Synthesis, Single Crystal X-Ray Structure, Hirshfeld Surface Analysis, DFT Computations, Docking Studies on Aurora Kinases and an Anticancer Property of 3-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-6-methoxy-4H-chromen-4-one

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Crystal Structure Determination
2.3. In Silico Docking with Aurora Kinases
3. Results and Discussion
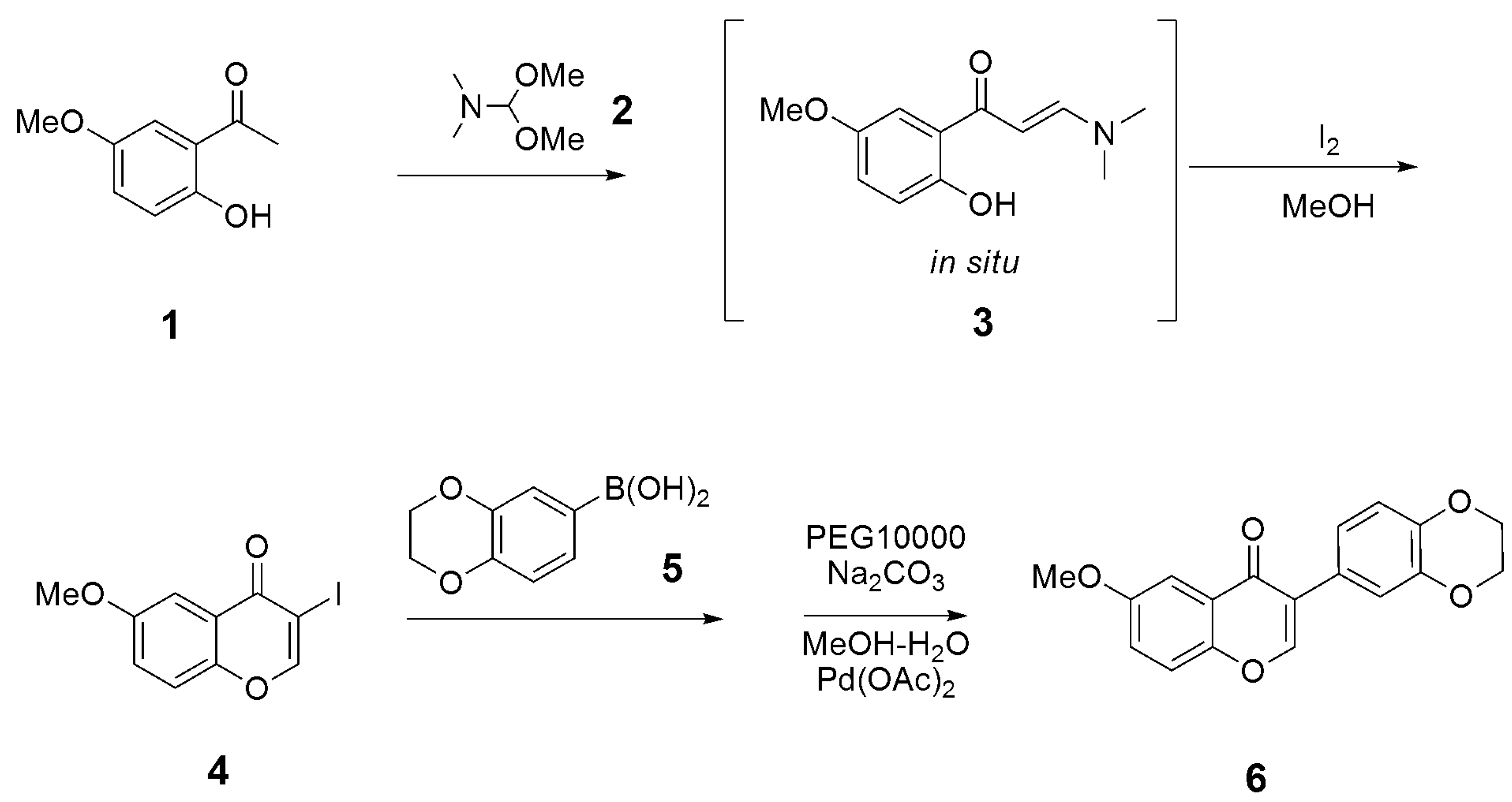
3.1. Synthesis
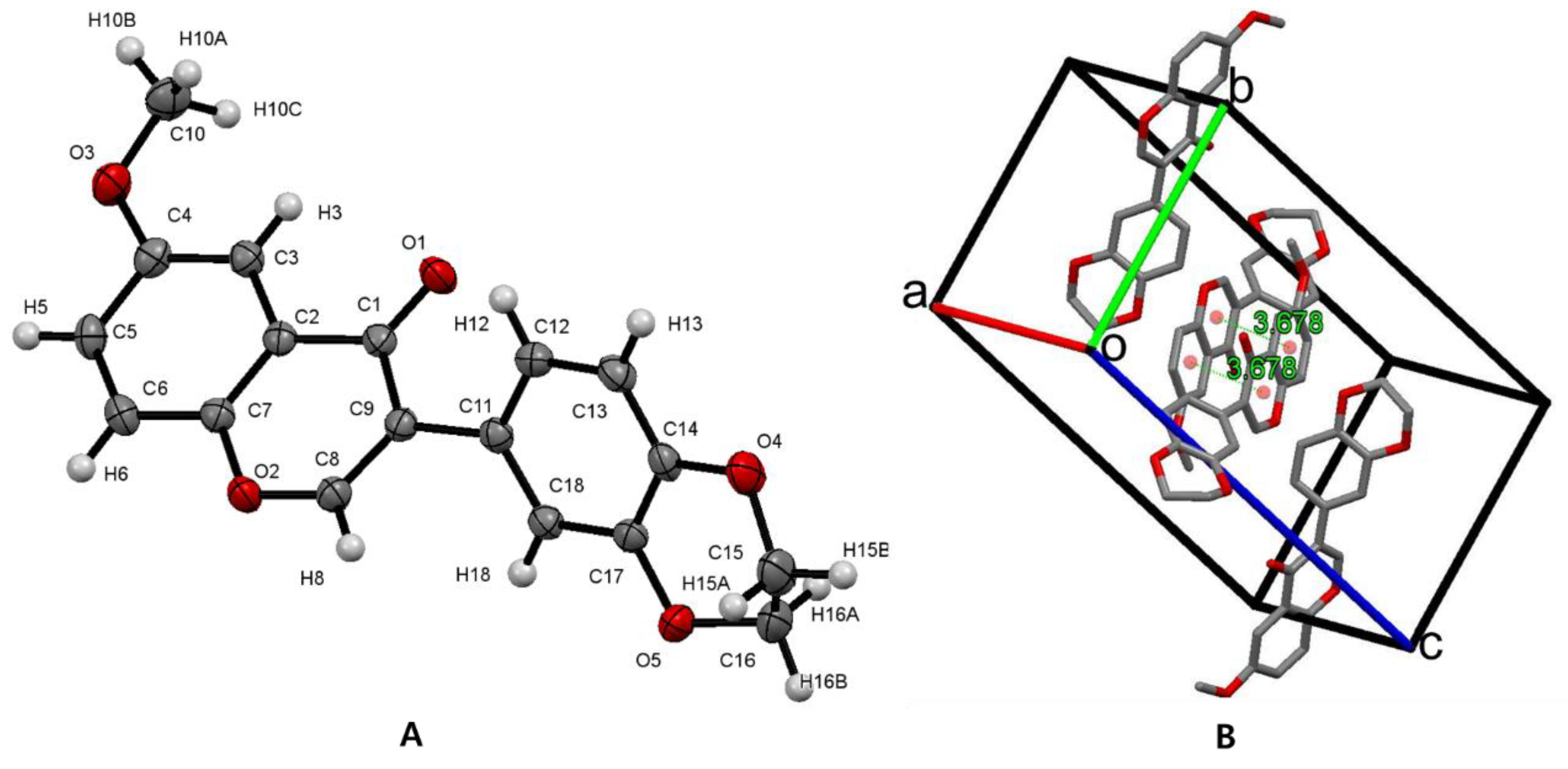
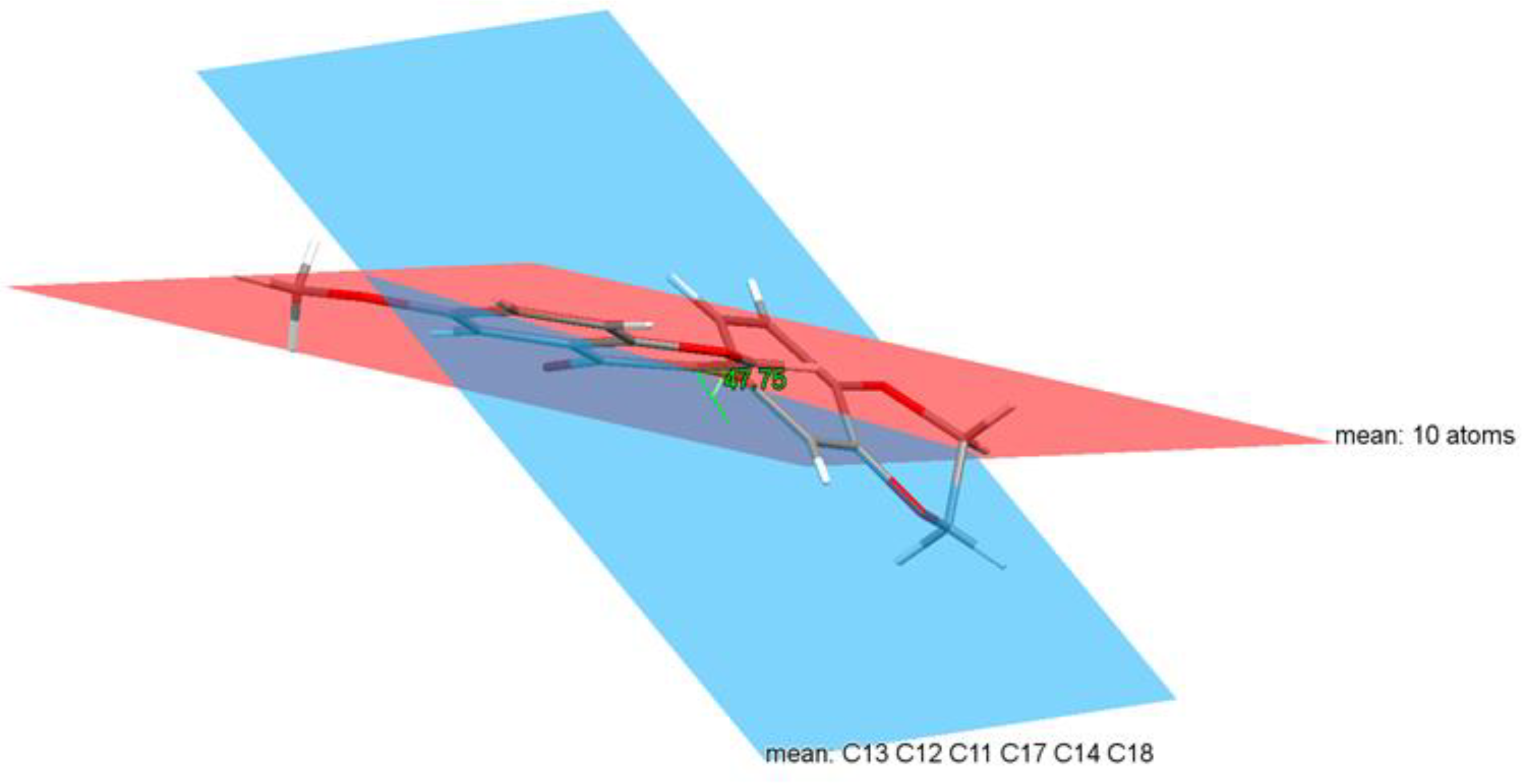
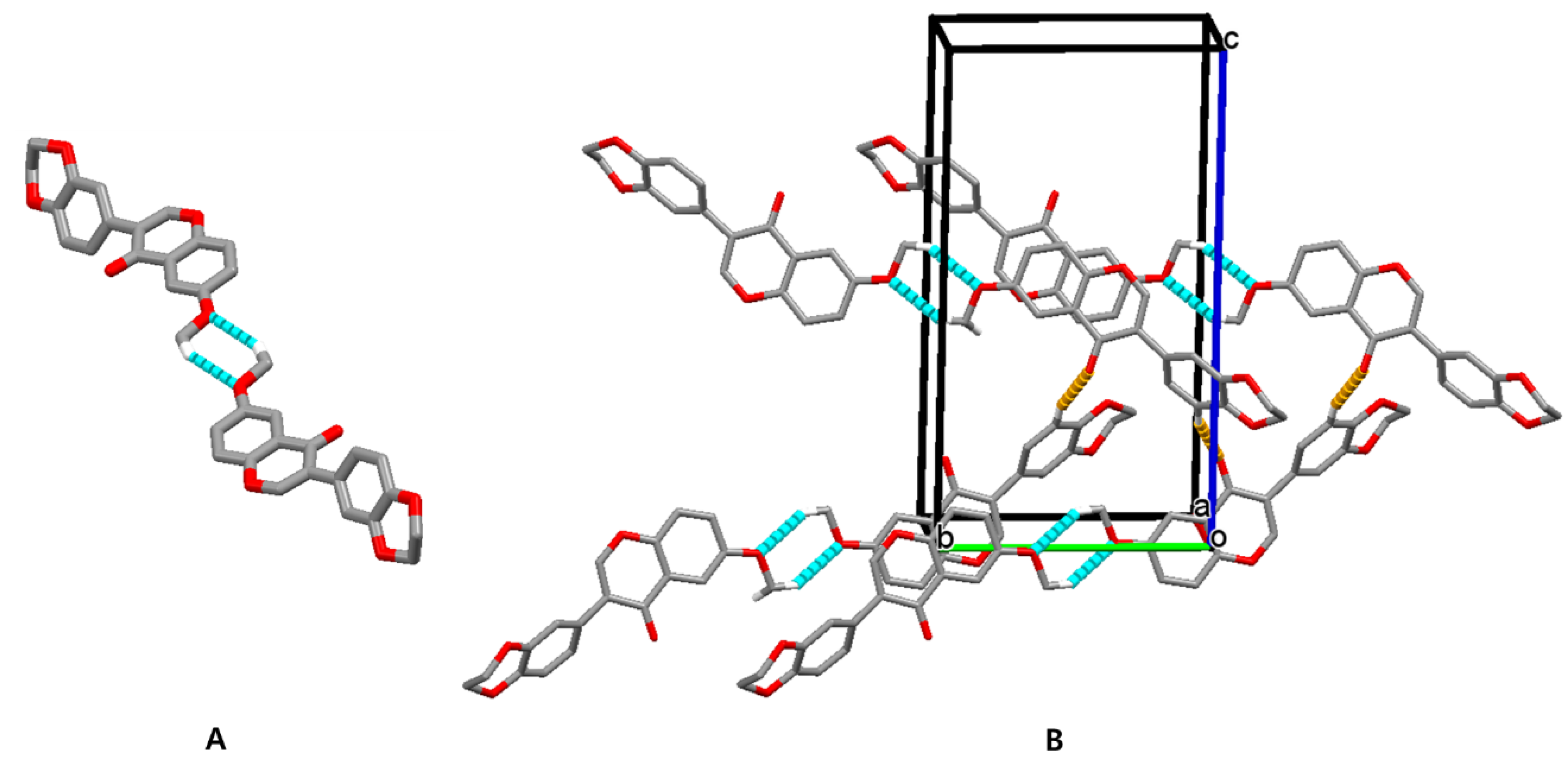
3.2. Crystal Structure of Isoflavone Compound 6
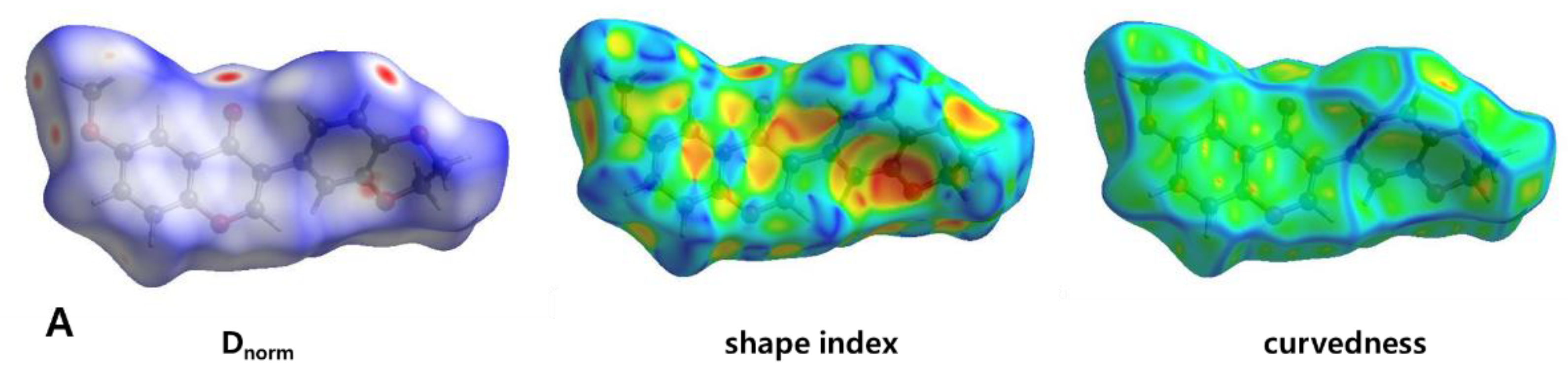
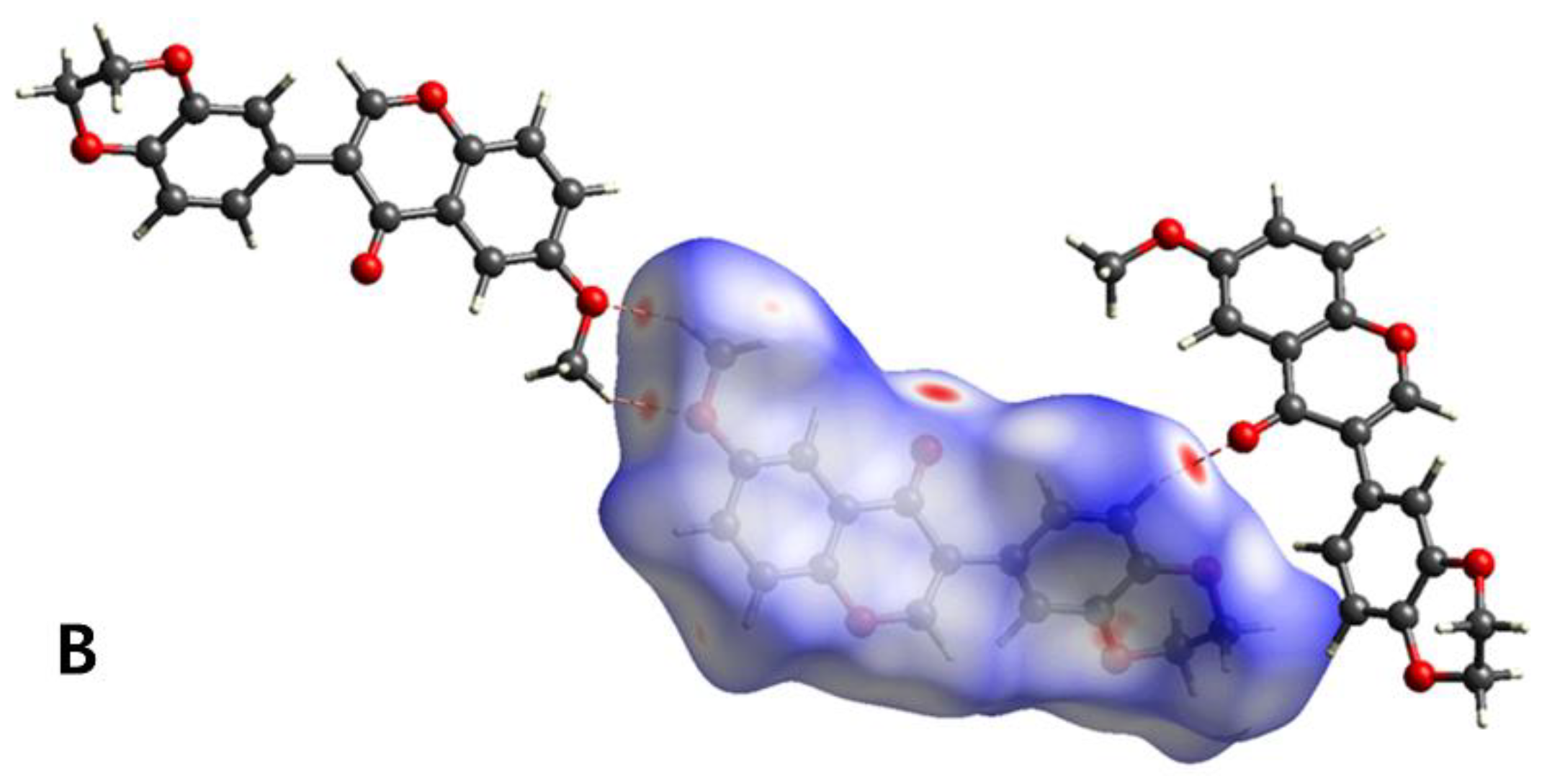
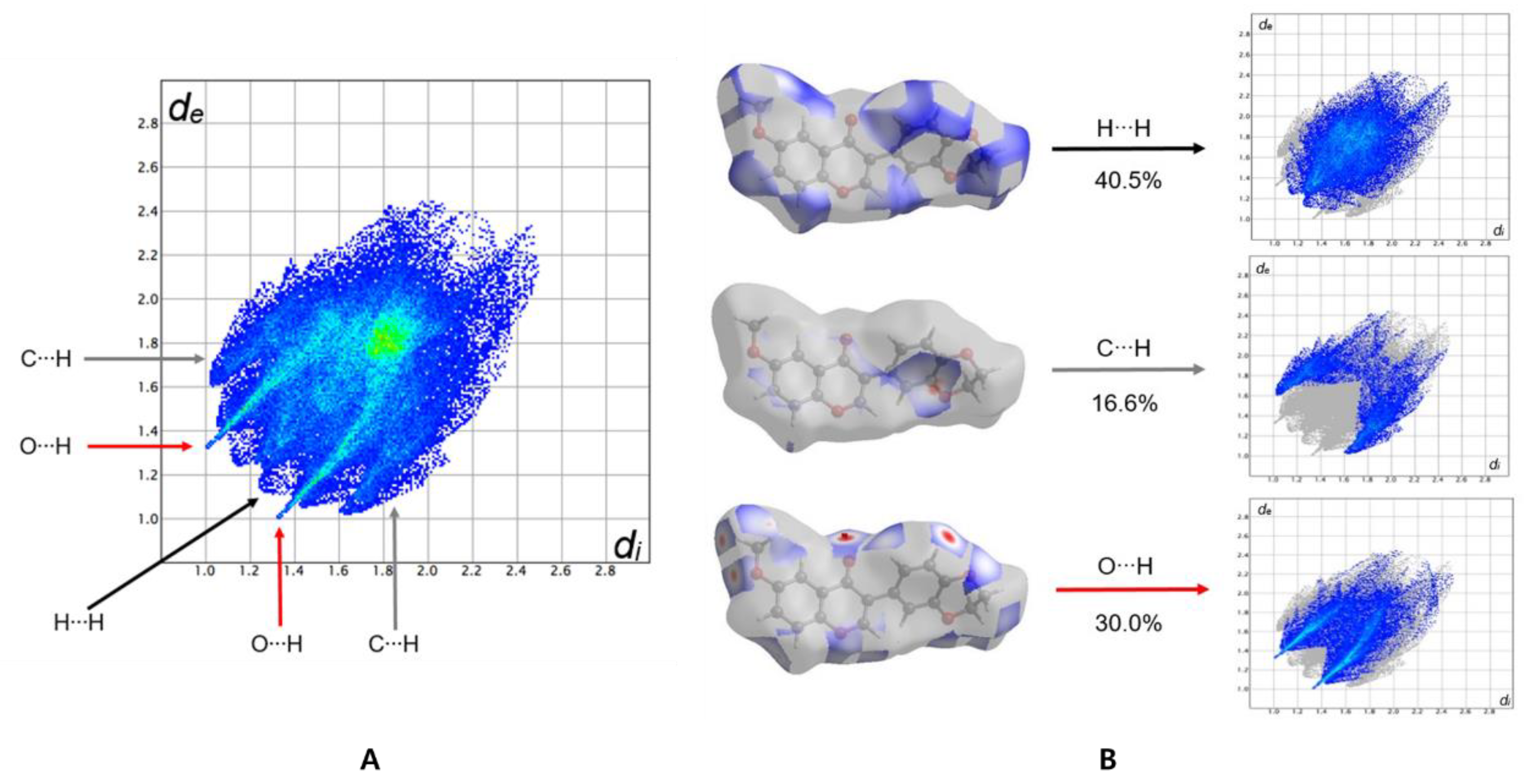
3.3. Hirshfeld Surface Analysis
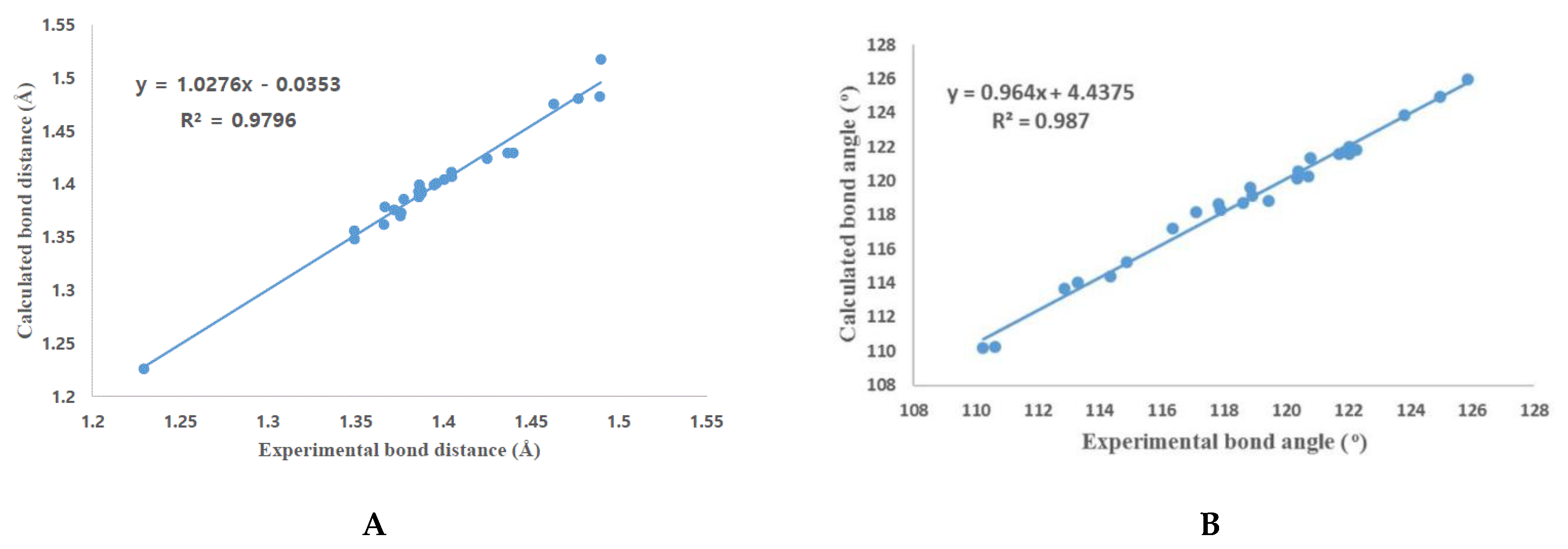
3.4. DFT Calculation
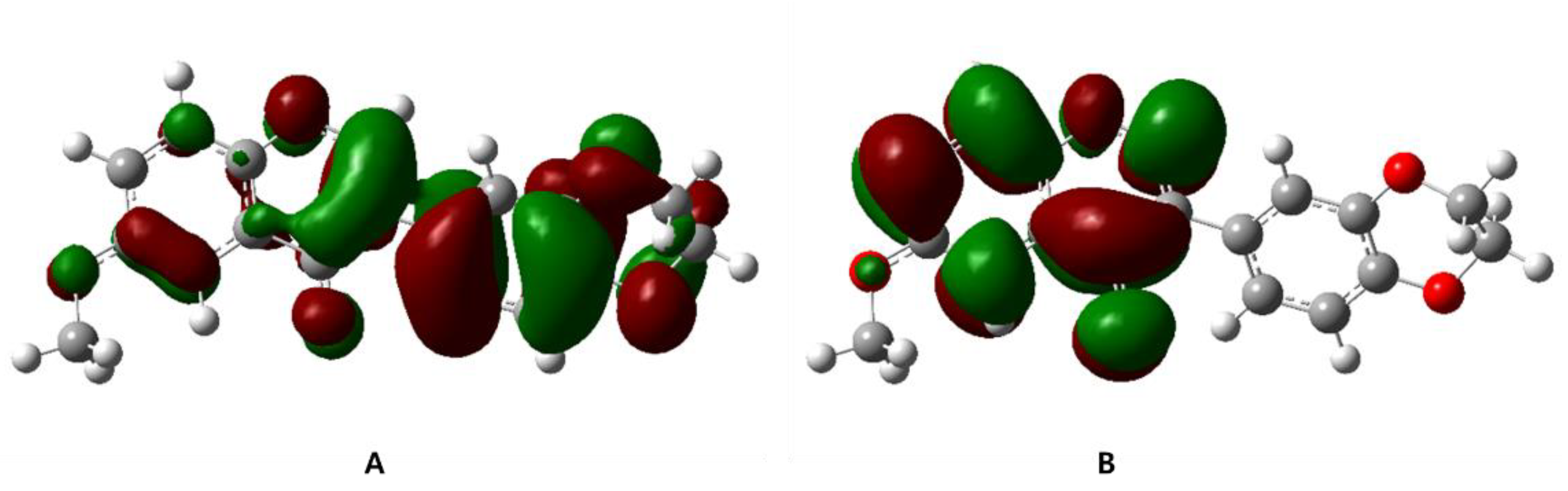
3.5. Frontier Molecular Orbital Calculation
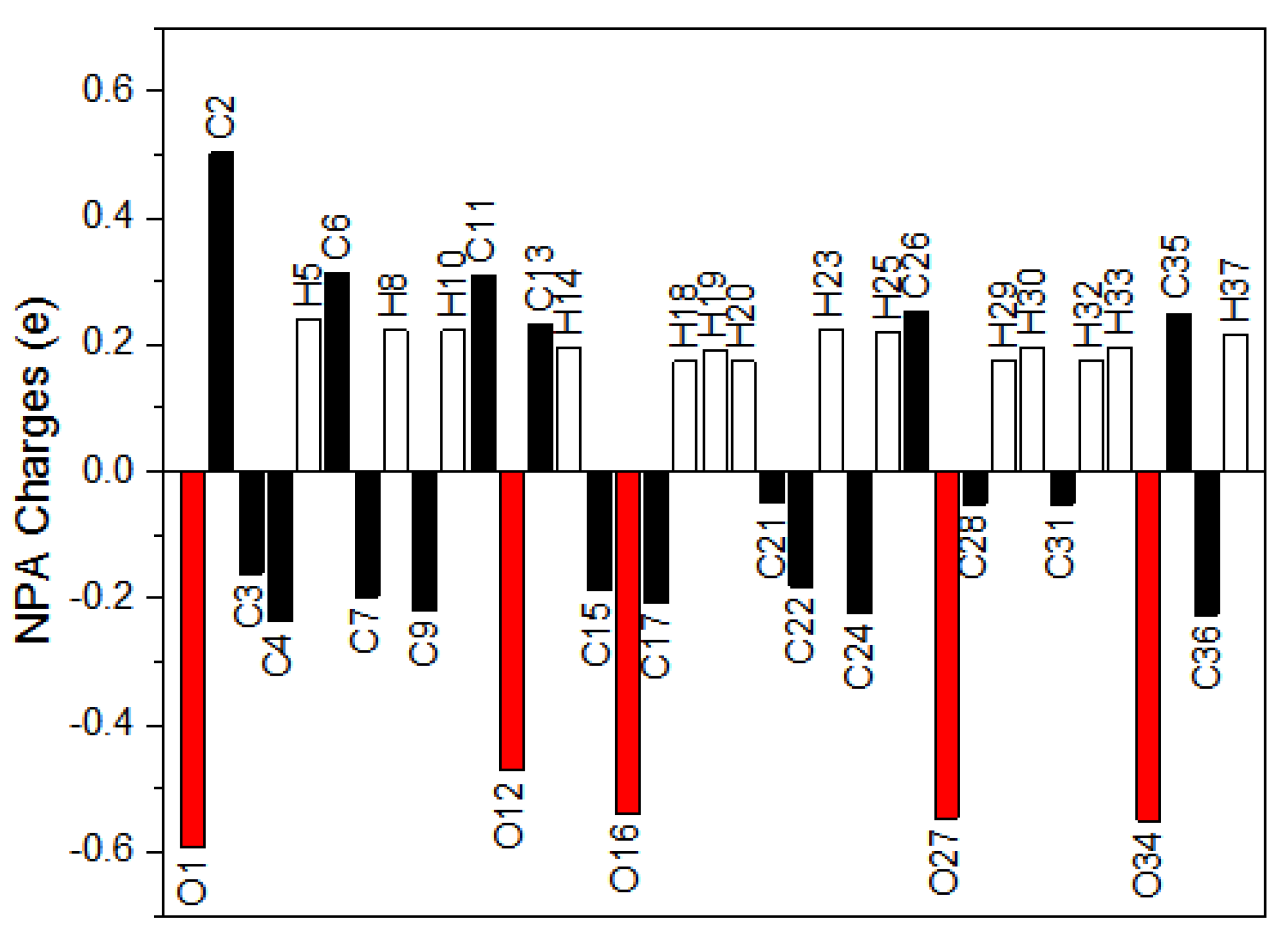
3.6. Natural Population Analysis (NPA)
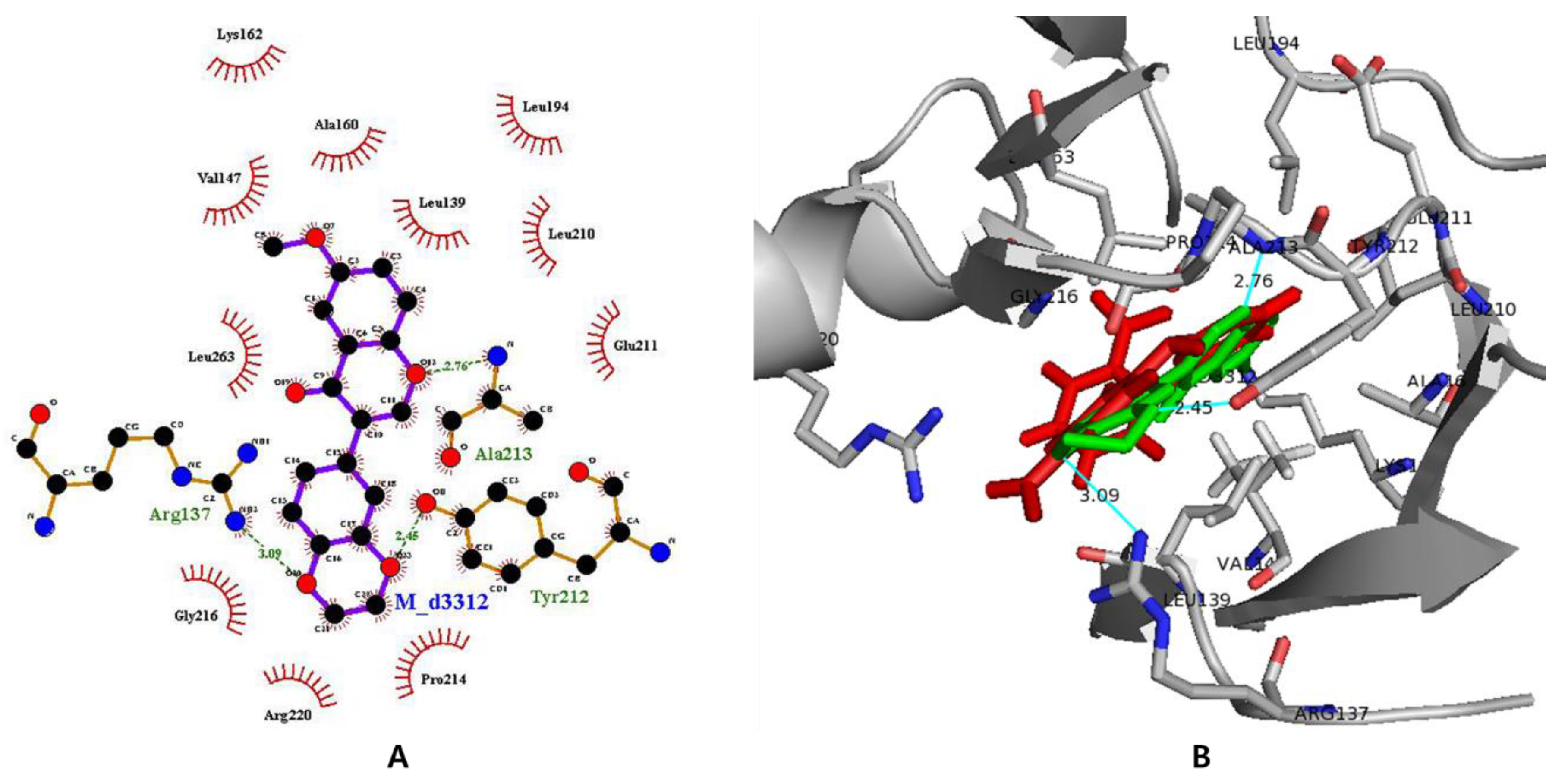
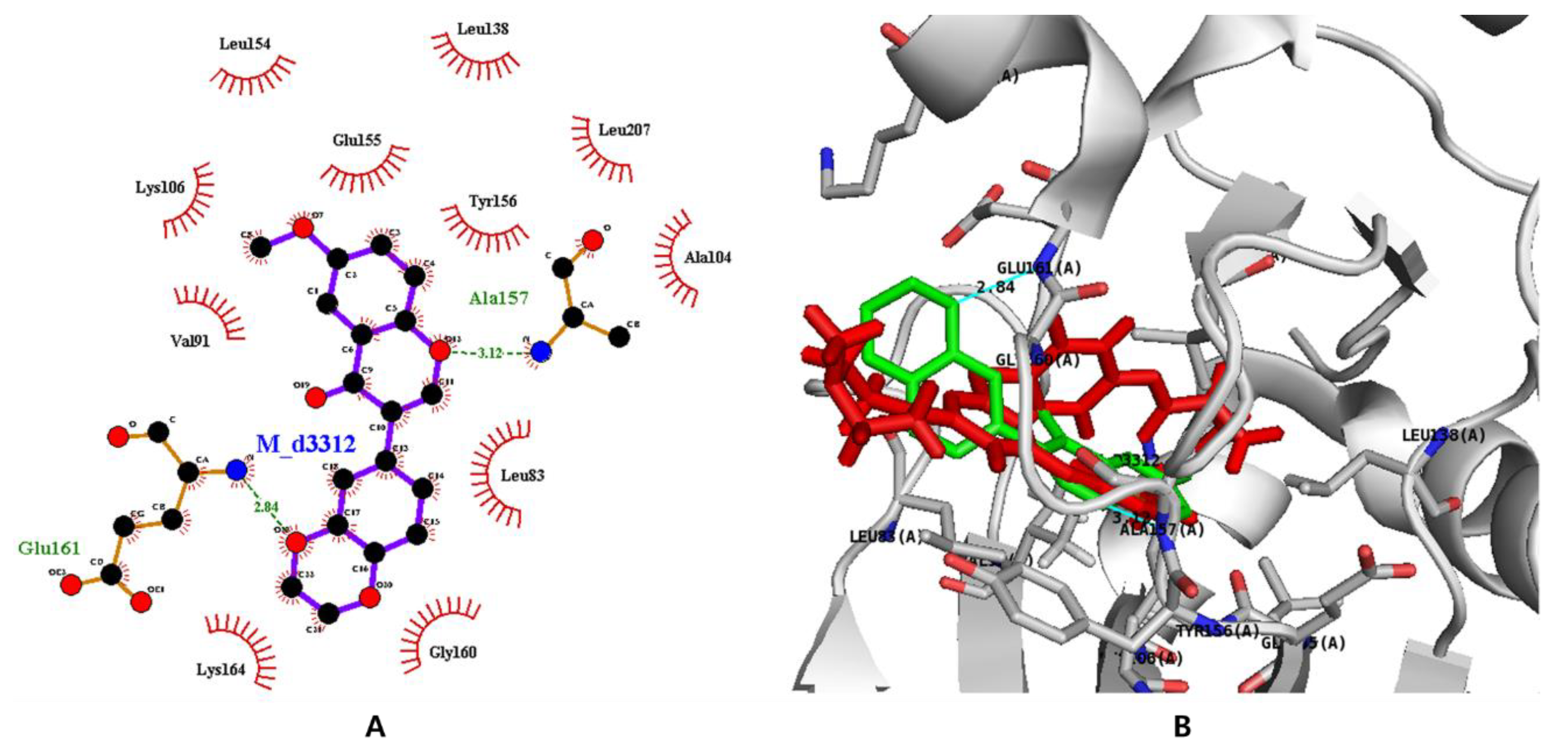
3.7. In Silico Docking with Aurora Kinases
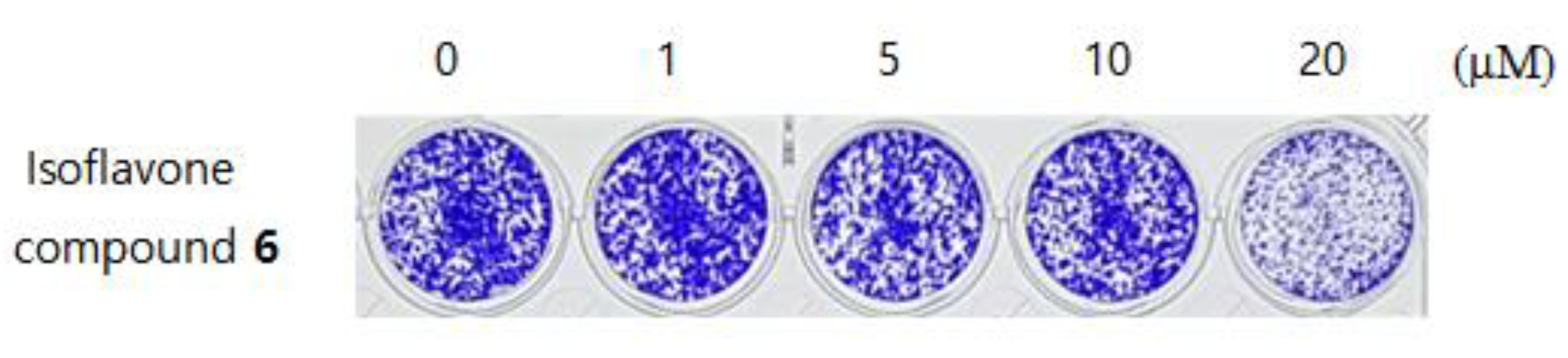
3.8. Anti-Cancer Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Liu, J.; Taylor, S.F.; Dupart, P.S.; Arnold, C.L.; Sridhar, J.; Jiang, Q.; Wang, Y.; Skripnikova, E.V.; Zhao, M.; Foroozesh, M. Pyranoflavones: A group of small-molecule probes for exploring the active site cavities of cytochrome P450 enzymes 1A1, 1A2, and 1B1. J. Med. Chem. 2013, 56, 4082–4092. [Google Scholar] [CrossRef] [Green Version]
