
Abstract
Perrault syndrome is a rare heterogeneous condition characterised by sensorineural hearing loss and premature ovarian insufficiency. Additional neuromuscular pathology is observed in some patients. There are six genes in which variants are known to cause Perrault syndrome; however, these explain only a minority of cases. We investigated the genetic cause of Perrault syndrome in seven affected individuals from five different families, successfully identifying the cause in four patients. This included previously reported and novel causative variants in known Perrault syndrome genes, CLPP and LARS2, involved in mitochondrial proteolysis and mitochondrial translation, respectively. For the first time, we show that pathogenic variants in PEX6 can present clinically as Perrault syndrome. PEX6 encodes a peroxisomal biogenesis factor, and we demonstrate evidence of peroxisomal dysfunction in patient serum. This study consolidates the clinical overlap between Perrault syndrome and peroxisomal disorders, and highlights the need to consider ovarian function in individuals with atypical/mild peroxisomal disorders. The remaining patients had variants in candidate genes such as TFAM, involved in mtDNA transcription, replication, and packaging, and GGPS1 involved in mevalonate/coenzyme Q10 biosynthesis and whose enzymatic product is required for mouse folliculogenesis. This genomic study highlights the diverse molecular landscape of this poorly understood syndrome.




Similar content being viewed by others
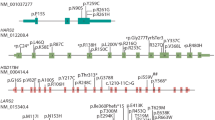
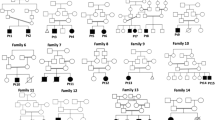
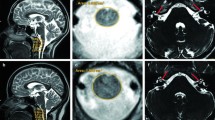
Data availability
Described variants are submitted to ClinVar. Further data generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Abyzov A, Urban AE, Snyder M, Gerstein M (2011) CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21:974–984. https://doi.org/10.1101/gr.114876.110
Bidet M, Bachelot A, Bissauge E, Golmard JL, Gricourt S, Dulon J, Coussieu C, Badachi Y, Touraine P (2011) Resumption of ovarian function and pregnancies in 358 patients with premature ovarian failure. J Clin Endocrinol Metab 96:3864–3872. https://doi.org/10.1210/jc.2011-1038
Blok MJ, van den Bosch BJ, Jongen E, Hendrickx A, de Die-Smulders CE, Hoogendijk JE, Brusse E, de Visser M, Poll-The BT, Bierau J, de Coo IF, Smeets HJ (2009) The unfolding clinical spectrum of POLG mutations. J Med Genet 46:776–785. https://doi.org/10.1136/jmg.2009.067686
Calvo SE, Clauser KR, Mootha VK (2016) MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44:D1251–D1257. https://doi.org/10.1093/nar/gkv1003
Chatzispyrou IA, Alders M, Guerrero-Castillo S, Zapata Perez R, Haagmans MA, Mouchiroud L, Koster J, Ofman R, Baas F, Waterham HR, Spelbrink JN, Auwerx J, Mannens MM, Houtkooper RH, Plomp AS (2017) A homozygous missense mutation in ERAL1, encoding a mitochondrial rRNA chaperone, causes Perrault syndrome. Hum Mol Genet 26:2541–2550. https://doi.org/10.1093/hmg/ddx152
Chen K, Yang K, Luo SS, Chen C, Wang Y, Wang YX, Li DK, Yang YJ, Tang YL, Liu FT, Wang J, Wu JJ, Sun YM (2017) A homozygous missense variant in HSD17B4 identified in a consanguineous Chinese Han family with type II Perrault syndrome. BMC Med Genet 18:91. https://doi.org/10.1186/s12881-017-0453-0
Cherot E, Keren B, Dubourg C, Carre W, Fradin M, Lavillaureix A, Afenjar A, Burglen L, Whalen S, Charles P, Marey I, Heide S, Jacquette A, Heron D, Doummar D, Rodriguez D, Billette de Villemeur T, Moutard ML, Guet A, Xavier J, Perisse D, Cohen D, Demurger F, Quelin C, Depienne C, Odent S, Nava C, David V, Pasquier L, Mignot C (2018) Using medical exome sequencing to identify the causes of neurodevelopmental disorders: experience of 2 clinical units and 216 patients. Clin Genet 93:567–576. https://doi.org/10.1111/cge.13102
