Redox-Sensitive Linear and Cross-Linked Cystamine-Based Polymers for Colon-Targeted Drug Delivery: Design, Synthesis, and Characterisation


Abstract
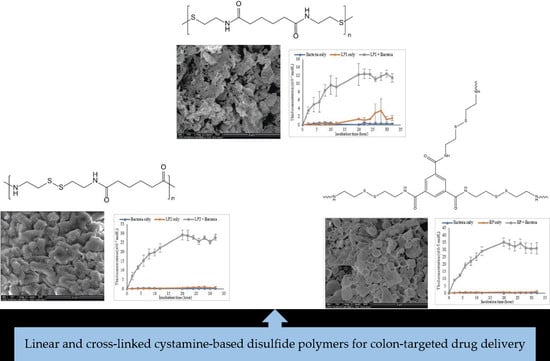
:
1. Introduction
2. Materials and Methods
2.1. Materials
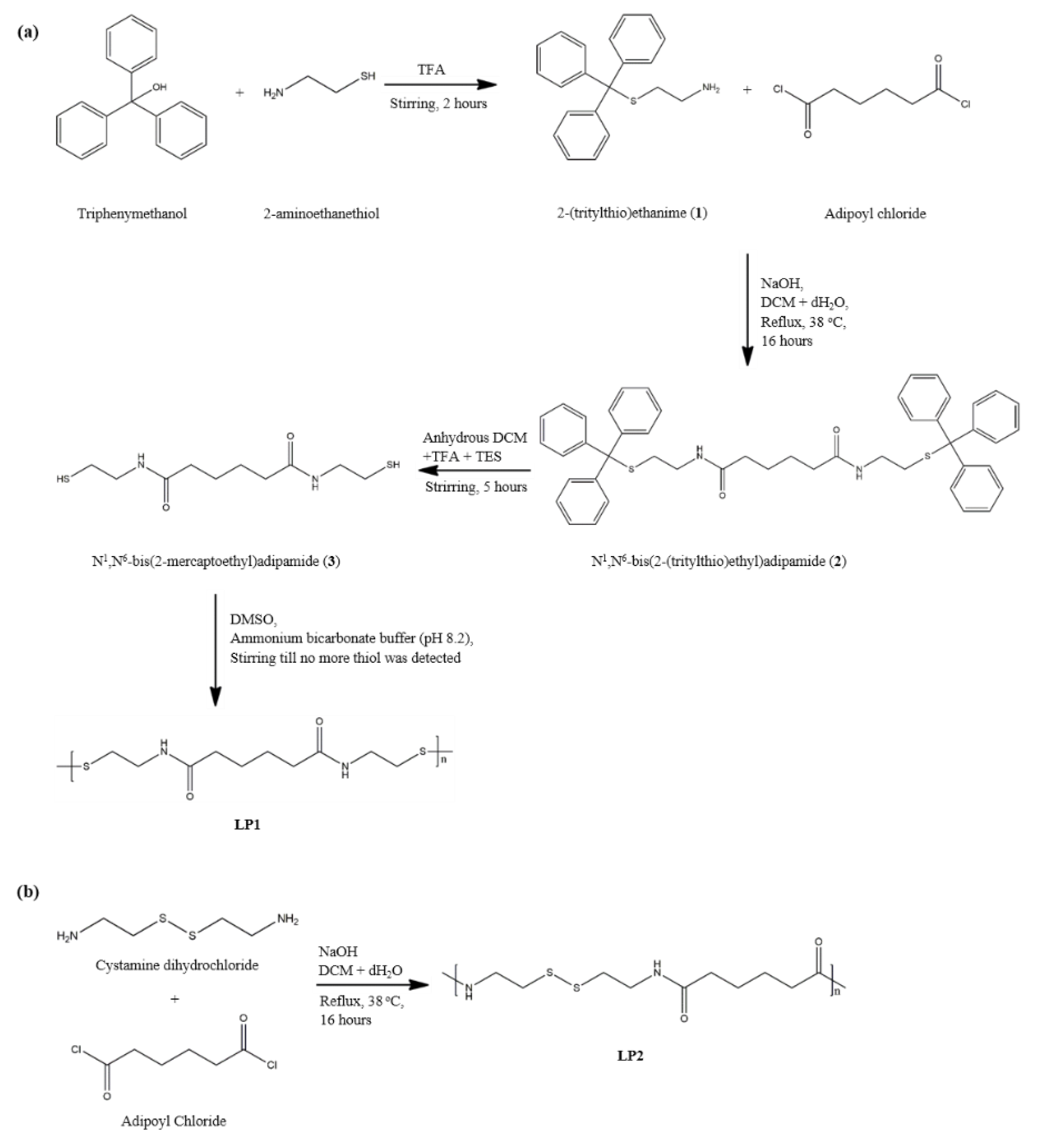
2.2. Preparation of Cysteamine-Based Dithiol Monomer
2.2.1. Synthesis of (triphenylmethyl)thioethylamine, 1
2.2.2. Synthesis of N1, N6-bis(2-(tritylthio)ethyl)adipamide, 2
2.2.3. Removal of Trityl Group, N1, N6-bis(2-mercaptoethyl)adipamide, 3
2.3. Synthesis of Cystamine-Based Polymer via Oxidative Polymerisation, LP1
2.4. Alternative Method of Synthesising Cystamine-Based Linear Disulfide Polymer, LP2
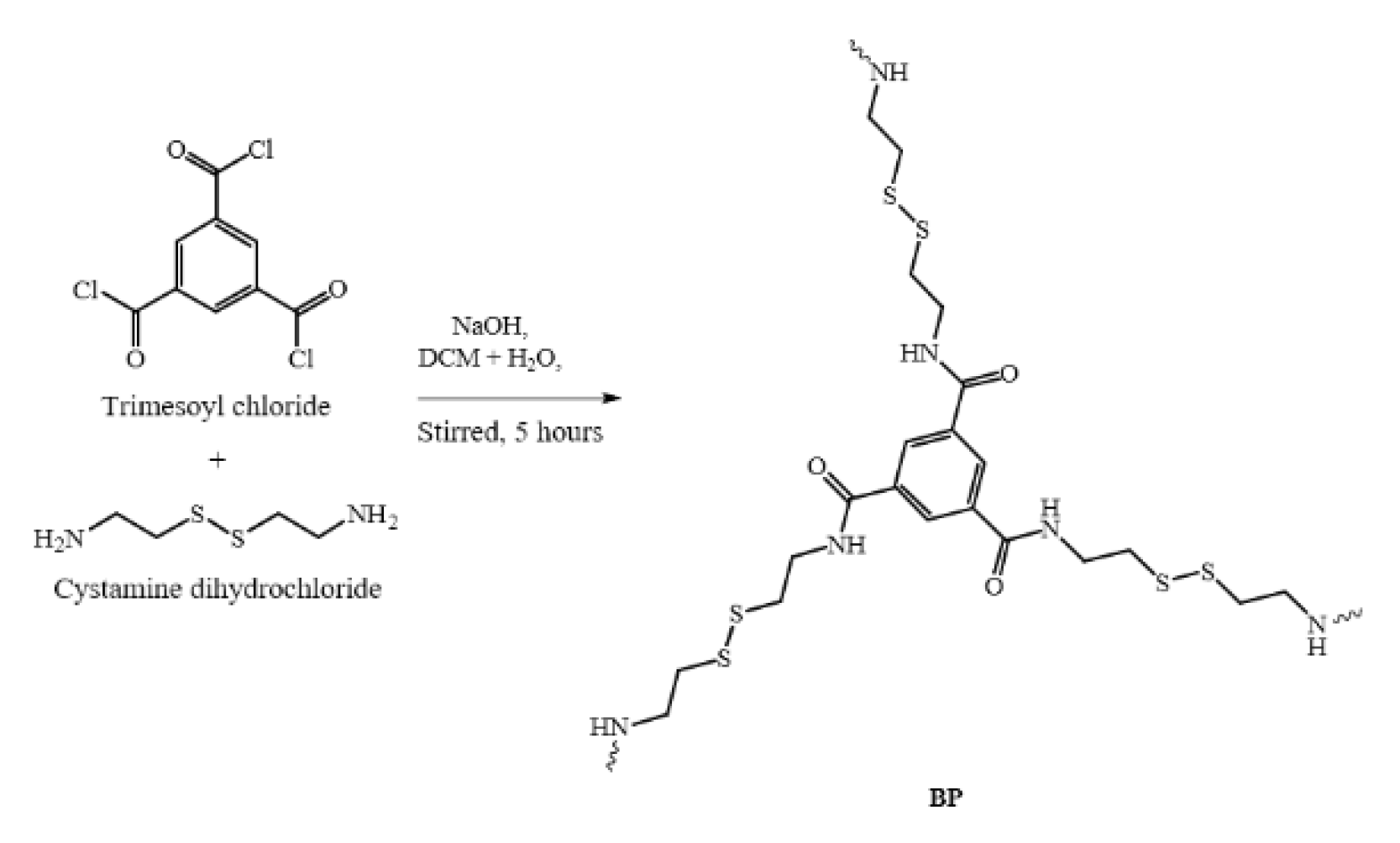
2.5. Synthesis of Cystamine-Based Crosslinked Polymer, BP
2.6. Physical Characterisation of the Synthesised Compounds
2.6.1. Fourier Transform Infrared Resonance (FTIR)
2.6.2. 1-Dimensional Nuclear Magnetic Resonance (1D-NMR)
2.6.3. Solid State Nuclear Magnetic Resonance (CP/MAS 13C NMR)
2.6.4. Solubility Test for Disulfide Cross-Linked Polymers
2.6.5. Melting Point Analysis
2.6.6. Scanning Electron Microscopy-Energy Dispersion X-Ray (SEM-EDX)
2.7. Chemical Reduction Studies of the Synthesised Disulfide Polymers
2.7.1. Measurement of Thiol Using Ellman’s Reagent
2.7.2. Chemical Reduction Studies
2.8. Degradation Studies of Disulfide Polymers
2.8.1. Preparation of de-Aerated SØrensen Phosphate Buffer, pH 7.4
2.8.2. Preparation of Polymers in Visking Dialysis Bags
2.8.3. Incubation of Polymers in Simulated Gastric Fluid
2.8.4. Incubation of Polymers in Simulated Intestinal Fluid
2.8.5. Incubation of Polymers with Lactobacillus in SØrensen’s Phosphate Buffer
2.8.6. Control Incubation
2.8.7. Statistical Analysis
3. Results and Discussion
3.1. Synthesis and Characterisation of 3
3.2. Synthesis and Characterisation of Polymers
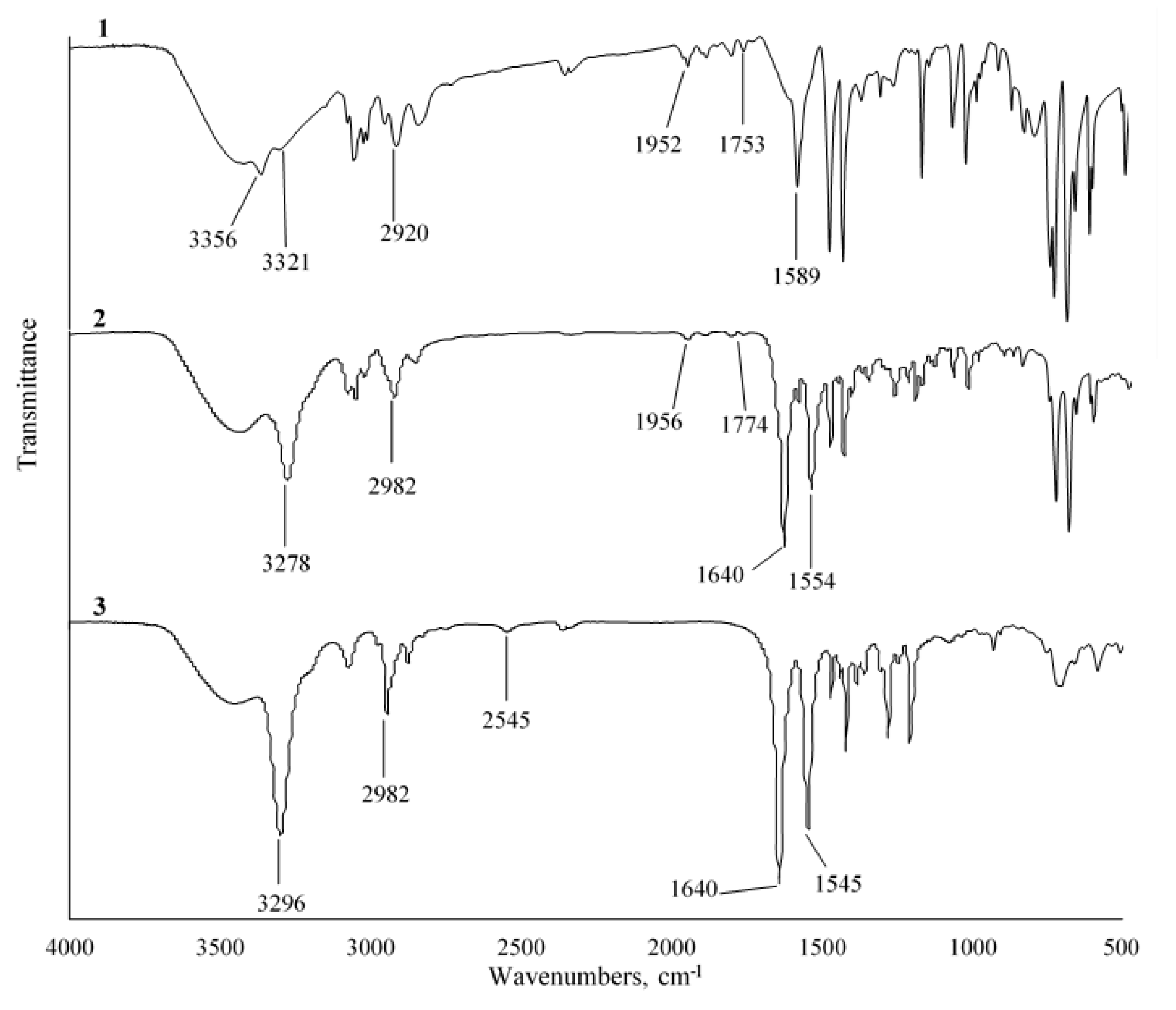
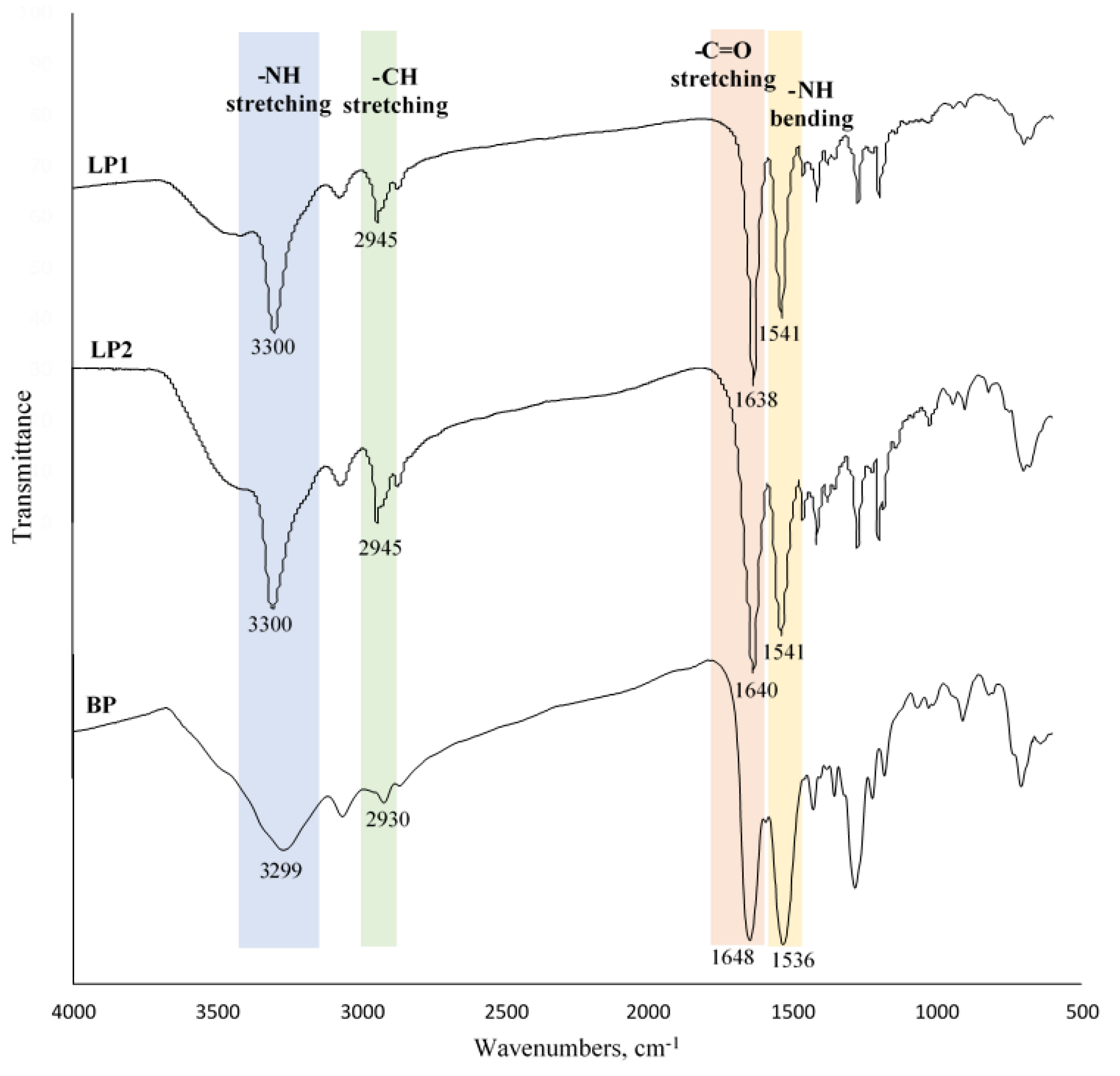
3.2.1. FTIR Analysis
3.2.2. NMR Analysis
3.2.3. Solubility of LP1, LP2, and BP
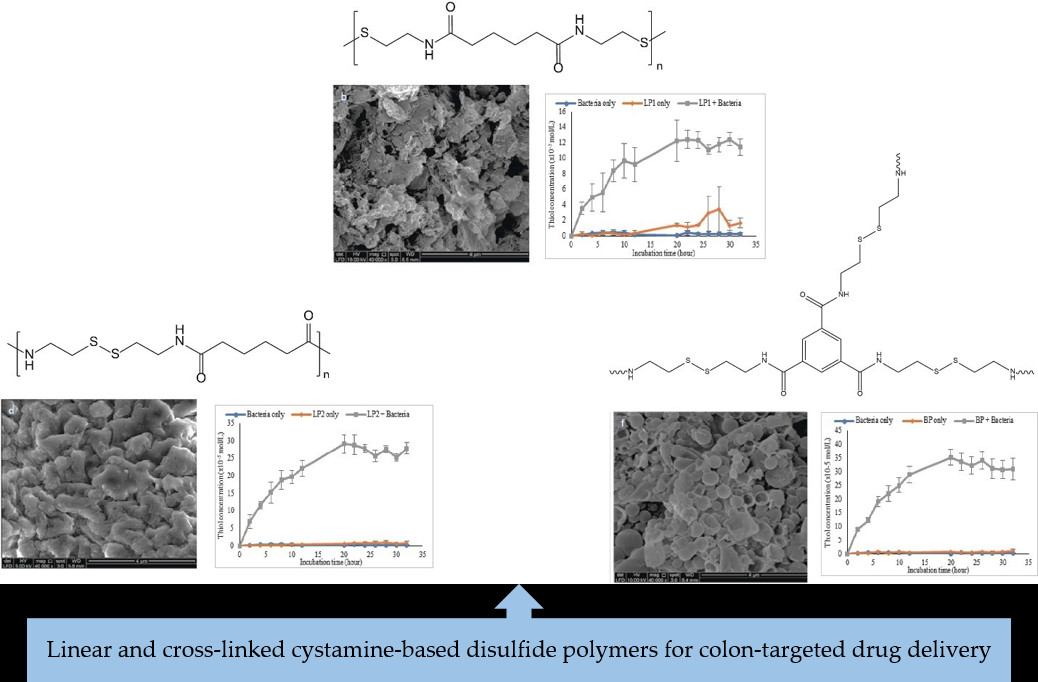
3.2.4. SEM Morphology of the Polymers
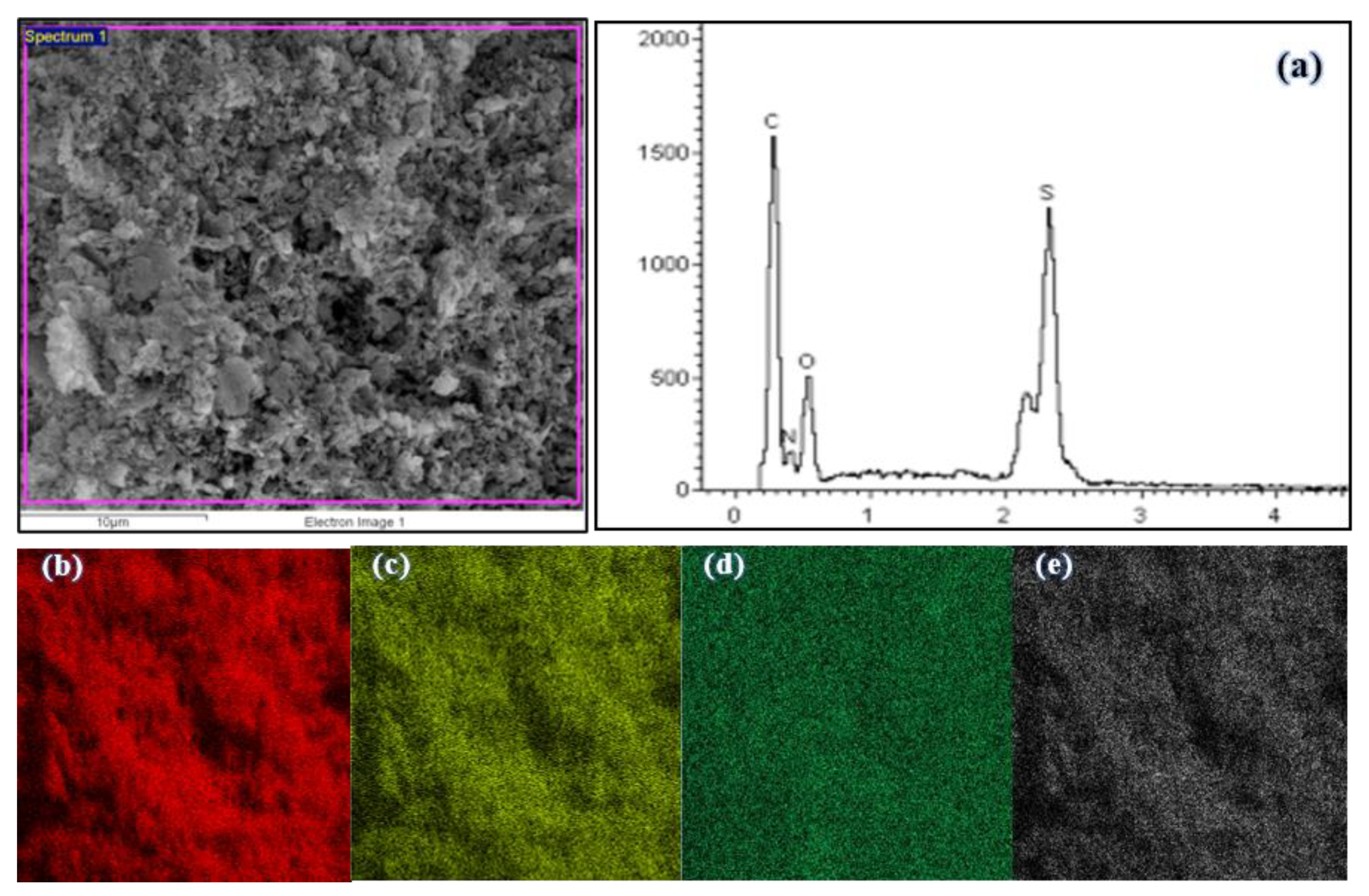
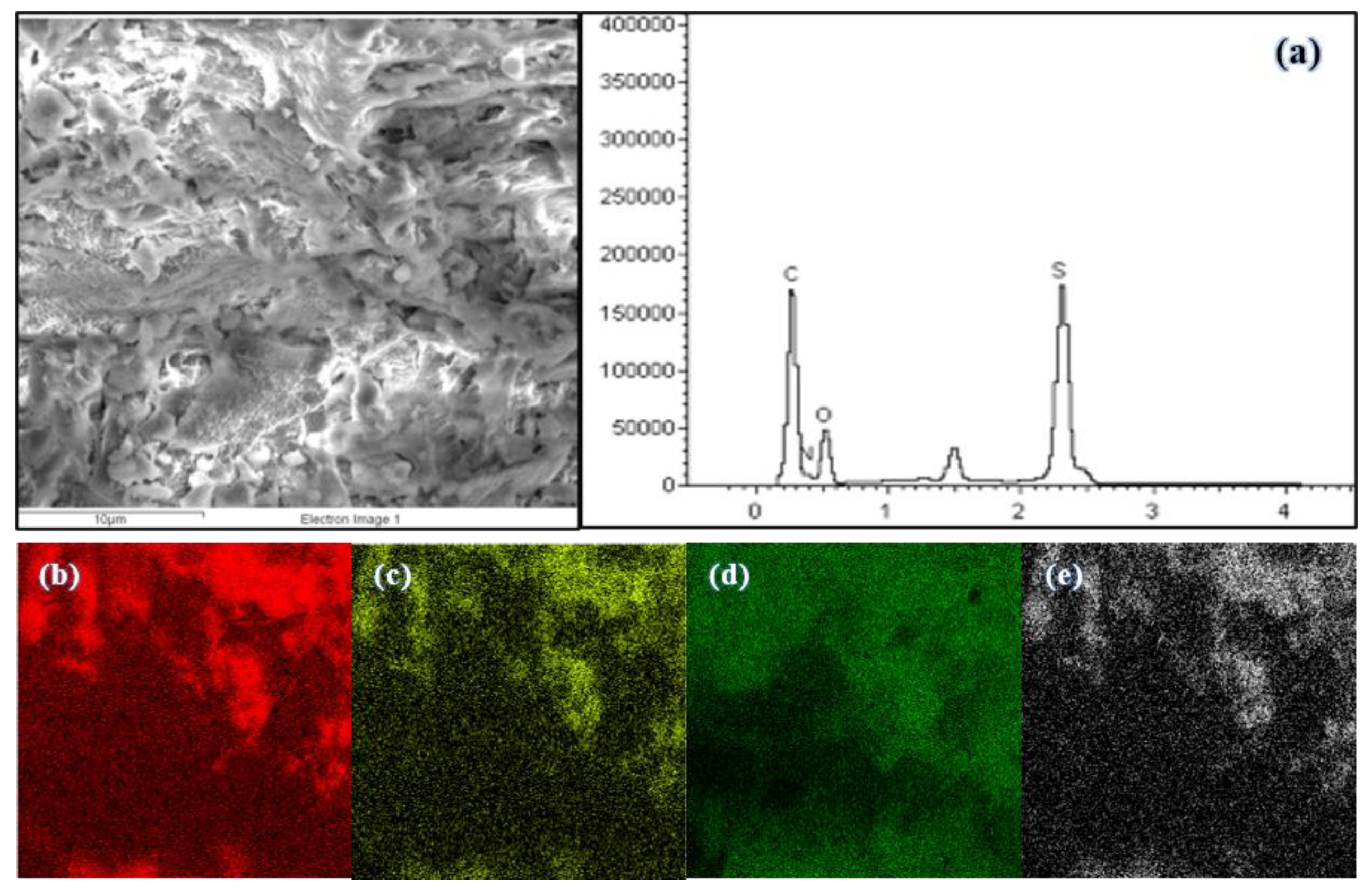