- Cushnie, T.P.T.; Lamb, A.J. Antimicrobial activity of flavonoids. Int. J. Antimicrob. Agents 2005, 26, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Nijveldt, R.J.; van Nood, E.; van Hoorn, D.E.; Boelens, P.G.; van Norren, K.; van Leeuwen, P.A. Flavonoids: A review of probable mechanisms of action and potential applications. Am. J. Clin. Nutr. 2001, 74, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Gupta, S. Apigenin: A promising molecule for cancer prevention. Pharm. Res. 2010, 27, 962–978. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, J.; Furman, C.; Bernier, J.L.; Duriez, P.; Teissier, E.; Cotelle, N. Antioxidant properties of di-tert-butylhydroxylated flavonoids. Free Radic. Biol. Med. 2000, 29, 900–912. [Google Scholar] [CrossRef]
- Park, W.H. MAPK inhibitors differentially affect gallic acid-induced human pulmonary fibroblast cell growth inhibition. Mol. Med. Rep. 2011, 4, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.Q.; Han, X.Z.; Li, X.; Ren, D.M.; Wang, X.N.; Lou, H.X. Flavonoids from Dracocephalum tanguticum and their cardioprotective effects against doxorubicin-induced toxicity in H9c2 cells. Bioorg. Med. Chem. Lett. 2010, 20, 6411–6415. [Google Scholar] [CrossRef]
