
Abstract
Introduction
The Pierre-Robin sequence (PRS) is a pattern of congenital facial abnormalities comprising micrognathia, glossoptosis, and airway obstruction. Associated spinal pathologies have rarely been reported with PRS.
Methods
We explore the molecular genetic basis of this association through a systematic review of spinal disease in patients with PRS. We also present an illustrative case of a PRS patient with tethered cord in the setting of chromosome 10q terminal deletion.
Results
Our systematic literature review of spinal disease in patients with PRS revealed several patterns in the underlying genetic syndromes causing these conditions to co-occur. These principles are illustrated in the case of a 6-month-old female with PRS and a 14.34-Mb terminal deletion of chromosome 10q, who was found to have a sacral dimple during a routine outpatient checkup. Magnetic resonance imaging of the spine revealed a lumbar syrinx associated with tethered spinal cord. Surgical de-tethering was undertaken, with subsequent improvement in motor function and decrease in the size of the syrinx. The deletion of chromosome 10q in our patient had not previously been described in association with tethered cord or PRS.
Conclusion
Spinal pathologies are understudied contributors to disease burden in patients with PRS. The range of predisposing syndromes and mutations in patients with both PRS and spinal disorders remains poorly characterized but may be more defined than previously conceived. Clinical screening is most critical during neonatal and adolescent developmental periods with continued neurological assessment. This study emphasizes the need for early genetic testing and counseling in this patient population, in parallel with research efforts to develop molecular classifications to guide clinical management.
Similar content being viewed by others
Background
The Pierre Robin sequence (PRS), also known as Robin sequence, is a pattern of congenital facial abnormalities comprising micrognathia, glossoptosis, and airway obstruction [1]. The reported incidence varies widely with an approximate occurrence of 1 in 8500 to 14,000 births. Approximately 50% of PRS cases are isolated (non-syndromic), while the remainder are associated with additional anomalies such as a genetic or acquired syndrome [2,3,4,5].
PRS is most commonly associated with hearing loss, dysmorphic facial features, global developmental delay/intellectual disability, and/or congenital heart defects [4]. Spinal pathologies have rarely been reported in association with PRS, often co-occurring with other congenital anomalies [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22]. The molecular genetic and clinical characteristics of spinal disease in PRS remain poorly characterized. We thus performed a systematic review of spinal disease in patients with PRS. We additionally report a case of a PRS patient presenting with tethered cord and lumbar syrinx in the setting of chromosome 10q terminal deletion.
Illustrative case
A 6-month-old female with a history of PRS was found to have a sacral dimple during a routine outpatient checkup. Neurological examination was notable for diffuse hypotonia and global developmental delay, but without focal deficit, incontinence, or prior urinary tract infections. No other cutaneous stigmata were noted. Medical history included premature birth at 31 weeks with grade I intraventricular hemorrhage. She had multiple congenital abnormalities including micrognathia, glossoptosis, and airway obstruction characteristic of PRS, as well as cleft palate, strabismus, patent ductus arteriosus, mild dilation of the aortic sinuses, clinodactyly of the fifth finger, and syndactyly involving the bilateral second and third toes.
Prior microarray analysis as a neonate had shown a 14.34 Mb terminal deletion of chromosome 10q. The deletion extended from band 10q26.11 to 10q26.3, comprising 112 genes including 62 OMIM genes (Supplementary Table 1). Genetic counseling was pursued, and the patient’s parents declined additional familial karyotyping.
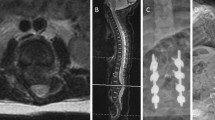
Magnetic resonance imaging (MRI) of the spine with and without contrast revealed a syrinx of the conus medullaris without other abnormalities (Fig. 1a). This was managed expectantly with repeat MRI at 12 months of age, which showed expansion of the syrinx and fatty infiltration of the filum terminale consistent with tethered spinal cord (Fig. 1b). Surgical release of the tethered cord was thus undertaken. The procedure was uneventful and the patient recovered well postoperatively. Follow-up MRI 8 months postoperatively showed slight decrease in the size of the syrinx (Fig. 1c). One year postoperatively, neurological examination remained nonfocal without any deficits referable to the tethered cord or surgical procedure. Motor and cognitive function were improved compared to initial presentation but with ongoing delay in meeting respective milestones.