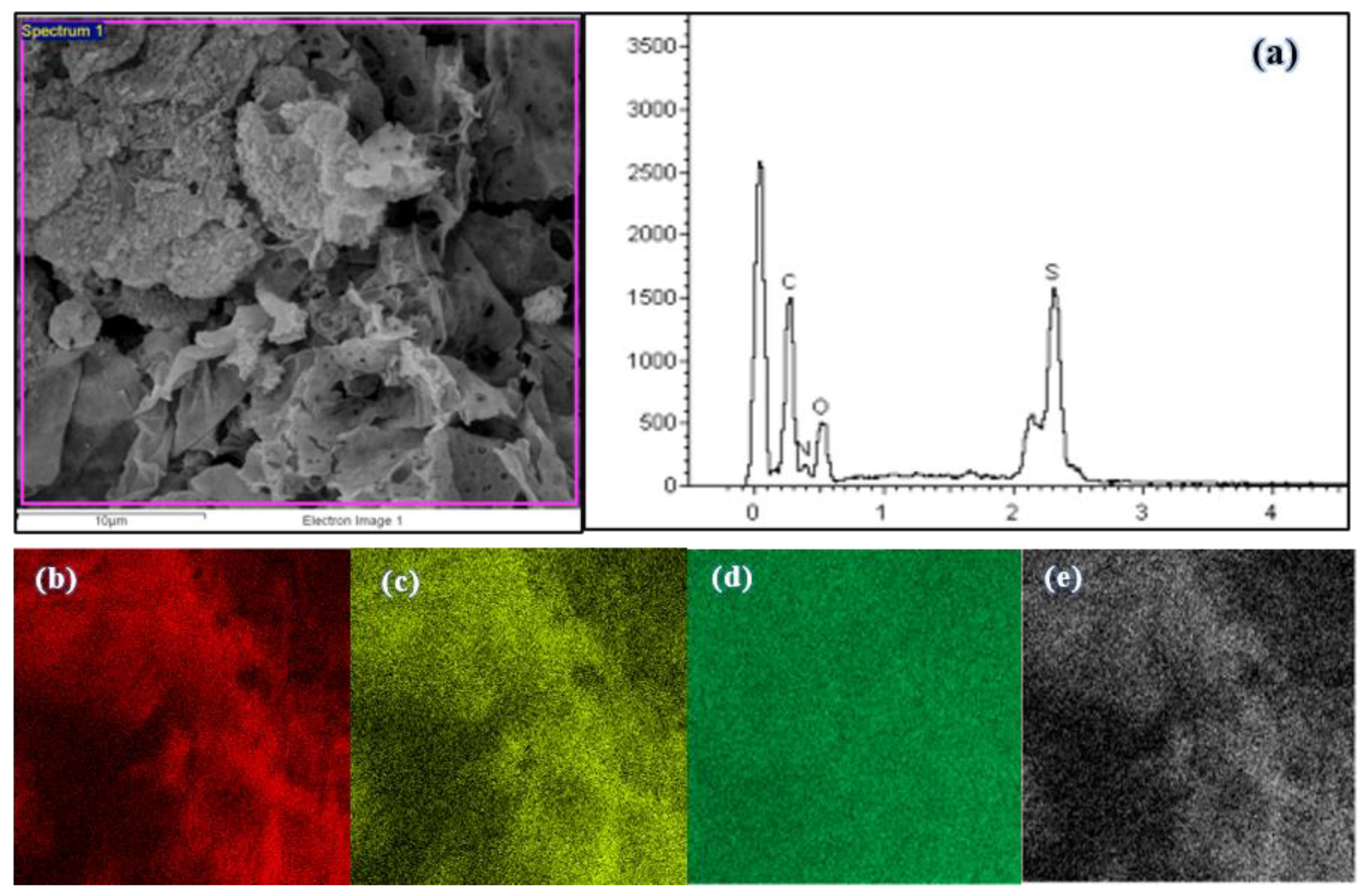
3.2.5. EDX Micrograph and Elemental Mapping of Polymers
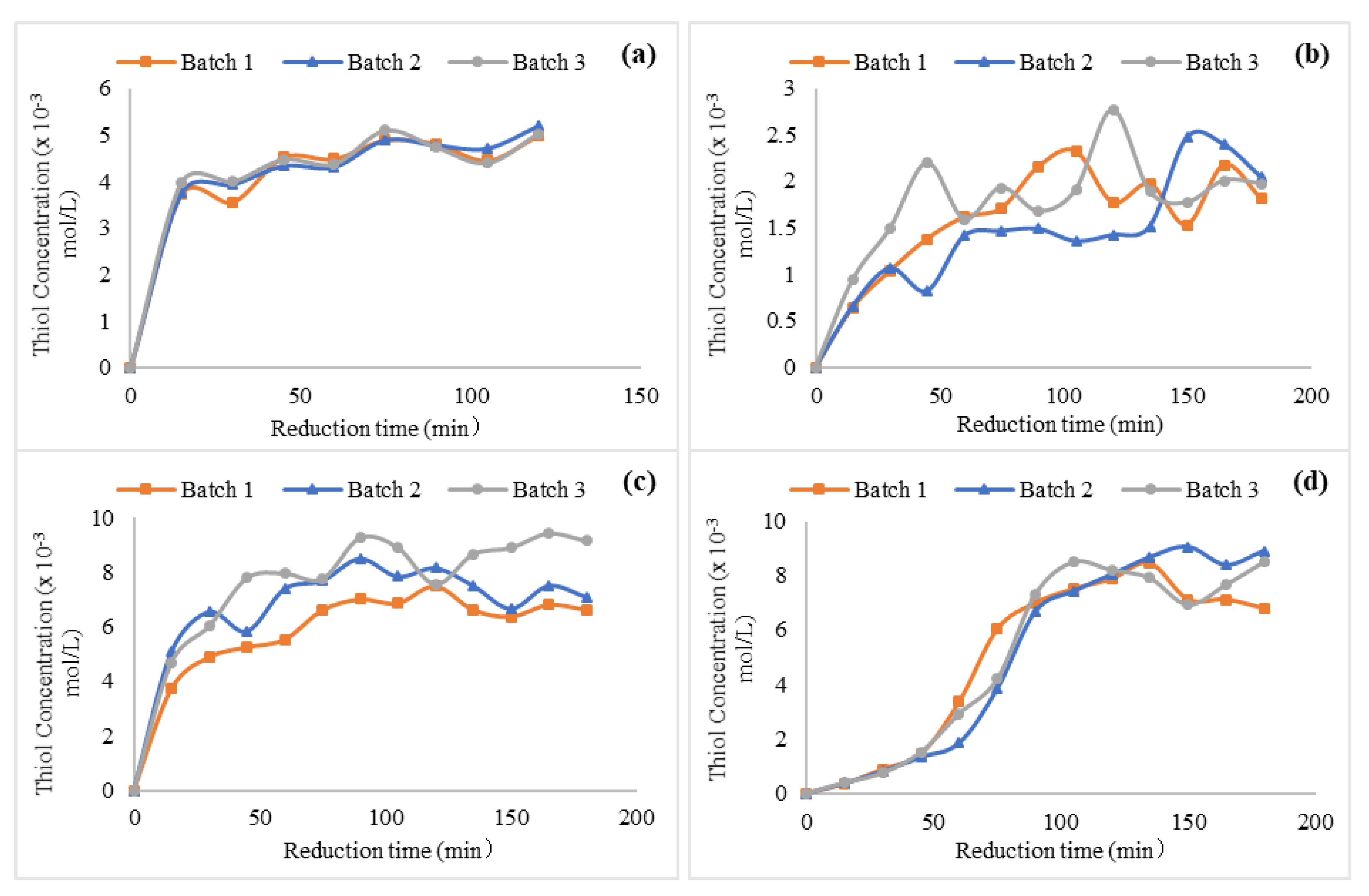
3.2.6. Chemical Reduction of Disulfide Polymers
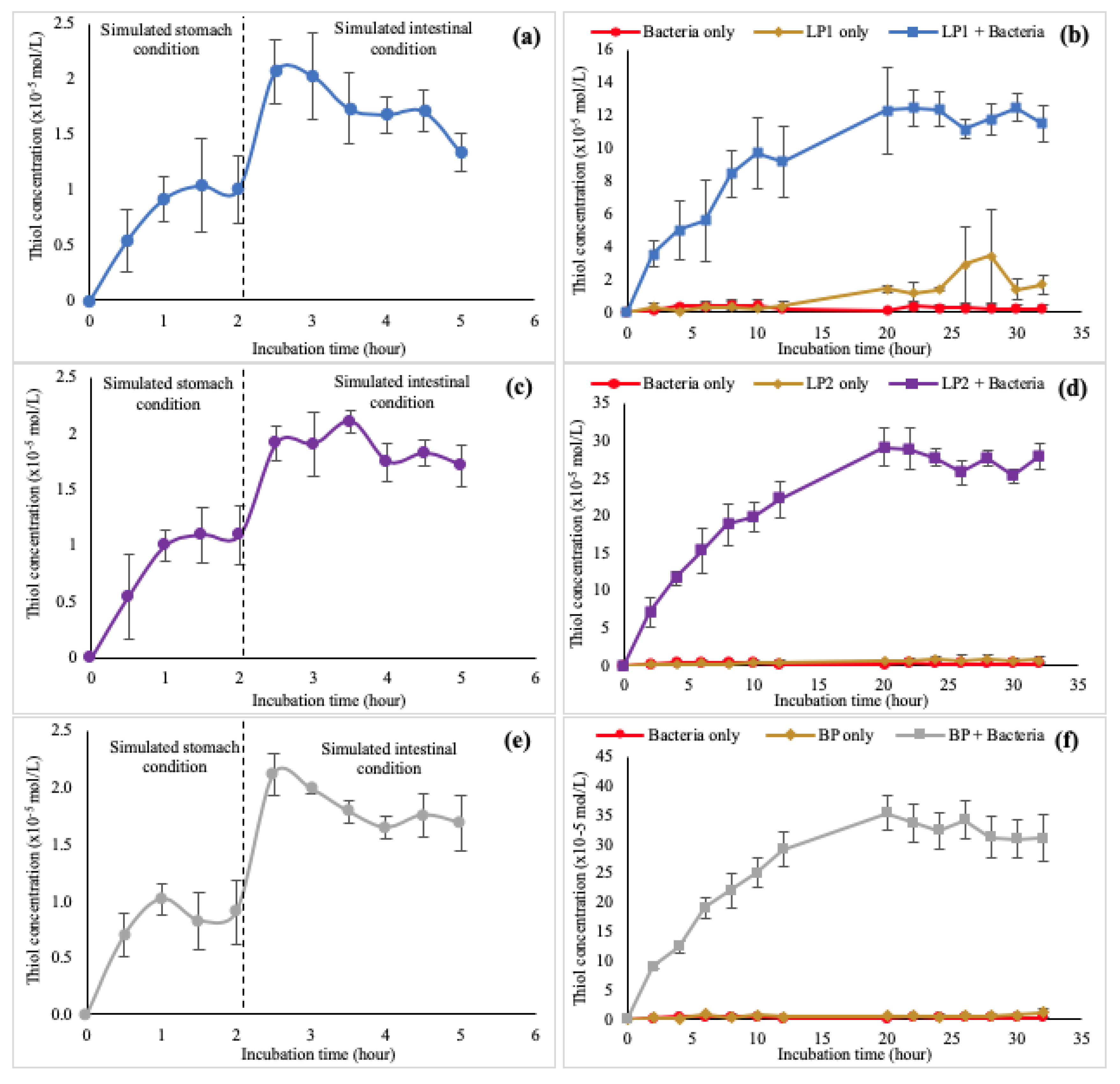
3.2.7. In Vitro Dissolution of Polymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Amidon, S.; Brown, J.E.; Dave, V.S. Colon-Targeted Oral Drug Delivery Systems: Design Trends and Approaches. AAPS PharmSciTech 2015, 16, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.R.; Kumria, R. Polysaccharides in colon-specific drug delivery. Int. J. Pharm. 2001, 224, 19–38. [Google Scholar] [CrossRef]
- Ensign, L.M.; Cone, R.; Hanes, J. Oral drug delivery with polymeric nanoparticles: The gastrointestinal mucus barriers. Adv. Drug Deliv. Rev. 2012, 64, 557–570. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A.M.; Gran, M.P.; Peppas, N.A. Designing the new generation of intelligent biocompatible carriers for protein and peptide delivery. Acta Pharm. Sin. B 2018, 8, 147–164. [Google Scholar] [CrossRef]
- Cheng, W.; Gu, L.; Ren, W.; Liu, Y. Stimuli-responsive polymers for anti-cancer drug delivery. Mater. Sci. Eng. C 2015, 45, 600–608. [Google Scholar] [CrossRef]
- Oshi, M.A.; Naeem, M.; Bae, J.; Kim, J.; Lee, J.; Hasan, N.; Kim, W.; Im, E.; Jung, Y.; Yoo, J.-W. Colon-targeted dexamethasone microcrystals with pH-sensitive chitosan/alginate/Eudragit S multilayers for the treatment of inflammatory bowel disease. Carbohydr. Polym. 2018, 198, 434–442. [Google Scholar] [CrossRef]
- Thapaliya, R.; Shrestha, K.; Sharma, A.; Dhakal, N.; Manandhar, P.; Shrestha, S.; Bhattarai, R. Physicochemical characterization of naproxen microcrystals for colon specific pulsatile drug delivery designed using pulsincap technique. J. Pharm. Investig. 2019, 49, 553–564. [Google Scholar] [CrossRef]
- Patel, D.M.; Jani, R.H.; Patel, C.N. Design and evaluation of colon targeted modified pulsincap delivery of 5-fluorouracil according to circadian rhythm. Int. J. Pharm. Investig. 2011, 1, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Naeem, M.; Awan, U.A.; Subhan, F.; Cao, J.; Hlaing, S.P.; Lee, J.; Im, E.; Jung, Y.; Yoo, J.-W. Advances in colon-targeted nano-drug delivery systems: Challenges and solutions. Arch. Pharmacal Res. 2020, 43, 153–169. [Google Scholar] [CrossRef]