- Gaspar, A.; Matos, M.J.; Garrido, J.; Uriarte, E.; Borges, F. Chromone: A valid scaffold in medicinal chemistry. Chem. Rev. 2014, 114, 4960–4992. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Kaur, M.; Silakari, O. Flavones: An important scaffold for medicinal chemistry. Eur. J. Med. Chem. 2014, 84, 206–239. [Google Scholar] [CrossRef]
- Barakat, A.; Soliman, A.M.; El-Faham, A.; Ali, M.; Al-Majid, A.M.; Yousuf, S.; Choudhary, M.I. Three multi-components reaction: Synthesis and X-ray single-crystal of hydroacridinone-based hydrazino-s-triazine derivative as a new class of urease inhibitor. Crystals 2020, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Al-Wabli, R.I.; Al-Ghamdi, A.R.; Aswathy, S.V.; Ghabbour, H.A.; Al-Agamy, M.H.; Hubert Joe, I.H.; Attia, M.I. Synthesis, single crystal X-ray structure, DFT computations, hirshfeld surface analysis and molecular docking simulations on ({[(1E)-1-(1,3-Benzodioxol-5-yl)-3-(1H-imidazol-1-yl) propylidene]amino}oxy)(furan-2-yl)methanone: A new antifungal agent. Crystals 2019, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Joubert, J. Synthesis, crystal structure, DFT studies, docking, studies, and fluorescent properties of 2-(Adamantan-1-yl)-2H-isoindole-1-carbonitrile. Crystals 2019, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.Y.; Lee, Y.; Kim, B.S.; Lee, J.; Ahn, S.; Koh, D.; Lim, Y.; Lee, Y.H. Inhibitory effect of synthetic flavone derivatives on pan-aurora kinases: Induction of G2/M cell-cycle arrest and apoptosis in HCT116 human colon cancer cells. Int. J. Mol. Sci. 2018, 19, 86. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.; Ahn, E.; Sung, J.; Koh, D.; Lim, Y.; Park, S. Synthetic polyphenol compounds inhibit β-catenin/Tcf signaling: Structure-activity relationship. J. Ind. Eng. Chem. 2017, 56, 258–269. [Google Scholar] [CrossRef]
- Lee, K.; Lee, D.H.; Jung, Y.J.; Shin, S.Y.; Koh, D.; Lee, Y.H. A methoxyflavanone derivative, 2′,3′,4′-trimethoxy-5,6-naphthoflavanone, inhibits proliferation of HCT116 human colon cancer cells by inducing G2/M cell cycle arrest and apoptosis. Appl. Biol. Chem. 2016, 59, 249–253. [Google Scholar] [CrossRef]
- Sophors, P.; Kim, Y.M.; Seo, G.Y.; Huh, J.S.; Lim, Y.; Koh, D.S.; Cho, M. A synthetic isoflavone, DCMF, promotes human keratinocyte migration by activating Src/FAK signaling pathway. Biochem. Biophys. Res. Commun. 2016, 472, 332–338. [Google Scholar] [CrossRef]
- Ahn, S.; Shin, S.Y.; Jung, Y.; Jung, H.; Kim, B.S.; Koh, D.; Lim, Y. (1) H and (13) C NMR spectral assignments of novel flavonoids bearing benzothiazepine. Magn. Reson. Chem. 2016, 54, 382–390. [Google Scholar] [CrossRef]