de Oliveira VC, Moreira GSA, Bressan FF, Gomes Mariano Junior C, Roballo KCS, Charpentier M, Concordet JP, Meirelles FV, Ambrosio CE (2019) Edition of TFAM gene by CRISPR/Cas9 technology in bovine model. PLoS ONE 14:e0213376. https://doi.org/10.1371/journal.pone.0213376
Demain LA, Urquhart JE, O'Sullivan J, Williams SG, Bhaskar SS, Jenkinson EM, Lourenco CM, Heiberg A, Pearce SH, Shalev SA, Yue WW, Mackinnon S, Munro KJ, Newbury-Ecob R, Becker K, Kim MJRTOK, Newman WG (2017) Expanding the genotypic spectrum of Perrault syndrome. Clin Genet 91:302–312. https://doi.org/10.1111/cge.12776
Diao F, Jiang C, Wang XX, Zhu RL, Wang Q, Yao B, Li CJ (2016) Alteration of protein prenylation promotes spermatogonial differentiation and exhausts spermatogonial stem cells in newborn mice. Sci Rep 6:28917. https://doi.org/10.1038/srep28917
Dominguez-Ruiz M, Garcia-Martinez A, Corral-Juan M, Perez-Alvarez AI, Plasencia AM, Villamar M, Moreno-Pelayo MA, Matilla-Duenas A, Menendez-Gonzalez M, Del Castillo I (2019) Perrault syndrome with neurological features in a compound heterozygote for two TWNK mutations: overlap of TWNK-related recessive disorders. J Transl Med 17:290. https://doi.org/10.1186/s12967-019-2041-x
Ebberink MS, Kofster J, Wanders RJ, Waterham HR (2010) Spectrum of PEX6 mutations in Zellweger syndrome spectrum patients. Hum Mutat 31:E1058–E1070. https://doi.org/10.1002/humu.21153
ESHRE (2015) European Society of Human Reproduction and Embryology (ESHRE) Guidelines: management of women with premature ovarian insufficiency
Faridi R, Rehman AU, Morell RJ, Friedman PL, Demain L, Zahra S, Khan AA, Tohlob D, Assir MZ, Beaman G, Khan SN, Newman WG, Riazuddin S, Friedman TB (2017) Mutations of SGO2 and CLDN14 collectively cause coincidental Perrault syndrome. Clin Genet 91:328–332. https://doi.org/10.1111/cge.12867
Golezar S, Ramezani Tehrani F, Khazaei S, Ebadi A, Keshavarz Z (2019) The global prevalence of primary ovarian insufficiency and early menopause: a meta-analysis. Climacteric. https://doi.org/10.1080/13697137.2019.1574738
Heeringa SF, Chernin G, Chaki M, Zhou W, Sloan AJ, Ji Z, Xie LX, Salviati L, Hurd TW, Vega-Warner V, Killen PD, Raphael Y, Ashraf S, Ovunc B, Schoeb DS, McLaughlin HM, Airik R, Vlangos CN, Gbadegesin R, Hinkes B, Saisawat P, Trevisson E, Doimo M, Casarin A, Pertegato V, Giorgi G, Prokisch H, Rotig A, Nurnberg G, Becker C, Wang S, Ozaltin F, Topaloglu R, Bakkaloglu A, Bakkaloglu SA, Muller D, Beissert A, Mir S, Berdeli A, Varpizen S, Zenker M, Matejas V, Santos-Ocana C, Navas P, Kusakabe T, Kispert A, Akman S, Soliman NA, Krick S, Mundel P, Reiser J, Nurnberg P, Clarke CF, Wiggins RC, Faul C, Hildebrandt F (2011) COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest 121:2013–2024. https://doi.org/10.1172/JCI45693
Heimer G, Keratar JM, Riley LG, Balasubramaniam S, Eyal E, Pietikainen LP, Hiltunen JK, Marek-Yagel D, Hamada J, Gregory A, Rogers C, Hogarth P, Nance MA, Shalva N, Veber A, Tzadok M, Nissenkorn A, Tonduti D, Renaldo F, University of Washington Center for Mendelian G, Kraoua I, Panteghini C, Valletta L, Garavaglia B, Cowley MJ, Gayevskiy V, Roscioli T, Silberstein JM, Hoffmann C, Raas-Rothschild A, Tiranti V, Anikster Y, Christodoulou J, Kastaniotis AJ, Ben-Zeev B, Hayflick SJ (2016) MECR mutations cause childhood-onset dystonia and optic atrophy, a mitochondrial fatty acid synthesis disorder. Am J Hum Genet 99:1229–1244. https://doi.org/10.1016/j.ajhg.2016.09.021
Jenkinson EM, Rehman AU, Walsh T, Clayton-Smith J, Lee K, Morell RJ, Drummond MC, Khan SN, Naeem MA, Rauf B, Billington N, Schultz JM, Urquhart JE, Lee MK, Berry A, Hanley NA, Mehta S, Cilliers D, Clayton PE, Kingston H, Smith MJ, Warner TT, University of Washington Center for Mendelian G, Black GC, Trump D, Davis JR, Ahmad W, Leal SM, Riazuddin S, King MC, Friedman TB, Newman WG (2013) Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am J Hum Genet 92:605–613. https://doi.org/10.1016/j.ajhg.2013.02.013