- Pinto, J.F. Site-specific drug delivery systems within the gastro-intestinal tract: From the mouth to the colon. Int. J. Pharm. 2010, 395, 44–52. [Google Scholar] [CrossRef]
- Ma, Z.-G.; Ma, R.; Xiao, X.-L.; Zhang, Y.-H.; Zhang, X.-Z.; Hu, N.; Gao, J.-L.; Zheng, Y.-F.; Dong, D.-L.; Sun, Z.-J. Azo polymeric micelles designed for colon-targeted dimethyl fumarate delivery for colon cancer therapy. Acta Biomater. 2016, 44, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N. Modified-release solid formulations for colonic delivery. Recent Pat. Drug Deliv. Formul. 2007, 1, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Ayub, A.D.; Chiu, H.I.; Mat Yusuf, S.N.A.; Abd Kadir, E.; Ngalim, S.H.; Lim, V. Biocompatible disulphide cross-linked sodium alginate derivative nanoparticles for oral colon- targeted drug delivery. Artif. Cells Nanomed. Biotechnol. 2019, 47, 353–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saphier, S.; Haft, A.; Margel, S. Bacterial reduction as means for colonic drug delivery: Can other chemical groups provide an alternative to the azo bond? J. Med. Chem. 2012, 55, 10781–10785. [Google Scholar] [CrossRef] [PubMed]
- Jose, S.; Dhanya, K.; Cinu, T.A.; Litty, J.; Chacko, A.J. Colon targeted drug delivery: Different approaches. J. Young Pharm. 2009, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Chiu; Ayub; Mat, Y.; Yahaya; Abbd, K.; Lim. Docetaxel-Loaded Disulfide Cross-Linked Nanoparticles Derived from Thiolated Sodium Alginate for Colon Cancer Drug Delivery. Pharmaceutics 2020, 12, 38. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Cheng, Y.; Zhao, X.; Luo, Y.; Chen, J.; Yuan, W.E. Advances in redox-responsive drug delivery systems of tumor microenvironment. J. Nanobiotechnol. 2018, 16, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, Y.; Lim, V. Colon targeted drug delivery of branch-chained disulphide cross-linked polymers: Design, synthesis, and characterisation studies. Chem. Cent. J. 2016, 10, 77. [Google Scholar] [CrossRef] [Green Version]