- Bruker. APEX2, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta. Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Martin, M.P.; Zhu, J.Y.; Lawrence, H.R.; Pireddu, R.; Luo, Y.; Alam, R.; Ozcan, S.; Sebti, S.M.; Lawrence, N.J.; Schonbrunn, E. A novel mechanism by which small molecule inhibitors induce the DFG flip in Aurora A. ACS Chem. Biol. 2012, 7, 698–706. [Google Scholar] [CrossRef] [Green Version]
- Fancelli, D.; Moll, J.; Varasi, M.; Bravo, R.; Artico, R.; Berta, D.; Bindi, S.; Cameron, A.; Candiani, I.; Cappella, P.; et al. 1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazoles: Identification of a potent Aurora kinase inhibitor with a favorable antitumor kinase inhibition profile. J. Med. Chem. 2006, 49, 7247–7251. [Google Scholar] [CrossRef] [PubMed]
- Elkins, J.M.; Santaguida, S.; Musacchio, A.; Knapp, S. Crystal structure of human aurora B in complex with INCENP and VX-680. J. Med. Chem. 2012, 55, 7841–7848. [Google Scholar] [CrossRef]
- Biegasiewicz, K.F.; Gordon, J.S.; Rodriguez, D.A.; Priefer, R. Development of a general approach to the synthesis of a library of isoflavonoid derivatives. Tetrahedron Lett. 2014, 55, 5210–5212. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Y.; Wang, Y. Phosphine-free palladium acetate catalyzed Suzuki reaction in water. J. Org. Chem. 2005, 70, 6122–6125. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Lim, Y.; Koh, D. Crystal structure of 2-(2,3-di-meth-oxy-naphthalen-1-yl)-3-hy-droxy-6-meth-oxy-4H-chromen-4-one. Acta. Cryst. E Cryst. Commun. 2015, 71, o842–o843. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.S.; Lim, Y.; Koh, D. Crystal structure of 2-(3,4-di-meth-oxy-phen-yl)-3-hy-droxy-4H-chromen-4-one. Acta. Cryst. Sect. E Struct. Rep. Online 2014, 70, o999–o1000. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 37, 3814–3816. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Petersson, G.A.; Allaham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6091. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1.; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Jung, K.Y.; Park, J.; Han, Y.S.; Lee, Y.H.; Shin, S.Y.; Lim, Y. Synthesis and biological evaluation of hesperetin derivatives as agents inducing apoptosis. Bioorg. Med. Chem. 2017, 25, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Shin, S.Y.; Ahn, S.; Koh, D.; Lee, Y.H.; Lim, Y. Biological evaluation of 2-pyrazolinyl-1-carbothioamide derivatives against HCT116 human colorectal cancer cell lines and elucidation on QSAR and molecular binding modes. Bioorg. Med. Chem. 2017, 25, 5423–5432. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical formula | C18H14O5 |
| Formula weight | 310.29 |
| Temperature | 223(2) K |
| Wavelength | 0.71073 Å |
| Crystal system | Monoclinic |
| Space group | P21/n |
| Unit cell dimensions | a = 7.1869(4) Å b = 10.2764(6) Å c = 19.6771(10) Å β = 99.442(2)° |
| Volume | 1433.57(14) Å3 |
| Z | 4 |
| Density (calculated) | 1.438 Mg/m3 |
