly cardioprotective therapy while waiting for the vaccine.

Simplified scheme showing the potential role and regulation of intracellular organelles as calcium stores involved in the activation of inflammasome and the following triggering of the cytokine storm. Inset (top): the complex molecular mechanism at the plasma membrane level, underlying the different functional outcomes of COVID-19 infection. VP viroporins, CaA calcium antagonists, TD tetrandrine, CC chloroquine, TPC two pore channels, TMPRSS2 serine protease, EL endolysosomes, ACE angiotensin-converting enzyme, AT angiotensin II receptor, Ang angiotensin, APJ apelin G protein coupled receptor, MAS angiopoietin 1–7 receptor, e-vWF endothelial von Willebrand factor, ET1 endothelin 1, NO nitric oxide
References
Afilalo J, Alexander KP, Mack MJ, et al. Frailty assessment in the cardiovascular care of older adults. J Am Coll Cardiol. 2014;63:747–62.
AlGhatrif M, Cingolani O, Lakatta EG. The dilemma of coronavirus disease 2019, aging, and cardiovascular disease. JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.1329.
Ando T, Mikawa K, Nishina K, et al. Hypocapnic alkalosis enhances oxidant-induced apoptosis of human alveolar epithelial type II cells. J Int Med Res. 2007;35:118–26. https://doi.org/10.1177/147323000703500113.
Auer RN. Hypoglycemic brain damage. Forensic Sci Int. 2004;146:105–10. https://doi.org/10.1016/j.forsciint.2004.08.001.
Bei Y, Zhou Q, Sun Q, Xiao J. Exercise as a platform for pharmacotherapy development in cardiac diseases. Curr Pharm Des. 2015;21:4409–16. https://doi.org/10.2174/1381612821666150803150008.
Bellumkonda L, Tyrrell D, Hummel SL, Goldstein DR. Pathophysiology of heart failure and frailty: a common inflammatory origin? Aging Cell. 2017;16:444–50. https://doi.org/10.1111/acel.12581.
Benderro GF, Sun X, Kuang Y, Lamanna JC. Decreased VEGF expression and microvascular density, but increased HIF-1 and 2α accumulation and EPO expression in chronic moderate hyperoxia in the mouse brain. Brain Res. 2012;1471:46–55. https://doi.org/10.1016/j.brainres.2012.06.055.
Bhatnagar A. Environmental cardiology. Circ Res. 2006;99:692–705. https://doi.org/10.1161/01.RES.0000243586.99701.cf.
Bodor GS. Biochemical markers of myocardial damage. EJIFCC. 2016;27:95–111.
Boengler K, Schulz R, Heusch G. Loss of cardioprotection with ageing. Cardiovasc Res. 2009;83:247–61. https://doi.org/10.1093/cvr/cvp033.
Bonow RO, Fonarow GC, O’Gara PT, Yancy CW. Association of coronavirus disease 2019 (COVID-19) with myocardial injury and mortality. JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.1105.
Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol. 2008;8:911–22.
Brook RD, Brook JR, Rajagopalan S. Air pollution: the “heart” of the problem. Curr Hypertens Rep. 2003;5:32–9. https://doi.org/10.1007/s11906-003-0008-y.
Brutus NA, Hanley S, Ashraf QM, et al. Effect of hyperoxia on serine phosphorylation of apoptotic proteins in mitochondrial membranes of the cerebral cortex of newborn piglets. Neurochem Res. 2009. https://doi.org/10.1007/s11064-008-9898-z.
Bullón P, Cano-García FJ, Alcocer-Gómez E, et al. Could NLRP3-inflammasome be a cardiovascular risk biomarker in acute myocardial infarction patients? Antioxidants Redox Signal. 2017;27:269–75. https://doi.org/10.1089/ars.2016.6970.
Burkard C, Verheije MH, Haagmans BL, et al. ATP1A1-mediated Src signaling inhibits coronavirus entry into host cells. J Virol. 2015. https://doi.org/10.1128/jvi.03274-14.
Burkard C, Verheije MH, Wicht O, et al. Coronavirus cell entry occurs through the Endo−/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 2014;10:e1004502. https://doi.org/10.1371/journal.ppat.1004502.
Cao B, Wang Y, Wen D, et al. A trial of lopinavir-ritonavir in adults hospitalized with severe Covid-19. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2001282.
Carro A, Bastiaenen R, Kaski JC. Age related issues in reperfusion of myocardial infarction. Cardiovasc Drugs Ther. 2011;25:139–48.
Castaño-Rodriguez C, Honrubia JM, Gutiérrez-Álvarez J, et al. Role of severe acute respiratory syndrome coronavirus viroporins E, 3a, and 8a in replication and pathogenesis. MBio. 2018;9. https://doi.org/10.1128/mBio.02325-17.
Cesaroni G, Forastiere F, Stafoggia M, et al. Long term exposure to ambient air pollution and incidence of acute coronary events: prospective cohort study and meta-analysis in 11 European cohorts from the ESCAPE Project. BMJ. 2014;348:f7412–2. https://doi.org/10.1136/bmj.f7412.
Chamsi-Pasha MAR, Shao Z, Tang WHW. Angiotensin-converting enzyme 2 as a therapeutic target for heart failure. Curr Heart Fail Rep. 2014;11:58–63. https://doi.org/10.1007/s11897-013-0178-0.
Chan JF-W, Kok K-H, Zhu Z, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9:221–36. https://doi.org/10.1080/22221751.2020.1719902.
Chen CC, Krüger J, Sramala I, et al. ORF8a of SARS-CoV forms an ion channel: experiments and molecular dynamics simulations. Biochim Biophys Acta Biomembr. 2011;1808:572–9. https://doi.org/10.1016/j.bbamem.2010.08.004.
Chen I-YY, Moriyama M, Chang M-FF, Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol. 2019;10:50. https://doi.org/10.3389/fmicb.2019.00050.
Chen L, Li X, Chen M, et al. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res. 2020a. https://doi.org/10.1093/cvr/cvaa078.
Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020b;395:507–13. https://doi.org/10.1016/S0140-6736(20)30211-7.
Chen P-S, Tsai FT, Lin CK, et al. Ambient influenza and avian influenza virus during dust storm days and background days. Environ Health Perspect. 2010;118:1211–6. https://doi.org/10.1289/ehp.0901782.
Chorin E, Dai M, Shulman E, et al. The QT interval in patients with COVID-19 treated with hydroxychloroquine and azithromycin. Nat Med. 2020. https://doi.org/10.1038/s41591-020-0888-2.
Ciencewicki J, Jaspers I. Air pollution and respiratory viral infection. Inhal Toxicol. 2007;19:1135–46.
Clegg A, Young J, Iliffe S, et al. Frailty in elderly people. Lancet. 2013;381:752–62. https://doi.org/10.1016/S0140-6736(12)62167-9.