- Jeitner, T.M.; Pinto, J.T.; Cooper, A.J.L. Cystamine and cysteamine as inhibitors of transglutaminase activity in vivo. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Ayyavu, M.; Shanmugarathinam, A.; Kandasamy, R. Synthesis and characterization of cystamine conjugated chitosan-SS-mPEG based 5-Fluorouracil loaded polymeric nanoparticles for redox responsive drug release. Eur. J. Pharm. Sci. 2018, 116, 37–47. [Google Scholar] [CrossRef]
- Fraser-Pitt, D.J.; Mercer, D.K.; Smith, D.; Kowalczuk, A.; Robertson, J.; Lovie, E.; Perenyi, P.; Cole, M.; Doumith, M.; Hill, R.L.R.; et al. Cysteamine, an Endogenous Aminothiol, and Cystamine, the Disulfide Product of Oxidation, Increase Pseudomonas aeruginosa Sensitivity to Reactive Oxygen and Nitrogen Species and Potentiate Therapeutic Antibiotics against Bacterial Infection. Infect. Immun. 2018, 86, e00947-00917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, V.; Khiang Peh, K.; Sahudin, S. Synthesis, characterisation, and evaluation of a cross-linked disulphide amide-anhydride-containing polymer based on cysteine for colonic drug delivery. Int. J. Mol. Sci. 2013, 14, 24670–24691. [Google Scholar] [CrossRef] [PubMed]
- Pearson, D.A.; Blanchette, M.; Baker, M.L.; Guindon, C.A. Trialkylsilanes as scavengers for the trifluoroacetic acid deblocking of protecting groups in peptide synthesis. Tetrahedron Lett. 1989, 30, 2739–2742. [Google Scholar] [CrossRef]
- Fearon, W.R. An Introduction to Biochemistry; Elsevier Science: London, UK, 2014. [Google Scholar]
- Broaders, K.E.; Pastine, S.J.; Fre, J.M.J. Acid-degradable solid-walled microcapsules for pH-responsive burst-release drug delivery. Chem. Commun. 2011, 47, 665–667. [Google Scholar] [CrossRef]
- Mat Yusuf, S.N.A.; Ng, Y.M.; Ayub, A.D.; Ngalim, S.H.; Lim, V. Characterisation and evaluation of trimesic acid derivatives as disulphide cross-linked polymers for potential colon targeted drug delivery. Polymers 2017, 9, 311. [Google Scholar] [CrossRef] [Green Version]
- British Pharmacopoeia. In Recommendation on Dissolution Testing; The Stationery Office: London, UK, 2016; pp. 358–359.
- Pavia, D.L.; Lampman, G.M.; Kriz, G.S.; Vyvyan, J.A. Introduction to Spectroscopy; Cengage Learning: Stanford, CA, USA, 2008. [Google Scholar]