Magnetic resonance imaging of the lumbosacral region. a Sagittal and axial T2-weighted scans at 6 months of age showing a syrinx of the conus medullaris. b Sagittal T2-weighted and axial T1-weighted scans at 12 months of age showing enlargement of the syrinx, borderline low-lying conus medullaris, and fatty infiltration of the filum terminale (arrowhead) consistent with tethered cord. c Sagittal T2-weighted scan 8 months after release of tethered cord showing decreased syrinx size
Systematic literature review
Methods
We performed a systematic search for all cases of spinal diseases in patients with PRS reported in the literature using the PubMed, Google Scholar, Trip, and MEDLINE databases. The search strategy and results are summarized in Fig. 2. Inclusion criteria were (1) confirmed PRS, (2) any spinal pathology, and (3) English language. Search strings included all combinations of the terms “Pierre Robin sequence” or “Robin sequence” with the terms “spine,” “spinal,” “vertebral,” “tethered,” and “scoliosis.” Abstracts and full-text articles were screened to identify reports that passed the inclusion criteria. Additional publications were identified from the references listed in each study. We excluded reports that lacked case-level descriptions of spinal pathology, clinical characteristics, or management. The Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) criteria were followed. Ethical approval for the case report was obtained from the Institutional Review Board of Rady Children’s Hospital.

Flow diagram showing the systematic process of selecting studies included in the analysis of spinal pathologies associated with Pierre Robin sequence
Results
Our literature review of spinal pathology in patients with PRS identified 16 previously reported cases of spinal diseases (Table 1). The majority of patients presented with more than one spinal pathology. The most common disorders included congenital kyphoscoliosis (n = 7), cervical instability (n = 5), tethered cord (n = 2), and caudal regression syndrome (n = 2). Two patients had neural tube defects including spina bifida and myelomeningocele (n = 1 each). Although the majority of patients had multiple other congenital anomalies, only six patients had a known underlying genetic abnormality. Of the remaining ten patients, four had reportedly normal genetic analysis such as karyotype and/or microarray, while genetic testing was not reported in six.
Patients predominantly presented at birth or adolescence, with two patients presenting with spinal deformities at 8 years of age. Clinical presentations corresponded to the spinal levels and pathologies involved. Surgical management was pursued in nine cases (53%). The remainder of cases were managed medically. The majority of patients managed surgically had complete resolution of symptoms without neurologic complications. One patient with congenital vertebral dislocation and severe congenital scoliosis who underwent posterior spinal fusion sustained an iatrogenic spinal cord injury that caused worsened scoliosis [17]. Four patients encountered anesthesia-related challenges, including prolonged intubation postoperatively (n = 3) [7, 10, 16], nasopharyngeal intubation requirement (n = 3) [7, 16, 20], and tracheostomy requirement (n = 1) [10].
Tubbs et al. [8] reported the first case of tethered cord in a neonate with PRS and normal karyotype, co-occurring with several congenital anomalies including Chiari I malformation and caudal regression syndrome [8]. Abraham et al. described a 15-year-old patient with tethered cord, co-occurring with myelomeningocele and kyphoscoliosis; genetic testing was not reported [16]. Both patients were treated surgically, but outcomes were not reported.
We also performed another literature search to identify all genetic syndromes associated with PRS that are also known to predispose to spinal pathologies. These conditions are described in Table 2. Although more than 50 genetic syndromes have been associated with PRS [23], only 15 have also been associated with spinal pathologies. In 9 of these 15 syndromes, no patients have yet been reported who are affected by both PRS and spinal disease. The underlying molecular genetic disturbances predominantly involve genes responsible for extracellular matrix synthesis and organization, ribosome biosynthesis, and Golgi complex function. These genes are most commonly located on chromosomes 5, 12, and 17, with isolated cases of mutations in genomic regions 6p, 7p, and 22q. To our knowledge, ours is the first reported case of PRS and spinal pathology involving abnormalities of chromosome 10.