Cohen AJ, Brauer M, Burnett R, et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet. 2017. https://doi.org/10.1016/S0140-6736(17)30505-6.
Collard RM, Boter H, Schoevers RA, Oude Voshaar RC. Prevalence of frailty in community-dwelling older persons: a systematic review. J Am Geriatr Soc. 2012;60:1487–92. https://doi.org/10.1111/j.1532-5415.2012.04054.x.
Colson P, Rolain J-M, Lagier J-C, et al. Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int J Antimicrob Agents. 2020. https://doi.org/10.1016/j.ijantimicag.2020.105932.
Corrales-Medina VF, Musher DM, Shachkina S, Chirinos JA. Acute pneumonia and the cardiovascular system. Lancet. 2013;381:496–505.
Coutard B, Valle C, de Lamballerie X, et al. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir Res. 2020;176:104742. https://doi.org/10.1016/j.antiviral.2020.104742.
Crackower MA, Sarao R, Oliveira-dos-Santos AJ, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–8. https://doi.org/10.1038/nature00786.
Csiszár A, Csiszar A, Pinto JT, et al. Resveratrol encapsulated in novel fusogenic liposomes activates Nrf2 and attenuates oxidative stress in cerebromicrovascular endothelial cells from aged rats. J Gerontol A Biol Sci Med Sci. 2015;70:303–13. https://doi.org/10.1093/gerona/glu029.
D’Adamo H, Yoshikawa T, Ouslander JG. Coronavirus disease 2019 in geriatrics and long-term care: the ABCDs of COVID-19. J Am Geriatr Soc. 2020. https://doi.org/10.1111/jgs.16445.
Dalekos GN, Elisaf M, Bairaktari E, et al. Increased serum levels of interleukin-1beta in the systemic circulation of patients with essential hypertension: additional risk factor for atherogenesis in hypertensive patients? J Lab Clin Med. 1997;129:300–8. https://doi.org/10.1016/s0022-2143(97)90178-5.
Davis BK, Wen H, Ting JP-Y. The Inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–35. https://doi.org/10.1146/annurev-immunol-031210-101405.
De Wit E, Van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14:523–34.
Dijkman R, Jebbink MF, Deijs M, et al. Replication-dependent downregulation of cellular angiotensin-converting enzyme 2 protein expression by human coronavirus NL63. J Gen Virol. 2012. https://doi.org/10.1099/vir.0.043919-0.
Doan TN, Gletsu N, Cole J, Bernstein KE. Genetic manipulation of the renin-angiotensin system. Curr Opin Nephrol Hypertens. 2001;10:483–91.
Dockery DW, Pope CA, Xu X, et al. An association between air pollution and mortality in six U.S. cities. N Engl J Med. 1993;329:1753–9. https://doi.org/10.1056/NEJM199312093292401.
Done AJ, Traustadóttir T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016;10:191–9.
Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ res 87. 2000. https://doi.org/10.1161/01.res.87.5.e1.
Dushpanova A, Agostini S, Ciofini E, et al. Gene silencing of endothelial von Willebrand factor attenuates angiotensin II-induced endothelin-1 expression in porcine aortic endothelial cells. Sci Rep. 2016;6:1–12. https://doi.org/10.1038/srep30048.
Epstein FH, Dinarello CA. Interleukin-1 and the pathogenesis of the acute-phase response. N Engl J Med. 1984;311:1413–8.
Esler M, Esler D. Can angiotensin receptor-blocking drugs perhaps be harmful in the COVID-19 pandemic? J Hypertens. 2020;38:1–2. https://doi.org/10.1097/HJH.0000000000002450.
Falzarano D, Feldmann H. Delineating Ebola entry. Science. 2015;347:947–8. https://doi.org/10.1126/science.aaa8121.
Fan Z, Wu G, Yue M, et al. Hypertension and hypertensive left ventricular hypertrophy are associated with ACE2 genetic polymorphism. Life Sci. 2019;225:39–45. https://doi.org/10.1016/j.lfs.2019.03.059.
Farag NS, Breitinger U, Breitinger HG, El Azizi MA. Viroporins and inflammasomes: a key to understand virus-induced inflammation. Int J Biochem Cell Biol. 2020;122:105738.
Farag NS, Breitinger U, El-Azizi M, Breitinger HG. The p7 viroporin of the hepatitis C virus contributes to liver inflammation by stimulating production of interleukin-1β. Biochim Biophys Acta - Mol Basis Dis. 2017;1863:712–20. https://doi.org/10.1016/j.bbadis.2016.12.006.
Faris P, Shekha M, Montagna D, et al. Endolysosomal Ca2+ signalling and cancer hallmarks: two-pore channels on the move, TRPML1 lags behind! Cancers (Basel). 2018;11:27. https://doi.org/10.3390/cancers11010027.
Favre S, Gambini E, Nigro P, et al. Sildenafil attenuates hypoxic pulmonary remodelling by inhibiting bone marrow progenitor cells. J Cell Mol Med. 2017;21:871–80. https://doi.org/10.1111/jcmm.13026.
Felderhoff-Mueser U, Bittigau P, Sifringer M, et al. Oxygen causes cell death in the developing brain. Neurobiol Dis. 2004;17:273–82. https://doi.org/10.1016/j.nbd.2004.07.019.
Feng C, Li J, Sun W, et al (2016) Impact of ambient fine particulate matter (PM2.5) exposure on the risk of influenza-like-illness: a time-series analysis in Beijing, China. Environ heal 15:17. https://doi.org/10.1186/s12940-016-0115-2.
Ferreira AJ, Jacoby BA, Araújo CAA, et al. The nonpeptide angiotensin-(1–7) receptor Mas agonist AVE-0991 attenuates heart failure induced by myocardial infarction. Am J Physiol Circ Physiol. 2007;292:H1113–9. https://doi.org/10.1152/ajpheart.00828.2006.
Fleg JL, Aronow WS, Frishman WH. Cardiovascular drug therapy in the elderly: benefits and challenges. Nat Rev Cardiol. 2011;8:13–28. https://doi.org/10.1038/nrcardio.2010.162.
Fontes JA, Rose NR, Čiháková D. The varying faces of IL-6: from cardiac protection to cardiac failure. Cytokine. 2015;74:62–8.
Franchi L, Núñez G. Orchestrating inflammasomes. Science. 2012;337:1299–300.
Frisk-Holmberg M, Bergqvist Y, Englund U. Chloroquine intoxication [letter]. Br J Clin Pharmacol. 1983;15:502–3. https://doi.org/10.1111/j.1365-2125.1983.tb01540.x.
Fulop GA, Kiss T, Tarantini S, et al. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. GeroScience. 2018;40:513–21. https://doi.org/10.1007/s11357-018-0047-6.