- Imagawa, H.; Tsuchihashi, T.; Singh, R.K.; Yamamoto, H.; Sugihara, T.; Nishizawa, M. Triethyl- (or trimethyl-) silyl triflate-catalyzed reductive cleavage of triphenylmethyl (trityl) ethers with triethylsilane. Org. Lett. 2003, 5, 153–155. [Google Scholar] [CrossRef]
- Williams, D.L.H. Nitrosation Reactions and the Chemistry of Nitric Oxide; Elsevier Science: Amsterdam, The Netherlands, 2004; pp. 161–169. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, L.; Chen, L.; Dastidar, S.G.; Verma, C.; Li, J.; Tan, D.; Beuerman, R. Effect of structural parameters of peptides on dimer formation and highly oxidized side products in the oxidation of thiols of linear analogues of human β-defensin 3 by DMSO. J. Pept. Sci. 2009, 15, 95–106. [Google Scholar] [CrossRef]
- García Ruano, J.L.; Parra, A.; Alemán, J. Efficient synthesis of disulfides by air oxidation of thiols under sonication. Green Chem. 2008, 10, 706. [Google Scholar] [CrossRef]
- Tam, J.P.; Wu, C.R.; Liu, W.; Zhang, J.W. Disulfide bond formation in peptides by dimethyl sulfoxide. Scope and applications. J. Am. Chem. Soc. 1991, 113, 6657–6662. [Google Scholar] [CrossRef]
- Chen, L.; Annis, I.; Barany, G. Disulfide bond formation in peptides. Curr. Protoc. Protein Sci. 2001, 23, 18.16.11–18.16.19. [Google Scholar] [CrossRef]
- Tangerman, A. Measurement and biological significance of the volatile sulfur compounds hydrogen sulfide, methanethiol and dimethyl sulfide in various biological matrices. J. Chromatogr. B 2009, 877, 3366–3377. [Google Scholar] [CrossRef] [PubMed]
- Oswald, A.A.; Noel, F.; Stephenson, A.J. Organic sulfur compounds. V. Alkylammonium thiolate and peroxide salts; possible intermediates in amine-catalyzed oxidation of mercaptans by hydroperoxides. J. Org. Chem. 1961, 26, 3969–3974. [Google Scholar] [CrossRef]
- Capozzi, G.; Modena, G. The Thiol Group; Wiley: London, UK, 2010; pp. 785–839. [Google Scholar]
- King, J.F.; Gill, M.S.; Klassen, D.F. Mechanisms of reactions of sulfonyl compounds with nucleophiles in protic media. Pure Appl. Chem. 1996, 68, 825–830. [Google Scholar] [CrossRef]
- Williams, J.C.; Meador, M.A.B.; McCorkle, L.; Mueller, C.; Wilmoth, N. Synthesis and properties of step-growth polyamide aerogels cross-linked with triacid chlorides. Chem. Mater. 2014, 26, 4163–4171. [Google Scholar] [CrossRef]