Discussion and conclusions
Pierre Robin sequence (PRS), also known as Robin sequence (RS), begins with micrognathic mandible, leading to retro-positioning of the tongue (glossoptosis) and increased likelihood of airway compression at the level of the glottis. The inability of the base of the tongue to descend from the nasopharyngeal roof in turn impairs palate formation, leading to cleft palate [5, 24]. The triad of micrognathia, glossoptosis, and cleft palate is termed syndromic PRS (or “RS-plus”) when it occurs in association with an underlying genetic or acquired syndrome, in contrast to non-syndromic PRS occurring in isolation. Syndromic PRS carries a higher mortality than isolated PRS, making it imperative to establish definitive early diagnosis [25]. Disorders commonly associated with PRS include Stickler syndrome (most common) as well as 22q11.2 deletion, Treacher Collins, campomelic dysplasia, and Marshall syndromes. Chromosomal loci harboring genes associated with pre- and postnatal growth, neurodevelopment, morphogenesis, and patterning have been implicated in the development of PRS; these include regions 2q24.1–33.3, 4q32-qter, 17q21–24.3, and 11q21–23.1, among others [26,27,28,29,30,31,32,33].
The molecular genetic understanding of PRS has primarily been derived from case reports or heterogeneous cohorts ranging from 66 to 191 subjects [4, 33,34,35]. These studies have been critical to elucidating the epidemiology of PRS but are limited by lack of uniformity in genetic testing of patients and family members. Patients predominantly undergo karyotyping by conventional methods or microarray analysis. Although these approaches are sensitive for detecting copy number gains and losses associated with chromosomal imbalances, they are unable to detect point mutations, mosaic conditions, small deletions or duplications, balanced structural rearrangements, or certain polyploidy patterns. Only a small minority of patients in existing cohorts have received targeted or whole exome sequencing (WES). Studies describing systematic next-generation genetic sequencing of PRS patients or family members remain lacking.
Conducting such studies is inherently challenging due to the low prevalence as well as clinical and genetic heterogeneity of PRS. In this context, recent studies of rare neurodevelopmental disorders such as congenital hydrocephalus (CH) serve as roadmaps for future investigation. For example, Kahle and colleagues have performed parent-offspring trio WES in CH cohorts of similar size to published PRS cohorts. This unbiased strategy has uncovered that a significant portion of CH cases are associated with mutations in a defined subset of genes involved in brain development. These insights have in turn enabled hypothesis-driven laboratory investigation to guide targeted therapy development, as well as molecularly guided clinical classification that could guide surgical treatment decisions [36].
In the absence of a large genetically sequenced cohort of PRS patients, retrospective clinical series may serve as de facto cohorts to illuminate the molecular underpinnings of this disorder and its relation to other pathologies of interest. Accordingly, our systematic review and case description consolidate the current understanding of the embryological and developmental processes that may be shared between PRS and spinal disorders.
Given that PRS is commonly caused by disorders of morphogenesis, it is unsurprising that the majority of spinal pathologies we found were structural in nature and often due to disorders of bone and cartilage development. Several cases were due to genetic abnormalities in genes such as COL2A1 that are critical for chondrogenesis (Table 1). Expression of these genes is regulated by a common transcription factor, SOX9, which has independently been associated with both PRS and spinal deformity (Table 2). Similarly, we identified another report of a patient with ischiospinal dysostosis (ISD), a genetic disorder due to defects in separate pathways regulating chondrogenesis (Table 1). Several patients in our series manifested signs of cervical instability and craniocervical junction pathology, likely due to the shared developmental programs regulating formation of the cervical and mandibular regions.