Fung G, Luo H, Qiu Y, et al. Myocarditis. Circ Res. 2016;118:496–514. https://doi.org/10.1161/CIRCRESAHA.115.306573.
Galesloot TE, Vermeulen SH, Geurts-Moespot AJ, et al. Serum hepcidin: reference ranges and biochemical correlates in the general population. Blood. 2011;117:e218–25. https://doi.org/10.1182/blood-2011-02-337907.
Galione A. NAADP receptors. Cold Spring Harb Perspect Biol. 2019;11:a035071. https://doi.org/10.1101/cshperspect.a035071.
Gao J, Tian Z, Yang X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci Trends. 2020;14:72–3. https://doi.org/10.5582/bst.2020.01047.
Garami AR. Preventing a covid-19 pandemic - is there a magic bullet to save COVID19 patients? We can give it a try! In: Br. Med. J; 2020. https://www.bmj.com/content/368/bmj.m810/rr-24.
Gassmann M, Muckenthaler MU. Adaptation of iron requirement to hypoxic conditions at high altitude. J Appl Physiol. 2015;119:1432–40.
Genazzani AA, Mezna M, Dickey DM, et al. Pharmacological properties of the ca 2+ −release mechanism sensitive to NAADP in the sea urchin egg. Br J Pharmacol. 1997;121:1489–95. https://doi.org/10.1038/sj.bjp.0701295.
Giannitsis E, Katus HA. Cardiac troponin level elevations not related to acute coronary syndromes. Nat Rev Cardiol. 2013;10:623–34.
Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–8.
Godfraind T. Discovery and development of calcium channel blockers. Front Pharmacol. 2017;8. https://doi.org/10.3389/fphar.2017.00286.
Goetze O, Schmitt J, Spliethoff K, et al. Adaptation of iron transport and metabolism to acute high-altitude hypoxia in mountaineers. Hepatology. 2013. https://doi.org/10.1002/hep.26581.
Golden EB, Cho HY, Hofman FM, et al. Quinoline-based antimalarial drugs: a novel class of autophagy inhibitors. Neurosurg Focus. 2015;38:E12. https://doi.org/10.3171/2014.12.FOCUS14748.
Gorla R, Erbel R, Eagle KA, Bossone E. Systemic inflammatory response syndromes in the era of interventional cardiology. Vasc Pharmacol. 2018;107:53–66.
Guan W, Ni Z, Hu Y, et al (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J med NEJMoa2002032. https://doi.org/10.1056/NEJMoa2002032.
Gui L, Liu B, Lv G. Hypoxia induces autophagy in cardiomyocytes via a hypoxia-inducible factor 1-dependent mechanism. Exp Ther Med. 2016;11:2233–9. https://doi.org/10.3892/etm.2016.3190.
Gunaratne GS, Johns ME, Hintz HM, et al. A screening campaign in sea urchin egg homogenate as a platform for discovering modulators of NAADP-dependent Ca2+ signaling in human cells. Cell Calcium. 2018a;75:42–52. https://doi.org/10.1016/j.ceca.2018.08.002.
Gunaratne GS, Yang Y, Li F, et al. NAADP-dependent Ca2+ signaling regulates Middle East respiratory syndrome-coronavirus pseudovirus translocation through the endolysosomal system. Cell Calcium. 2018b;75:30–41. https://doi.org/10.1016/j.ceca.2018.08.003.
Guo H-C, Jin Y, Zhi X-Y, et al (2015) NLRP3 inflammasome activation by viroporins of animal viruses. Viruses 7:3380–3391. https://doi.org/10.3390/v7072777.
Guo T, Fan Y, Chen M, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020a. https://doi.org/10.1001/jamacardio.2020.1017.
Guo Y-R, Cao Q-D, Hong Z-S, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak – an update on the status. Mil Med Res. 2020b;7:1–10. https://doi.org/10.1186/s40779-020-00240-0.
Gurwitz D (2020) Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug dev res ddr.21656. https://doi.org/10.1002/ddr.21656.
Gyongyosi A, Terraneo L, Bianciardi P, et al. The impact of moderate chronic hypoxia and hyperoxia on the level of apoptotic and autophagic proteins in myocardial tissue. Oxidative Med Cell Longev. 2018;2018:1–12. https://doi.org/10.1155/2018/5786742.
Hamilton S, Terentyev D. Altered intracellular calcium homeostasis and arrhythmogenesis in the aged heart. Int J Mol Sci. 2019;20:2386. https://doi.org/10.3390/ijms20102386.
Hamming I, Timens W, Bulthuis MLC, et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–7. https://doi.org/10.1002/path.1570.
Hao P-P, Chen Y-G, Liu Y-P, et al. Association of plasma angiotensin-(1–7) level and left ventricular function in patients with type 2 diabetes mellitus. PLoS One. 2013;8:e62788. https://doi.org/10.1371/journal.pone.0062788.
Harmer D, Gilbert M, Borman R, Clark KL. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett. 2002;532:107–10. https://doi.org/10.1016/S0014-5793(02)03640-2.
Henry BM, Vikse J, Lippi G. COVID-19 induced renin–angiotensin system (RAS) imbalance may drive acute lung injury: the evidence and therapeutic options. Br Med J. 2020; https://www.bmj.com/content/368/bmj.m406/rr-19.
Henry C, Zaizafoun M, Stock E, et al. Impact of angiotensin-converting enzyme inhibitors and statins on viral pneumonia. Baylor Univ Med Cent Proc. 2018;31:419–23. https://doi.org/10.1080/08998280.2018.1499293.
Hilliard LM, Sampson AK, Brown RD, Denton KM. The “his and hers” of the renin-angiotensin system. Curr Hypertens Rep. 2013;15:71–9. https://doi.org/10.1007/s11906-012-0319-y.
Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020. https://doi.org/10.1016/j.cell.2020.02.052.
Holappa M, Valjakka J, Vaajanen A. Angiotensin(1-7) and ACE2, “the hot spots” of renin-angiotensin system, detected in the human aqueous humor. Open Ophthalmol J. 2015;9:28–32. https://doi.org/10.2174/1874364101509010028.
Holshue ML, DeBolt C, Lindquist S, et al. First case of 2019 novel coronavirus in the United States. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2001191.
Horng T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014;35:253–61.
Hu H, Ma F, Wei X, Fang Y. Coronavirus fulminant myocarditis treated with glucocorticoid and human immunoglobulin. Eur Heart J. 2020. https://doi.org/10.1093/eurheartj/ehaa190.
Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. https://doi.org/10.1016/S0140-6736(20)30183-5.
Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol. 2010;11:404–10. https://doi.org/10.1038/ni.1861.
Imai Y, Kuba K, Rao S, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–6. https://doi.org/10.1038/nature03712.
Inciardi RM, Lupi L, Zaccone G, et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.1096.