| Absorption coefficient | 0.106 mm−1 |
| F(000) | 648 |
| Crystal size | 0.190 × 0.150 × 0.100 mm3 |
| Theta range for data collection | 2.098 to 28.296° |
| Index ranges | −9 ≤ h ≤ 9, −13 ≤ k ≤ 13, −26 ≤ l ≤26 |
| Reflections collected | 39243 |
| Independent reflections | 3573 [R(int) = 0.0491] |
| Completeness to theta = 25.242° | 100.00% |
| Refinement method | Full-matrix least-squares on F2 |
| Data / restraints / parameters | 3573 / 0 / 209 |
| Goodness-of-fit on F2 | 1.039 |
| Final R indices [I>2sigma(I)] | R1 = 0.0396, wR2 = 0.0990 |
| R indices (all data) | R1 = 0.0577, wR2 = 0.1083 |
| Largest diff. peak and hole | 0.347 and −0.183 e.Å−3 |
| C(10)-O(3)-C(4)-C(3) | −5.8(2) |
| C(17)-C(14)-O(4)-C(15) | −12.8(2) |
| C(14)-C(17)-O(5)-C(16) | −15.9(2) |
| C(10)-O(3)-C(4)-C(3) | −5.8(2) |
| C(7)-C(2)-C(1)-C(9) | −3.1(2) |
| H(16A)-C(16)-C(15)-H(15A) | 179.1 |
| H(15A)-C(15)-C(16)-H(16B) | −62.3 |
| H(16A)-C(16)-C(15)-H(15B) | −62.5 |
| H(16B)-C(16)-C(15)-H(15B) | 56.1 |
| D-H···A | d(D-H) | d(H···A) | d(D···A) | <(DHA) |
|---|---|---|---|---|
| C(13)-H(13)···O(1)#1 | 0.94 | 2.48 | 3.3970(17) | 163.8 |
| C(10)-(10B)···O(3)#2 | 0.97 | 2.55 | 3.4005(19) | 146.9 |
| C(6)-H(6)···O(4)#3 | 0.94 | 2.61 | 3.2639(17) | 127.0 |
| C(16)-(16A)···O(1)#4 | 0.98 | 2.62 | 3.581(2) | 168.2 |
| Dihedral Angle | Experimental | Calculated | |
|---|---|---|---|
| Crystal | Vacuum | DMSO | |