We identified three patients in our literature review with both Chiari type I malformation and PRS (patients 2, 12, and 14 in Table 1). Intriguingly, one of these cases occurred in the setting of neurofibromatosis type 2 (NF2) (patient 12). Another patient had phenotypic features similar to DiGeorge syndrome (patient 2), while the final patient had congenital craniocervical vertebral fusion anomalies (patient 14). Given that NF2 and DiGeorge syndromes occur due to mutations at adjacent loci on the long arm of chromosome 22, we speculate that genes in this region may be responsible for shared developmental programs underlying morphogenesis of the mandible and spine, ultimately leading to PRS and disorders of the craniocervical junction, respectively. For example, preclinical studies have shown that the TBX1 gene on chromosome 22q11.2 is expressed in the pharyngeal arches, pouches, vertebral column, and tooth bud [23]. In this context, it is possible that deeper genetic characterization of the aforementioned patients may have revealed mutations in coding regions or regulatory elements of genes in these regions. Although neurofibromatosis type I (NF1) is also known to be associated with Chiari type I malformation [37], we did not encounter any patients with NF1 and PRS in our literature review (Table 1), nor any previously described association between NF1 and PRS (Table 2). However, the SOX9 and COG1 genes that underlie several developmental syndromes associated with both PRS and spinal pathologies are located in proximity to the NF1 gene on the long arm of chromosome 17. It is thus possible that, as in the case of NF2, patients with genetic abnormalities leading to NF1 and Chiari I malformation may also have increased rates of PRS.
We found two prior reports of tethered cord co-occurring with PRS (Table 1, patients 2 and 10). Genetic analysis and diagnostic information regarding underlying syndromes were not reported. Both patients had multiple spinal deformities such as scoliosis that are known to be associated with tethered cord. Although tethered cord has not previously been linked to the terminal region of chromosome 10q deleted in our patient, she displayed many phenotypic features previously associated with this segment, including facial abnormalities, growth and psychomotor delay, and digital anomalies [38]. Deletions of 10q26 have been reported in less than 30 prior cases; this segment contains multiple genes and regions critical for a variety of developmental processes [39] but has not previously been associated with PRS or tethered cord. It remains unclear which specific gene(s) in this region or deleted in our patient’s case (listed in Supplementary Table 1) may be responsible for either condition.
Roberti and colleagues described a 22-year-old patient with PRS and spinal deformity who harbored an interstitial chromosome 12q microdeletion covering more than 20 known genes and causing a complex malformative syndrome [21]. As in our case, genotype-phenotype correlations were not clear due to the dearth of reported similar cases and multiple genes affected. These included genes involved in extracellular matrix synthesis and cellular processes such as vesicular trafficking, similar to the other patients in our series. Collectively, these cases highlight the need for obtaining early genetic testing, establishing individualized genotype-phenotype correlations, and carefully evaluating patients for signs of underlying spinal pathology such as sacral dimples, clubfoot deformities, and frequent urinary tract infections. Detecting conditions such as tethered cord requires a high degree of clinical suspicion in patients with complex syndromes that may mask classic presenting symptoms early in the disease course.
Structural conditions such as occipitocervical instability, Chiari I malformation, and Klippel-Feil syndrome were highly represented in our literature review among reported PRS cases with spinal pathologies present at birth (Table 1), suggesting dysregulation of a common embryological process(es) underlying mandibular and spinal development. We speculate that this may take place as early as the sixth week of embryogenesis. During this week, neural crest-derived cells form the osteogenic membrane of the mandible [23], while concurrently the notochord and neural tube induce chondrification of developing vertebral structures [40]. In both of these phenomena, intercellular signaling leads to synthesis of collagen and proteoglycans, the two primary components of cartilage, which in turn serves as the “mold” for bone formation [41]. As stated above, the SOX9 transcription factor regulates many steps in chondrification including the expression of several genes involved in collagen and proteoglycan synthesis [23]. Collectively, these observations suggest that SOX9 pathway mutations leading to disordered chondrification during the sixth developmental week may be a parsimonious genetic explanation for the concomitant development of PRS and congenital structural spinal pathologies.