Jenq CC, Tsai FC, Tsai TY, et al. Effect of Anemia on prognosis in patients on extracorporeal membrane oxygenation. Artif Organs. 2018. https://doi.org/10.1111/aor.13123.
Jin X, Lian J-S, Hu J-H, et al. Epidemiological, clinical and virological characteristics of 74 cases of coronavirus-infected disease 2019 (COVID-19) with gastrointestinal symptoms. Gut gutjnl-2020-320926. 2020. https://doi.org/10.1136/gutjnl-2020-320926.
Kerr MH, Paton JY. Surfactant protein levels in severe respiratory syncytial virus infection. Am J Respir Crit Care Med. 1999;159:1115–8. https://doi.org/10.1164/ajrccm.159.4.9709065.
Kiely DG, Cargill RI, Wheeldon NM, et al. Haemodynamic and endocrine effects of type 1 angiotensin II receptor blockade in patients with hypoxaemic cor pulmonale. Cardiovasc Res. 1997;33:201–8. https://doi.org/10.1016/S0008-6363(96)00180-0.
Kiss T, Balasubramanian P, Valcarcel-Ares MN, et al. Nicotinamide mononucleotide (NMN) treatment attenuates oxidative stress and rescues angiogenic capacity in aged cerebromicrovascular endothelial cells: a potential mechanism for the prevention of vascular cognitive impairment. GeroScience. 2019;41:619–30. https://doi.org/10.1007/s11357-019-00074-2.
Komatsu T, Suzuki Y, Imai J, et al. Molecular cloning, mRNA expression and chromosomal localization of mouse angiotensin-converting enzyme-related carboxypeptidase (mACE2). Mitochondrial DNA. 2002;13:217–20. https://doi.org/10.1080/1042517021000021608.
Kramer BK, Ritthaler T, Schweda F, et al. Effects of hypoxia on renin secretion and renal renin gene expression. In: Kidney international. Supplement: Elsevier; 1998. p. S155–8.
Kreü S, Jazrawi A, Miller J, et al. Alkalosis in critically ill patients with severe sepsis and septic shock. PLoS One. 2017;12:e0168563. https://doi.org/10.1371/journal.pone.0168563.
Krishnan SM, Sobey CG, Latz E, et al. IL-1β and IL-18: inflammatory markers or mediators of hypertension? Br J Pharmacol. 2014;171:5589–602.
Kruse RL (2020) Therapeutic strategies in an outbreak scenario to treat the novel coronavirus originating in Wuhan, China. F1000Research 9:72.
Kuba K, Imai Y, Penninger JM. Multiple functions of angiotensin-converting enzyme 2 and its relevance in cardiovascular diseases. Circ J. 2013;77:301–8. https://doi.org/10.1253/circj.cj-12-1544.
Kuster GM, Pfister O, Burkard T, et al. SARS-CoV2: should inhibitors of the renin–angiotensin system be withdrawn in patients with COVID-19? Eur Heart J. 2020. https://doi.org/10.1093/eurheartj/ehaa235.
Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020:1–9. https://doi.org/10.1038/s41586-020-2180-5.
Lasocki S, Baron G, Driss F, et al. Diagnostic accuracy of serum hepcidin for iron deficiency in critically ill patients with anemia. Intensive Care Med. 2010. https://doi.org/10.1007/s00134-010-1794-8.
Lee YJ, Lee H, Park JSJS, et al. Cardiac troponin I as a prognostic factor in critically ill pneumonia patients in the absence of acute coronary syndrome. J Crit Care. 2015;30:390–4. https://doi.org/10.1016/j.jcrc.2014.12.001.
Li G, De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat Rev Drug Discov. 2020;19:149–50.
Li Q, Han X, Lan X, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI insight. 2017;2:e90777. https://doi.org/10.1172/jci.insight.90777.
Li W, Zhang C, Sui J, et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24:1634–43. https://doi.org/10.1038/sj.emboj.7600640.
Liang F, Liu F, Huang K, et al. Long-term exposure to fine particulate matter and cardiovascular disease in China. J Am Coll Cardiol. 2020;75:707–17. https://doi.org/10.1016/j.jacc.2019.12.031.
Lindsey ML. Assigning matrix metalloproteinase roles in ischaemic cardiac remodelling. Nat Rev Cardiol. 2018;15:471–9.
Lionetti V, Tuana SB, Casieri V, et al. Importance of functional food compounds in cardioprotection through action on the epigenome. Eur Heart J. 2019;40:575–82. https://doi.org/10.1093/eurheartj/ehy597.
Lionetti V, Recchia FA, Ranieri VM. Overview of ventilator-induced lung injury mechanisms. Curr Opin Crit Care. 2005;11:82–6.
Liu D, Zeng X, Li X, et al. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. 2018;113:5.
Liu J, Cao R, Xu M, et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020a;6:1–4.
Liu J, Wei T, Kwang J. Avian encephalomyelitis virus nonstructural protein 2C induces apoptosis by activating cytochrome c/caspase-9 pathway. Virology. 2004;318:169–82. https://doi.org/10.1016/j.virol.2003.09.012.
Liu Y, Chen X, Huang S, et al. Association between air pollutants and cardiovascular disease mortality in Wuhan, China. Int J Environ Res Public Health. 2015;12:3506–16. https://doi.org/10.3390/ijerph120403506.
Liu Y, Yang Y, Zhang C, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020b;63:364–74. https://doi.org/10.1007/s11427-020-1643-8.
Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–74. https://doi.org/10.1016/S0140-6736(20)30251-8.
Madjid M, Safavi-Naeini P, Solomon SD, Vardeny O. Potential effects of coronaviruses on the cardiovascular system. JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.1286.
Mæhle K, Haug B, Flaatten H, Nielsen E. Metabolic alkalosis is the most common acid–base disorder in ICU patients. Crit Care. 2014;18:420. https://doi.org/10.1186/cc13802.
Mahallawi WH, Khabour OF, Zhang Q, et al. MERS-CoV infection in humans is associated with a pro-inflammatory Th1 and Th17 cytokine profile. Cytokine. 2018;104:8–13. https://doi.org/10.1016/j.cyto.2018.01.025.
Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. 2015;116:1254–68.
Mann JK, Tager IB, Lurmann F, et al. Air pollution and hospital admissions for ischemic heart disease in persons with congestive heart failure or arrhythmia. Environ Health Perspect. 2002. https://doi.org/10.1289/ehp.021101247.
Marconi GD, Zara S, De Colli M, et al. Postnatal hyperoxia exposure differentially affects hepatocytes and liver haemopoietic cells in newborn rats. PLoS One. 2014. https://doi.org/10.1371/journal.pone.0105005.
Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14:10–22.