- Mathias, L.J.; Johnson, C.G. Solid-state NMR investigation of nylon-12. Macromolecules 1991, 24, 6114–6122. [Google Scholar] [CrossRef]
- Hatfield, G.R.; Glans, J.H.; Hammond, W.B. Characterization of structure and morphology in nylon 6 by solid-state carbon-13 and nitrogen-15 NMR. Macromolecules 1990, 23, 1654–1658. [Google Scholar] [CrossRef]
- Curran, S.A.; Laclair, C.P.; Aharoni, S.M. Solid-state NMR of aromatic polyamides. Macromolecules 1991, 24, 5903–5909. [Google Scholar] [CrossRef]
- Jayakannan, M.; Annu, S.; Ramalekshmi, S. Structural effects of dopants and polymerization methodologies on the solid-state ordering and morphology of polyaniline. J. Polym. Sci. Part B-Polym. Phys. 2005, 43, 1321–1331. [Google Scholar] [CrossRef]
- Bouchemal, K.; Briancon, S.; Perrier, E.; Fessi, H.; Bonnet, I.; Zydowicz, N. Synthesis and characterization of polyurethane and poly(ether urethane) nanocapsules using a new technique of interfacial polycondensation combined to spontaneous emulsification. Int. J. Pharm. 2004, 269, 89–100. [Google Scholar] [CrossRef]
- Karode, S.; Kulkarni, S.; Suresh, A.; Mashelkar, R. New insights into kinetics and thermodynamics of interfacial polymerization. Chem. Eng. Sci. 1998, 53, 2649–2663. [Google Scholar] [CrossRef]
- Aboubakar, M.; Puisieux, F.; Couvreur, P.; Deyme, M.; Vauthier, C. Study of the mechanism of insulin encapsulation in poly(isobutylcyanoacrylate) nanocapsules obtained by interfacial polymerization. J. Biomed. Mater. Res. 1999, 47, 568–576. [Google Scholar] [CrossRef]
- El-Gibaly, I.; Anwar, M. Hemolysate-filled polyethyleneimine and polyurea microcapsules as potential red blood cell substitutes: Effect of aqueous monomer type on properties of the prepared microcapsules. Int. J. Pharm. 2004, 278, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Montasser, I.; Briançon, S.; Fessi, H. The effect of monomers on the formulation of polymeric nanocapsules based on polyureas and polyamides. Int. J. Pharm. 2007, 335, 176–179. [Google Scholar] [CrossRef]
- Piradashvili, K.; Alexandrino, E.M.; Wurm, F.R.; Landfester, K. Reactions and polymerizations at the liquid–liquid interface. Chem. Rev. 2015, 116, 2141–2169. [Google Scholar] [CrossRef]