The management of PRS requires multidisciplinary collaboration, particularly in the setting of underlying genetic syndromes. Our systematic review and case description demonstrate that spinal pathologies are important but understudied contributors to disease burden in this patient population. Our findings raise the possibility that in patients with both PRS and spinal disorders, the range of predisposing syndromes and mutations may be more defined than previously anticipated, with pathways such as SOX9 of particular importance. Clinical screening may be most critical during specified ages such as the neonatal and adolescent periods, with emphasis on longitudinal assessment of deformity, cervical stability, and tethered cord symptomatology. Providers must encourage early genetic testing and counseling, including both microarray karyotyping and next-generation sequencing; the latter may involve whole exome sequencing for research purposes, in order to uncover novel regions of interest such as the terminal 10q deletion in our patient. Ultimately, these efforts will lead to development of targeted panels for deep sequencing at the individual level. However, developing clinically meaningful screening and management recommendations will require cohort-level genetic testing of patients and family members with rigorous genotype-phenotype correlation. In this context, the limitations of existing genetic testing, both in terms of sensitivity and clinical utility, must also be clearly discussed with patients and families.
Data availability
All data collected, generated, and analyzed in this study are included in the published article and accompanying Supplementary information.
Abbreviations
- OMIM:
-
Online Mendelian inheritance in man
- PRS:
-
Pierre Robin sequence
- WES:
-
Whole exome sequencing
References
Breugem CC, Evans KN, Poets CF, Suri S, Picard A, Filip C, Paes EC, Mehendale FV, Saal HM, Basart H, Murthy J, Joosten KFM, Speleman L, Collares MVM, van den Boogaard MJH, Muradin M, Andersson MEM, Kogo M, Farlie PG, Don Griot P, Mossey PA, Slator R, Abadie V, Hong P (2016) Best practices for the diagnosis and evaluation of infants with Robin sequence. JAMA Pediatr 170:894–902
Vatlach S, Maas C, Poets CF (2014) Birth prevalence and initial treatment of Robin sequence in Germany: a prospective epidemiologic study. Orphanet J Rare Dis 9:9
Printzlau A, Andersen M (2004) Pierre Robin sequence in Denmark: a retrospective population-based epidemiological study. Cleft Palate Craniofac J 41:47–52
Gomez-Ospina N, Bernstein JA (2016) Clinical, cytogenetic, and molecular outcomes in a series of 66 patients with Pierre Robin sequence and literature review: 22q11.2 deletion is less common than other chromosomal anomalies. Am J Med Genet A 170A:870–880
Hsieh ST, Woo AS (2019) Pierre Robin Sequence. Clin Plast Surg 46:249–259
Côté A, Fanous A, Almajed A, Lacroix Y (2015) Pierre Robin sequence: review of diagnostic and treatment challenges. Int J Pediatr Otorhinolaryngol 79:451–464
Barr RM, Khan SA, Shah MN, Roy S, Teichgraeber JF, Greives MR (2016) Cervical instability in Pierre Robin sequence: an addition to the algorithm. J Craniofac Surg 27:1674–1676
Tubbs RS, Oakes WJ (2006) Chiari I malformation, caudal regression syndrome, and Pierre Robin syndrome: a previously unreported combination. Childs Nerv Syst 22:1507–1508
Abu-Ghname A, Masoumy M, Monson LA (2019) Cervical stenosis in non-syndromic Pierre Robin sequence. J Craniofac Surg 30:1866–1868