Matsuda A, Kishi T, Jacob A, et al. Association between insertion/deletion polymorphism in angiotensin-converting enzyme gene and acute lung injury/acute respiratory distress syndrome: a meta-analysis. BMC Med Genet. 2012;13:76. https://doi.org/10.1186/1471-2350-13-76.
Mazidi M, Speakman JR. Impact of obesity and ozone on the association between particulate air pollution and cardiovascular disease and stroke mortality among US adults. J Am Heart Assoc. 2018. https://doi.org/10.1161/JAHA.117.008006.
McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114:1417–31.
McPhee JS, French DP, Jackson D, et al. Physical activity in older age: perspectives for healthy ageing and frailty. Biogerontology. 2016;17:567–80.
Milano G, Corno AF, Samaja M, et al. Daily reoxygenation decreases myocardial injury and improves post-ischaemic recovery after chronic hypoxia☆. Eur J Cardio-Thoracic Surg. 2010;37:942–9. https://doi.org/10.1016/j.ejcts.2009.10.030.
Millet JK, Whittaker GR. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci. 2014;111:15214–9. https://doi.org/10.1073/pnas.1407087111.
Mills NL, Donaldson K, Hadoke PW, et al. Adverse cardiovascular effects of air pollution. Nat Clin Pract Cardiovasc Med. 2009;6:36–44. https://doi.org/10.1038/ncpcardio1399.
Mitjà O, Clotet B. Use of antiviral drugs to reduce COVID-19 transmission. Lancet Glob Heal. 2020. https://doi.org/10.1016/S2214-109X(20)30114-5.
Moccia F, Lim D, Nusco GA, et al. NAADP activates a Ca2+ current that is dependent on F-actin cytoskeleton. FASEB J. 2003;17:1907–9. https://doi.org/10.1096/fj.03-0178fje.
Moccia F, Lucariello A, Guerra G. TRPC3-mediated Ca 2+ signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: the role of autologous endothelial colony forming cells. J Cell Physiol. 2018;233:3901–17. https://doi.org/10.1002/jcp.26152.
Morgan AJ, Platt FM, Lloyd-Evans E, Galione A. Molecular mechanisms of endolysosomal Ca 2+ signalling in health and disease. Biochem J. 2011;439:349–74.
Mullard A. NLRP3 inhibitors stoke anti-inflammatory ambitions. Nat Rev Drug Discov. 2019;18:405–7.
Muñoz-Planillo R, Kuffa P, Martínez-Colón G, et al. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–53. https://doi.org/10.1016/j.immuni.2013.05.016.
Murtas R, Russo AG. Effects of pollution, low temperature and influenza syndrome on the excess mortality risk in winter 2016–2017. BMC Public Health. 2019;19:1445. https://doi.org/10.1186/s12889-019-7788-8.
Nacarelli T, Azar A, Altinok O, et al. Rapamycin increases oxidative metabolism and enhances metabolic flexibility in human cardiac fibroblasts. GeroScience. 2018;40:243–56. https://doi.org/10.1007/s11357-018-0030-2.
Nagatomo F, Fujino H, Kondo H, Ishihara A. Oxygen concentration-dependent oxidative stress levels in rats. Oxidative Med Cell Longev. 2012;2012:1–5. https://doi.org/10.1155/2012/381763.
Netzer NC, Strohl KP, Högel J, et al. Right ventricle dimensions and function in response to acute hypoxia in healthy human subjects. Acta Physiol. 2017;219:478–85. https://doi.org/10.1111/apha.12740.
Newby AC. Metalloproteinase production from macrophages - a perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp Physiol. 2016;101:1327–37. https://doi.org/10.1113/EP085567.
Nguyen B-V, Bota DP, Mélot C, Vincent J-L. Time course of hemoglobin concentrations in nonbleeding intensive care unit patients. Crit Care Med. 2003;31:406–10. https://doi.org/10.1097/01.CCM.0000048623.00778.3F.
Nieto-Torres JL, Verdiá-Báguena C, Castaño-Rodriguez C, et al. Relevance of viroporin ion channel activity on viral replication and pathogenesis. Viruses. 2015;7:3552–73.
Nieva JL, Madan V, Carrasco L. Viroporins: structure and biological functions. Nat Rev Microbiol. 2012;10:563–74.
Nydegger C, Corno AF, von Segesser LK, et al. Effects of PDE-5 inhibition on the cardiopulmonary system after 2 or 4 weeks of chronic hypoxia. Cardiovasc Drugs Ther. 2019;33:407–14. https://doi.org/10.1007/s10557-019-06887-9.
Nydegger C, Martinelli C, Di Marco F, et al. Phosphodiesterase-5 inhibition alleviates pulmonary hypertension and basal lamina thickening in rats challenged by chronic hypoxia. Front Physiol. 2018;9:289. https://doi.org/10.3389/fphys.2018.00289.
Obas V, Vasan RS. The aging heart. Clin Sci. 2018;132:1367–82.
Okada M, Matsuzawa A, Yoshimura A, Ichijo H. The lysosome rupture-activated TAK1-JNK pathway regulates NLRP3 inflammasome activation. J Biol Chem. 2014;289:32926–36. https://doi.org/10.1074/jbc.M114.579961.
Onder G, Rezza G, Brusaferro S. Case-fatality rate and characteristics of patients dying in relation to COVID-19 in Italy. JAMA - J Am Med Assoc doi. 2020. https://doi.org/10.1001/jama.2020.4683.
Oparil S, Acelajado MC, Bakris GL, et al. Hypertension. Nat Rev Dis Prim. 2018;4:18014.
Ottolenghi S, Maria Rubino F, Sabbatini G, et al. Oxidative stress markers to investigate the effects of hyperoxia in anesthesia. Int J Mol Sci. 2019;20. https://doi.org/10.3390/ijms20215492.
Ottolenghi S, Sabbatini G, Brizzolari A, et al (2020) Hyperoxia and oxidative stress in anesthesia and critical care medicine. Minerva Anestesiol 86:64–75. https://doi.org/10.23736/S0375-9393.19.13906-5.
Oudit GY, Herzenberg AM, Kassiri Z, et al. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclerosis. Am J Pathol. 2006;168:1808–20. https://doi.org/10.2353/ajpath.2006.051091.
Oudit GY, Kassiri Z, Jiang C, et al. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Investig. 2009;39:618–25. https://doi.org/10.1111/j.1365-2362.2009.02153.x.
Packer M, McMurray JJV. Importance of endogenous compensatory vasoactive peptides in broadening the effects of inhibitors of the renin-angiotensin system for the treatment of heart failure. Lancet. 2017;389:1831–40. https://doi.org/10.1016/S0140-6736(16)30969-2.
Pagliaro P, Moro F, Tullio F, et al. Cardioprotective pathways during reperfusion: focus on redox signaling and other modalities of cell signaling. Antioxid Redox Signal. 2011;14:833–50. https://doi.org/10.1089/ars.2010.3245.