| C(1)-C(9)-C(11)-C(12) | 49.3755 | 42.4779 | 49.5214 |
| C(1)-C(9)-C(11)-C(18) | −134.245 | −138.276 | −131.476 |
| C(8)-C(9)-C(11)-C(12) | −129.908 | −137.454 | −131.062 |
| C(8)-C(9)-C(11)-C(18) | 46.4713 | 41.7922 | 47.9409 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, S.; Sung, J.; Lee, J.H.; Yoo, M.; Lim, Y.; Shin, S.Y.; Koh, D. Synthesis, Single Crystal X-Ray Structure, Hirshfeld Surface Analysis, DFT Computations, Docking Studies on Aurora Kinases and an Anticancer Property of 3-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-6-methoxy-4H-chromen-4-one. Crystals 2020, 10, 413. https://doi.org/10.3390/cryst10050413
Ahn S, Sung J, Lee JH, Yoo M, Lim Y, Shin SY, Koh D. Synthesis, Single Crystal X-Ray Structure, Hirshfeld Surface Analysis, DFT Computations, Docking Studies on Aurora Kinases and an Anticancer Property of 3-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-6-methoxy-4H-chromen-4-one. Crystals. 2020; 10(5):413. https://doi.org/10.3390/cryst10050413
Chicago/Turabian StyleAhn, Seunghyun, Jiha Sung, Ji Hye Lee, Miri Yoo, Yoongho Lim, Soon Young Shin, and Dongsoo Koh. 2020. "Synthesis, Single Crystal X-Ray Structure, Hirshfeld Surface Analysis, DFT Computations, Docking Studies on Aurora Kinases and an Anticancer Property of 3-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-6-methoxy-4H-chromen-4-one" Crystals 10, no. 5: 413. https://doi.org/10.3390/cryst10050413