Zhu X, Evans KN, El-Gharbawy A, Lee JY, Brooker JE, Jabbour N, Tyler-Kabara EC, Madan-Khertarpal S, Losee JE, Goldstein JA (2018) Cervical spine injury from unrecognized craniocervical instability in severe Pierre Robin sequence associated with skeletal dysplasia. Cleft Palate Craniofac J 55:773–777
Ilyas M, Bashir I, Ellahi I, Shaheen F, Choh N (2018) Pierre Robin syndrome with caudal regression syndrome—a rare combination of congenital syndromes. Childs Nerv Syst 34:1105–1106. https://doi.org/10.1007/s00381-018-3800-3
Cirillo Silengo M, Davi GF, Bianco R, DeMarco A, Franceschini P (1983) Kniest disease with Pierre Robin syndrome and hydrocephalus. Pediatr Radiol 13:106–109
Al Kaissi A, Ganger R, Klaushofer K, Grill F (2011) Cervico-thoracic kyphosis in a girl with Pierre Robin sequence. Ger Med Sci 9:Doc06
Gamble JG, Rinsky LA (1985) Combined occipitoatlantoaxial hypermobility with anterior and posterior arch defects of the atlas in Pierre-Robin syndrome. J Pediatr Orthop 5:475–478. https://doi.org/10.1097/01241398-198507000-00018
Molnár S, Szappanos L, Körmendi Z, Veres R (2007) Occipitoatlantoaxial instability and congenital thoracic vertebral deformity in Pierre Robin sequence: a case report. Spine 32:E501–E504
Abraham V, Grewal S, Bhatia G, Kaur N, Raghav W, Jain P, Gupta N, Singh M, George C (2018) Pierre robin sequence with cervicothoracic kyphoscoliosis: an anesthetic challenge. J Anaesthesiol Clin Pharmacol 34:128–129
Almasri M, Kishta W, Abduljabbar FH, Arlet V, Saran N, Oullet J (2017) Ischiospinal Dysostosis in a Child with Pierre-Robin Syndrome. Case Rep Orthop 2017:1–5. https://doi.org/10.1155/2017/8263536
Davidson TB, Sanchez-Lara PA, Randolph LM, Krieger MD, Wu S-Q, Panigrahy A, Shimada H, Erdreich-Epstein A (2012) Microdeletion del(22)(q12.2) encompassing the facial development-associated gene, MN1 (meningioma 1) in a child with Pierre-Robin sequence (including cleft palate) and neurofibromatosis 2 (NF2): a case report and review of the literature. BMC Med Genet 13:19. https://doi.org/10.1186/1471-2350-13-19
Kunakornsawat S, Sathira-Angkura V, Prasartritha T, Sudprasert W, Tungsiripat R (2015) The treatment of flexion myelopathy of the thoracic spine in Pierre Robin sequence: a case report. J Med Assoc Thail 98:1041–1044
Lee J, Hida K, Seki T, Kitamura J, Iwasaki Y (2003) Pierre-Robin syndrome associated with Chiari type I malformation. Childs Nerv Syst 19:380–383
Roberti D, Conforti R, Giugliano T, Brogna B, Tartaglione I, Casale M, Piluso G, Perrotta S (2018) A novel 12q13.2-q13.3 microdeletion syndrome with combined features of Diamond Blackfan anemia, Pierre Robin sequence and Klippel Feil deformity. Front Genet 9:549. https://doi.org/10.3389/fgene.2018.00549
Rymer AN, Porteous GH, Neal JM (2015) Anesthetic challenges in an adult with Pierre Robin sequence, severe juvenile scoliosis, and respiratory failure. A A Case Rep 5:95–98
Logjes RJH, Breugem CC, Van Haaften G, Paes EC, Sperber GH, van den Boogaard M-JH, Farlie PG (2018) The ontogeny of Robin sequence. Am J Med Genet A 176:1349–1368
Gangopadhyay N, Mendonca DA, Woo AS (2012) Pierre robin sequence. Semin Plast Surg 26:76–82
Evans KN, Sie KC, Hopper RA, Glass RP, Hing AV, Cunningham ML (2011) Robin sequence: from diagnosis to development of an effective management plan. Pediatrics 127:936–948
Jakobsen LP, Knudsen MA, Lespinasse J, García Ayuso C, Ramos C, Fryns J-P, Bugge M, Tommerup N (2006) The genetic basis of the Pierre Robin sequence. Cleft Palate Craniofac J 43:155–159
Gorlin RJ, Cohen MM Jr, RCM H (2001) Syndromes of the head and neck. Oxford University Press, Oxford
Borgaonkar DS (1997) Chromosomal variation in man: a catalog of chromosomal variants and anomalies. Wiley, Hoboken