Pagliaro P, Penna C. Rethinking the renin-angiotensin system and its role in cardiovascular regulation. Cardiovasc Drugs Ther. 2005;19:77–87. https://doi.org/10.1007/s10557-005-6900-8.
Park J-E, Li K, Barlan A, et al. Proteolytic processing of Middle East respiratory syndrome coronavirus spikes expands virus tropism. Proc Natl Acad Sci. 2016;113:12262–7. https://doi.org/10.1073/pnas.1608147113.
Pasqua T, Pagliaro P, Rocca C, et al. Role of NLRP-3 inflammasome in hypertension: a potential therapeutic target. Curr Pharm Biotechnol. 2018;19:708–14. https://doi.org/10.2174/1389201019666180808162011.
Patel S (2015) Function and dysfunction of two-pore channels. Sci signal 8:re7–re7. https://doi.org/10.1126/scisignal.aab3314.
Patel VB, Zhong J-C, Grant MB, Oudit GY. Role of the ACE2/angiotensin 1–7 axis of the renin–angiotensin system in heart failure. Circ Res. 2016;118:1313–26. https://doi.org/10.1161/CIRCRESAHA.116.307708.
Paz Ocaranza M, Riquelme JA, García L, et al. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nat Rev Cardiol. 2020;17:116–29.
Penny CJ, Vassileva K, Jha A, et al. Mining of Ebola virus entry inhibitors identifies approved drugs as two-pore channel pore blockers. Biochim Biophys Acta - Mol Cell Res. 2019;1866:1151–61. https://doi.org/10.1016/j.bbamcr.2018.10.022.
Peters A, Dockery DW, Muller JE, Mittleman MA. Increased particulate air pollution and the triggering of myocardial infarction. Circulation. 2001;103:2810–5. https://doi.org/10.1161/01.CIR.103.23.2810.
Piperno A, Galimberti S, Mariani R, et al. Modulation of hepcidin production during hypoxia-induced erythropoiesis in humans in vivo: data from the HIGHCARE project. Blood. 2011;117:2953–9. https://doi.org/10.1182/blood-2010-08-299859.
Pooladanda V, Thatikonda S, Bale S, et al. Nimbolide protects against endotoxin-induced acute respiratory distress syndrome by inhibiting TNF-α mediated NF-κB and HDAC-3 nuclear translocation. Cell Death Dis. 2019;10:1–17. https://doi.org/10.1038/s41419-018-1247-9.
Porcelli S, Marzorati M, Healey B, et al. Lack of acclimatization to chronic hypoxia in humans in the Antarctica. Sci Rep. 2017;7:1–6. https://doi.org/10.1038/s41598-017-18212-1.
Rabkin SW. The role of interleukin 18 in the pathogenesis of hypertension-induced vascular disease. Nat Clin Pract Cardiovasc Med. 2009;6:192–9.
Rahman T, Cai X, Brailoiu GC, et al (2014) Two-pore channels provide insight into the evolution of voltage-gated Ca2+and Na+ channels. Sci signal 7:ra109. https://doi.org/10.1126/scisignal.2005450.
Raj VS, Mou H, Smits SL, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495:251–4. https://doi.org/10.1038/nature12005.
Ramchandran R, Pilipenko E, Bach L, et al. Hypoxic regulation of pulmonary vascular smooth muscle cyclic guanosine monophosphate-dependent kinase by the ubiquitin conjugating system. Am J Respir Cell Mol Biol. 2012;46:323–30. https://doi.org/10.1165/rcmb.2011-0165OC.
Romaní-Pérez M, Outeiriño-Iglesias V, Moya CM, et al. Activation of the GLP-1 receptor by liraglutide increases ACE2 expression, reversing right ventricle hypertrophy, and improving the production of SP-A and SP-B in the lungs of type 1 diabetes rats. Endocrinology. 2015;156:3559–69. https://doi.org/10.1210/en.2014-1685.
Rose NR. Critical cytokine pathways to cardiac inflammation. J Interf Cytokine Res. 2011;31:705–10. https://doi.org/10.1089/jir.2011.0057.
Rosenfeldt Z, Conklen K, Jones B, et al. Comparison of nicardipine with clevidipine in the management of hypertension in acute cerebrovascular diseases. J Stroke Cerebrovasc Dis. 2018;27:2067–73. https://doi.org/10.1016/j.jstrokecerebrovasdis.2018.03.001.
Ruan H, Li W, Wang J, et al. Propofol alleviates ventilator-induced lung injury through regulating the Nrf2/NLRP3 signaling pathway. Exp Mol Pathol. 2020a;114:104427. https://doi.org/10.1016/j.yexmp.2020.104427.
Ruan Q, Yang K, Wang W, et al (2020b) Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive care med 1–3. https://doi.org/10.1007/s00134-020-05991-x.
Sakurai Y, Kolokoltsov AA, Chen CC, et al. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science. 2015;347:995–8. https://doi.org/10.1126/science.1258758.
Samaja M, Mariani C, Prestini A, Cerretelli P. Acid-base balance and O 2 transport at high altitude. Acta Physiol Scand. 1997;159:249–56. https://doi.org/10.1046/j.1365-201X.1997.574342000.x.
Samet JM, Dominici F, Curriero FC, et al. Fine particulate air pollution and mortality in 20 U.S. cities, 1987–1994. N Engl J Med. 2000;343:1742–9. https://doi.org/10.1056/NEJM200012143432401.
Schindler C, Bramlage P, Kirch W, Ferrario CM. Role of the vasodilator peptide angiotensin-(1-7) in cardiovascular drug therapy. Vasc Health Risk Manag. 2007;3:125–37.
Schnabel E, Karrasch S, Schulz H, et al. High blood pressure, antihypertensive medication and lung function in a general adult population. Respir Res. 2011a;12:50. https://doi.org/10.1186/1465-9921-12-50.
Schnabel E, Nowak D, Brasche S, et al. Association between lung function, hypertension and blood pressure medication. Respir Med. 2011b;105:727–33. https://doi.org/10.1016/j.rmed.2010.12.023.
Schroder K, Tschopp J. The Inflammasomes. Cell. 2010;140:821–32.
Scott C, Griffin S. Viroporins: structure, function and potential as antiviral targets. J Gen Virol. 2015;96:2000–27.
Setti L, Passarini F, de Gennaro G, et al (2020) Evaluation of the potential relationship between particulate matter (PM) pollution and COVID-19 infection spread in Italy. Soc Ital di med ambient 1–6.
Silva-Palacios A, Königsberg M, Zazueta C (2016) Nrf2 signaling and redox homeostasis in the aging heart: a potential target to prevent cardiovascular diseases? Ageing res. Rev. 26:81–95.