- Freitas, S.; Merkle, H.P.; Gander, B. Microencapsulation by solvent extraction/evaporation: Reviewing the state of the art of microsphere preparation process technology. J. Control. Release 2005, 102, 313–332. [Google Scholar] [CrossRef]
- Budhian, A.; Siegel, S.J.; Winey, K.I. Haloperidol-loaded PLGA nanoparticles: Systematic study of particle size and drug content. Int. J. Pharm. 2007, 336, 367–375. [Google Scholar] [CrossRef]
- Witt, D. Recent developments in disulfide bond formation. Synthesis 2008, 2008, 2491–2509. [Google Scholar] [CrossRef]
- Liu, H.-N.; Wu, H.; Chen, Y.-Z.; Chen, Y.-J.; Shen, X.-Z.; Liu, T.-T. Altered molecular signature of intestinal microbiota in irritable bowel syndrome patients compared with healthy controls: A systematic review and meta-analysis. Dig. Liver Dis. 2017, 49, 331–337. [Google Scholar] [CrossRef]
- Zhuang, X.; Xiong, L.; Li, L.; Li, M.; Chen, M. Alterations of gut microbiota in patients with irritable bowel syndrome: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2017, 32, 28–38. [Google Scholar] [CrossRef]
- Wilding, I.; Davis, S.; O’Hagan, D. Targeting of drugs and vaccines to the gut. Pharmacol. Ther. 1994, 62, 97–124. [Google Scholar] [CrossRef]
- Scheline, R.R. Metabolism of foreign compounds by gastrointestinal microorganisms. Pharmacol. Rev. 1973, 25, 451–523. [Google Scholar] [PubMed]
- Mathiowitz, E.; Cohen, M.D. Polyamide microcapsules for controlled release. I. Characterization of the membranes. J. Membr. Sci. 1989, 40, 1–26. [Google Scholar] [CrossRef]
- Mathiowitz, E.; Cohen, M.D. Polyamide microcapsules iv. effects of swelling for controlled release. J. Membr. Sci. 1989, 40, 55–65. [Google Scholar] [CrossRef]
- Kawabata, N.; Uchihori, D.; Fukuda, S.; Funahashi, H. Digestion of cross-linked poly(vinylpyridinium halide) by activated-sludge, and application to make poly(methyl methacrylate) biodegradable by incorporation of a pyridinium group into the main-chain. J. Appl. Polym. Sci. 1994, 51, 33–42. [Google Scholar] [CrossRef]
- Maitra, J.; Shukla, V.K. Cross-linking in hydrogels-a review. Am. J. Polym. Sci. 2014, 4, 25–31. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Solvent | Polymers | ||