Strehle EM, Bantock HM (2003) The phenotype of patients with 4q-syndrome. Genet Couns 14:195–205
Losty PD, Flake AW, Rintala RJ, Hutson JM, Lwai N (2018) Rickham’s neonatal surgery. Springer, Berlin
Benko S, Fantes JA, Amiel J, Kleinjan DJ, Thomas S, Ramsay J, Jamshidi N, Essafi A, Heaney S, Gordon CT, McBride D, Golzio C, Fisher M, Perry P, Abadie V, Ayuso C, Holder-Espinasse M, Kilpatrick N, Lees MM, Picard A, Temple IK, Thomas P, Vazquez MP, Vekemans M, Crollius HR, Hastie ND, Munnich A, Etchevers HC, Pelet A, Farlie PG, FitzPatrick DR, Lyonnet S (2009) Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat Genet 41:359–364
Amarillo IE, Dipple KM, Quintero-Rivera F (2013) Familial microdeletion of 17q24.3 upstream of SOX9 is associated with isolated Pierre Robin sequence due to position effect. Am J Med Genet A 161A:1167–1172
Basart H, Paes EC, Maas SM et al (2015) Etiology and pathogenesis of robin sequence in a large Dutch cohort. Am J Med Genet A 167:1983–1992
Izumi K, Konczal LL, Mitchell AL, Jones MC (2012) Underlying genetic diagnosis of Pierre Robin sequence: retrospective chart review at two children’s hospitals and a systematic literature review. J Pediatr 160:645–650.e2
Bütow K-W, Hoogendijk CF, Zwahlen RA (2009) Pierre Robin sequence: appearances and 25 years of experience with an innovative treatment protocol. J Pediatr Surg 44:2112–2118
Furey CG, Zeng X, Dong W, Jin SC, Choi J, Timberlake AT, Dunbar AM, Allocco AA, Günel M, Lifton RP, Kahle KT (2018) Human genetics and molecular mechanisms of congenital hydrocephalus. World Neurosurg 119:441–443
Pozetti M, Belsuzarri TAB, Belsuzarri NCB, Seixas NB, Araujo JFM (2016) Neurofibromatosis type 1 and Chiari type 1 malformation: a case report and literature review of a rare association. Surg Neurol Int 7:S469–S472
Chang Y-T, Chou I-C, Wang C-H, Chin Z-N, Kuo H-T, Lin C-C, Tsai C-H, Tsai F-J (2013) Chromosome 10q deletion del (10)(q26.1q26.3) is associated with cataract. Pediatr Neonatol 54:132–136
Lin S, Zhou Y, Fang Q, Wu J, Zhang Z, Ji Y, Luo Y (2016) Chromosome 10q26 deletion syndrome: two new cases and a review of the literature. Mol Med Rep 14:5134–5140
Kaplan KM, Spivak JM, Bendo JA (2005) Embryology of the spine and associated congenital abnormalities. Spine J 5:564–576
Pang D, Thompson DNP (2011) Embryology and bony malformations of the craniovertebral junction. Childs Nerv Syst 27:523–564
Author information
Authors and Affiliations
Contributions
MG, DRSD, and ML conceptualized the review and collected clinical data. AY and MG performed literature review and data analysis. All authors participated in study design, critical analysis of systematic literature review, and manuscript writing.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest relating to the research, authorship, and/or publication of this work.
Ethics approval and consent to participate
Ethical approval for the case report was obtained from the Institutional Review Board of Rady Children’s Hospital (protocol #120518).
Consent for publication
Written informed consent for publication of clinical details and clinical images was obtained from the parent of the patient.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 15 kb).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yekula, A., Grant, C., Gupta, M. et al. Clinical and genetic characterization of patients with Pierre Robin sequence and spinal disease: review of the literature and novel terminal 10q deletion. Childs Nerv Syst 36, 1367–1377 (2020). https://doi.org/10.1007/s00381-020-04642-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-020-04642-2