Silver MR. Anemia in the long-term ventilator-dependent patient with respiratory failure. Chest. 2005;128:568S–75S. https://doi.org/10.1378/chest.128.5_suppl_2.568S.
Siu K, Yuen K, Castano-Rodriguez C, et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019;33:8865–77. https://doi.org/10.1096/fj.201802418R.
Smith G, Nielsen M. Criteria for admission. BMJ. 1999;318:1544. https://doi.org/10.1136/bmj.318.7197.1544.
Sodhi CP, Nguyen J, Yamaguchi Y, et al. A dynamic variation of pulmonary ACE2 is required to modulate neutrophilic inflammation in response to Pseudomonas aeruginosa lung infection in mice. J Immunol. 2019;203:3000–12. https://doi.org/10.4049/jimmunol.1900579.
Sodhi CP, Wohlford-Lenane C, Yamaguchi Y, et al. Attenuation of pulmonary ACE2 activity impairs inactivation of des-arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am J Physiol - Lung Cell Mol Physiol. 2018;314:L17–31. https://doi.org/10.1152/ajplung.00498.2016.
Soler MJ, Wysocki J, Batlle D. ACE2 alterations in kidney disease. Nephrol Dial Transplant. 2013;28:2687–97. https://doi.org/10.1093/ndt/gft320.
Soler MJ, Wysocki J, Ye M, et al. ACE2 inhibition worsens glomerular injury in association with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int. 2007;72:614–23. https://doi.org/10.1038/sj.ki.5002373.
Steg PG, James SK, Atar D, et al. ESC guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J. 2012;33:2569–619.
Sungnak W, Huang N, Bécavin C, et al. SARS-CoV-2 entry genes are Most highly expressed in nasal goblet and ciliated cells within human airways Nat med. 2020. https://doi.org/10.1038/s41591-020-0868-6.
Tatarkova Z, Engler I, Calkovska A, et al. Effect of long-term normobaric hyperoxia on oxidative stress in mitochondria of the guinea pig brain. Neurochem Res. 2011;36:1475–81. https://doi.org/10.1007/s11064-011-0473-7.
Terraneo L, Bianciardi P, Caretti A, et al. Chronic systemic hypoxia promotes LNCaP prostate cancer growth in vivo. Prostate. 2010;70:1243–54. https://doi.org/10.1002/pros.21160.
Terraneo L, Paroni R, Bianciardi P, et al. Brain adaptation to hypoxia and hyperoxia in mice. Redox Biol. 2017;11:12–20. https://doi.org/10.1016/j.redox.2016.10.018.
Terraneo L, Samaja M. Comparative response of brain to chronic hypoxia and hyperoxia. Int J Mol Sci. 2017;18:1914. https://doi.org/10.3390/ijms18091914.
Terraneo L, Virgili E, Caretti A, et al. In vivo hyperoxia induces hypoxia-inducible factor-1α overexpression in LNCaP tumors without affecting the tumor growth rate. Int J Biochem Cell Biol. 2014;51:65–74. https://doi.org/10.1016/j.biocel.2014.03.019.
The Novel Coronavirus Pneumonia Emergency Response Epidemiology team, Novel Coronavirus Pneumonia Emergency Response Epidemiology Team. The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) in China. China CDC Wkly. 2020;41:145–51. https://doi.org/10.3760/cma.j.issn.0254-6450.2020.02.003.
Thomson G. COVID-19: social distancing, ACE 2 receptors, protease inhibitors and beyond? Int J Clin Pract e13503. 2020. https://doi.org/10.1111/ijcp.13503.
Tikellis C, Bernardi S, Burns WC. Angiotensin-converting enzyme 2 is a key modulator of the renin-angiotensin system in cardiovascular and renal disease. Curr Opin Nephrol Hypertens. 2011;20:62–8. https://doi.org/10.1097/MNH.0b013e328341164a.
Too KKW, Hung IFN, Li IWS, et al. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clin Infect Dis. 2010;50:850–9. https://doi.org/10.1086/650581.
Tocchetti C, Molinaro M, Angelone T, et al. Nitroso-redox balance and modulation of basal myocardial function: an update from the Italian Society of Cardiovascular Research (SIRC). Curr Drug Targets. 2015;16:895–903. https://doi.org/10.2174/1389450116666150304103517.
Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018;15:203–14.
Tseng HHL, Vong CT, Kwan YW, et al. Lysosomal Ca2+ signaling regulates high glucose-mediated interleukin-1β secretion via transcription factor EB in human monocytic cells. Front Immunol 8. 2017. https://doi.org/10.3389/fimmu.2017.01161.
Turner AJ, Hiscox JA, Hooper NM. ACE2: from vasopeptidase to SARS virus receptor. Trends Pharmacol Sci. 2004;25:291–4.
Ungvari Z, Tarantini S, Donato AJ, et al. Mechanisms of vascular aging. Circ Res. 2018;123:849–67. https://doi.org/10.1161/CIRCRESAHA.118.311378.
Ungvari Z, Tarantini S, Nyúl-Tóth Á, et al. Nrf2 dysfunction and impaired cellular resilience to oxidative stressors in the aged vasculature: from increased cellular senescence to the pathogenesis of age-related vascular diseases. GeroScience. 2019;41:727–38.
Valcarcel-Ares MN, Gautam T, Warrington JP, et al. Disruption of Nrf2 signaling impairs angiogenic capacity of endothelial cells: implications for microvascular aging. Journals Gerontol Ser A Biol Sci Med Sci. 2012;67:821–9. https://doi.org/10.1093/gerona/glr229.
Vangjeli C, Dicker P, Tregouet D-A, et al. A polymorphism in ACE2 is associated with a lower risk for fatal cardiovascular events in females: the MORGAM project. J Renin-Angiotensin-Aldosterone Syst. 2011;12:504–9. https://doi.org/10.1177/1470320311405557.
Vincent MJ, Bergeron E, Benjannet S, et al (2005) Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J https://doi.org/10.1186/1743-422X-2-69
Walls AC, Park YJ, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020. https://doi.org/10.1016/j.cell.2020.02.058.
Wan Y, Shang J, Graham R, et al (2020) Receptor recognition by novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS. J Virol 94:. https://doi.org/10.1128/jvi.00127-20.
Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA - J Am Med Assoc. 2020a;323:1061–9. https://doi.org/10.1001/jama.2020.1585.
Wang H, Yang P, Liu K, et al. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008. https://doi.org/10.1038/cr.2008.15.
Wang K, Xie S, Sun B. Viral proteins function as ion channels. Biochim Biophys Acta Biomembr. 2011;1808:510–5.
Wang L, He W, Yu X, et al (2020b) Coronavirus disease 2019 in elderly patients: characteristics and prognostic factors based on 4-week follow-up. J infect 0: https://doi.org/10.1016/j.jinf.2020.03.019.
Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020c;30:269–71. https://doi.org/10.1038/s41422-020-0282-0.
Wang T, Du Z, Zhu F, et al. Comorbidities and multi-organ injuries in the treatment of COVID-19. Lancet. 2020d;395:e52.
Warner FJ, Smith AI, Hooper NM, Turner AJ. Angiotensin-converting enzyme-2: a molecular and cellular perspective. Cell Mol Life Sci. 2004;61:2704–13.
Wikenheiser J, Wolfram JA, Gargesha M, et al. Altered hypoxia-inducible factor-1 alpha expression levels correlate with coronary vessel anomalies. Dev Dyn. 2009;238:2688–700. https://doi.org/10.1002/dvdy.22089.
Wong CK, Lam CWK, Wu AKL, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. 2004;136:95–103. https://doi.org/10.1111/j.1365-2249.2004.02415.x.
Wong DW, Oudit GY, Reich H, et al. Loss of angiotensin-converting enzyme-2 (Ace2) accelerates diabetic kidney injury. Am J Pathol. 2007;171:438–51. https://doi.org/10.2353/ajpath.2007.060977.
Wösten-Van Asperen RM, Lutter R, Specht PA, et al. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J Pathol. 2011;225:618–27. https://doi.org/10.1002/path.2987.
Wu JT, Leung K, Bushman M, et al. Estimating clinical severity of COVID-19 from the transmission dynamics in Wuhan. China Nat Med. 2020a:1–5. https://doi.org/10.1038/s41591-020-0822-7.
Wu P, Duan F, Luo C, et al. Characteristics of ocular findings of patients with coronavirus disease 2019 (COVID-19) in Hubei Province. China JAMA Ophthalmol. 2020b. https://doi.org/10.1001/jamaophthalmol.2020.1291.
Xing YF, Xu YH, Shi MH, Lian YX. The impact of PM2.5 on the human respiratory system. J Thorac Dis. 2016;8:E69–74.
Xu H, Zhong L, Deng J, et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. 2020a;12:1–5. https://doi.org/10.1038/s41368-020-0074-x.
Xu RY, Zhu XF, Yang Y, Ye P. High-sensitive cardiac troponin T. J Geriatr Cardiol. 2013;10:102–9.
Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020b. https://doi.org/10.1016/S2213-2600(20)30076-X.
Yagil C, Sapojnikov M, Kreutz R, et al. Role of chromosome X in the sabra rat model of salt-sensitive hypertension. Hypertension. 1999;33:261–5. https://doi.org/10.1161/01.hyp.33.1.261.
Yang Z-Y, Huang Y, Ganesh L, et al. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J Virol. 2004;78:5642–50. https://doi.org/10.1128/JVI.78.11.5642-5650.2004.
Yao W-X, Jiang M-X. Effects of tetrandrine on cardiovascular electrophysiologic properties. Acta Pharmacol Sin. 2002;23:1069–74.
Zaki AM, van Boheemen S, Bestebroer TM, et al. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814–20. https://doi.org/10.1056/NEJMoa1211721.
Zeng W, Chen W, Leng X, et al. Impairment of cardiac function and remodeling induced by myocardial infarction in rats are attenuated by the nonpeptide angiotensin-(1-7) analog AVE 0991. Cardiovasc Ther. 2012;30:152–61. https://doi.org/10.1111/j.1755-5922.2010.00255.x.
Zhang H, Baker A. Recombinant human ACE2: acing out angiotensin II in ARDS therapy. Crit Care. 2017;21:305.
Zhang J, Dong X, Cao Y, et al (2020) Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy all.14238. https://doi.org/10.1111/all.14238.
Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. 2020;1–2.
Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020a;395:1054–62. https://doi.org/10.1016/S0140-6736(20)30566-3.
Zhou P, Yang X, Lou, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020b;579:270–3. https://doi.org/10.1038/s41586-020-2012-7.
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–6. https://doi.org/10.1038/nature09663.
Zhou Y qun, Liu D qiang, Chen S ping, et al (2020c) Nrf2 activation ameliorates mechanical allodynia in paclitaxel-induced neuropathic pain. Acta Pharmacol Sin 1–8. https://doi.org/10.1038/s41401-020-0394-6
Zhu J, Yang Y, Hu SG, et al. T-lymphocyte Kv1.3 channel activation triggers the NLRP3 inflammasome signaling pathway in hypertensive patients. Exp Ther Med. 2017;14:147–54. https://doi.org/10.3892/etm.2017.4490.
Ziello JE, Jovin IS, Huang Y. Hypoxia-inducible factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med. 2007;80:51–60.
Zoja C, Cattaneo S, Fiordaliso F, et al. Distinct cardiac and renal effects of ET A receptor antagonist and ACE inhibitor in experimental type 2 diabetes. Am J Physiol Physiol. 2011;301:F1114–23. https://doi.org/10.1152/ajprenal.00122.2011.
Zumla A, Hui DS, Perlman S. Middle East respiratory syndrome. Lancet. 2015;386:995–1007.
Acknowledgments
The Italian Society of Cardiovascular Researches is gratefully thankful to the Fondazione Cassa di Risparmio di Imola for continuous support to our scientific and dissemination activities. T.P. and C.R. gratefully acknowledge DiSS_PSR2019_DIP_013 and PNRA18_00071 n. 1314 (M.S.); PON “Ricerca e Innovazione” 2014-2020: AIM “Attraction and International Mobility” (European Union & MIUR, Italy) and POR Calabria FESR-FSE 2014/2020: Azione 10.5.12 - LineaB (European Union & Calabria Region).
Funding
Research conducted in Authors’ laboratories is supported by: Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Program (2018–2022) - Dept. of Biology and Biotechnology “L. Spallanzani”, University of Pavia, and the EU Horizon 2020 FETOPEN-2018-2020 Program under Grant Agreement N. 828984 (LION-HEARTED) (F.M.); FIL_2018_Miragoli (M.M.); ETHERNA project (Prog. n. 161/16, Fondazione Pisa, Italy) (V.L.). Funding source had no such involvement in study design, in the collection, analysis, interpretation of data, in the writing of the report; and in the decision to submit the paper for publication.
Author information
Authors and Affiliations
Contributions
F.M. and T.A. designed and directed the project. Other authors contributed equally and are listed in alphabetical order. All authors discussed and commented on the manuscript, provided critical feedback and helped shape the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Consent for publication
All the authors have read and approved the revised manuscript, and they are willing to publish it.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Moccia, F., Gerbino, A., Lionetti, V. et al. COVID-19-associated cardiovascular morbidity in older adults: a position paper from the Italian Society of Cardiovascular Researches. GeroScience 42, 1021–1049 (2020). https://doi.org/10.1007/s11357-020-00198-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-020-00198-w