|---|---|---|---|
| LP1 | LP2 | BP | |
| Trifluoroacetic acid | √ | √ | √ |
| Acetic acid | – | – | – |
| Formic acid | √ | √ | √ |
| Acetone | – | – | – |
| Chloroform | – | – | – |
| Cyclohexane | – | – | – |
| Dichloromethane | – | – | – |
| Dimethylsulfoxide | – | – | – |
| Ethanol | – | – | – |
| Toluene | – | – | – |
| Water | – | – | – |
| Buffer system of gastric (without pepsin) | – | – | – |
| Buffer system of intestinal (without pancreatin) | – | – | – |
| Buffer system of colon (without Lactobacillus) | – | – | – |
| Incubation Medium | LP1 | LP2 | BP |
|---|---|---|---|
| Gastric (a) | 1.00 ± 0.30 | 0.90 ± 0.28 | 1.09 ± 0.25 |
| Small intestinal (b) | 1.34 ± 0.17 | 1.70 ± 0.18 | 1.68 ± 0.24 |
| Colon (c) | 11.47 ± 1.08 | 27.93 ± 1.65 | 31.00 ± 3.92 |
| Statistical analysis | p < 0.05 | p < 0.05 | p < 0.05 |
| Dunnett’s (significance) | a & ci b & ci | a & cii b & cii | a & ci b & ci |
| Incubation Medium | LP1 | LP2 | BP |
|---|---|---|---|
| Bacteria only (a) | 0.22 ± 0.21 | 0.22 ± 0.21 | 0.22 ± 0.21 |
| Polymer only (b) | 1.67 ± 0.62 | 0.98 ± 0.52 | 1.11 ± 0.76 |
| Polymer + bacteria (c) | 11.46 ± 1.08 | 27.93 ± 1.65 | 31.00 ± 3.92 |
| Statistical analysis | p < 0.05 | p < 0.05 | p < 0.05 |
| Dunnett’s T (2-sided) (significance) | a & c b & c | a & c b & c | a & c b & c |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ng, Y.M.; Mat Yusuf, S.N.A.; Chiu, H.I.; Lim, V. Redox-Sensitive Linear and Cross-Linked Cystamine-Based Polymers for Colon-Targeted Drug Delivery: Design, Synthesis, and Characterisation. Pharmaceutics 2020, 12, 461. https://doi.org/10.3390/pharmaceutics12050461
Ng YM, Mat Yusuf SNA, Chiu HI, Lim V. Redox-Sensitive Linear and Cross-Linked Cystamine-Based Polymers for Colon-Targeted Drug Delivery: Design, Synthesis, and Characterisation. Pharmaceutics. 2020; 12(5):461. https://doi.org/10.3390/pharmaceutics12050461
Chicago/Turabian StyleNg, Yoke Mooi, Siti Nur Aishah Mat Yusuf, Hock Ing Chiu, and Vuanghao Lim. 2020. "Redox-Sensitive Linear and Cross-Linked Cystamine-Based Polymers for Colon-Targeted Drug Delivery: Design, Synthesis, and Characterisation" Pharmaceutics 12, no. 5: 461. https://doi.org/10.3390/pharmaceutics12050461