Jiang C, Diao F, Sang YJ, Xu N, Zhu RL, Wang XX, Chen Z, Tao WW, Yao B, Sun HX, Huang XX, Xue B, Li CJ (2017) GGPP-Mediated protein geranylgeranylation in oocyte is essential for the establishment of oocyte-granulosa cell communication and primary-secondary follicle transition in mouse ovary. PLoS Genet 13:e1006535. https://doi.org/10.1371/journal.pgen.1006535
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44:D862–D868. https://doi.org/10.1093/nar/gkv1222
Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA (1998) Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet 18:231–236. https://doi.org/10.1038/ng0398-231
Layer RM, Chiang C, Quinlan AR, Hall IM (2014) LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15:R84. https://doi.org/10.1186/gb-2014-15-6-r84
Lerat J, Jonard L, Loundon N, Christin-Maitre S, Lacombe D, Goizet C, Rouzier C, Van Maldergem L, Gherbi S, Garabedian EN, Bonnefont JP, Touraine P, Mosnier I, Munnich A, Denoyelle F, Marlin S (2016) An application of NGS for molecular investigations in Perrault syndrome: study of 14 families and review of the literature. Hum Mutat 37:1354–1362. https://doi.org/10.1002/humu.23120
Luborsky JL, Meyer P, Sowers MF, Gold EB, Santoro N (2003) Premature menopause in a multi-ethnic population study of the menopause transition. Hum Reprod 18:199–206. https://doi.org/10.1093/humrep/deg005
Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, Oldfors A, Rautakorpi I, Peltonen L, Majamaa K, Somer H, Suomalainen A (2004) Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet 364:875–882. https://doi.org/10.1016/S0140-6736(04)16983-3
Machiela MJ, Chanock SJ (2015) LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31:3555–3557. https://doi.org/10.1093/bioinformatics/btv402
Mollet J, Giurgea I, Schlemmer D, Dallner G, Chretien D, Delahodde A, Bacq D, de Lonlay P, Munnich A, Rotig A (2007) Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest 117:765–772. https://doi.org/10.1172/JCI29089
Morino H, Pierce SB, Matsuda Y, Walsh T, Ohsawa R, Newby M, Hiraki-Kamon K, Kuramochi M, Lee MK, Klevit RE, Martin A, Maruyama H, King MC, Kawakami H (2014) Mutations in twinkle primase-helicase cause Perrault syndrome with neurologic features. Neurology 83:2054–2061. https://doi.org/10.1212/WNL.0000000000001036
Newman WG, Friedman TB, Conway GS, Demain LAM (1993) Perrault syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds) GeneReviews ((R)), Seattle (WA)
Newman WG, Friedman TB, Conway GS, Demain LAM (2018) Perrault syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds) GeneReviews((R)), Seattle (WA)
Pagnamenta AT, Taanman JW, Wilson CJ, Anderson NE, Marotta R, Duncan AJ, Bitner-Glindzicz M, Taylor RW, Laskowski A, Thorburn DR, Rahman S (2006) Dominant inheritance of premature ovarian failure associated with mutant mitochondrial DNA polymerase gamma. Hum Reprod 21:2467–2473. https://doi.org/10.1093/humrep/del076
Pierce SB, Walsh T, Chisholm KM, Lee MK, Thornton AM, Fiumara A, Opitz JM, Levy-Lahad E, Klevit RE, King MC (2010) Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrome. Am J Hum Genet 87:282–288. https://doi.org/10.1016/j.ajhg.2010.07.007
Pierce SB, Chisholm KM, Lynch ED, Lee MK, Walsh T, Opitz JM, Li W, Klevit RE, King MC (2011) Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci USA 108:6543–6548. https://doi.org/10.1073/pnas.1103471108
Pierce SB, Gersak K, Michaelson-Cohen R, Walsh T, Lee MK, Malach D, Klevit RE, King MC, Levy-Lahad E (2013) Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am J Hum Genet 92:614–620. https://doi.org/10.1016/j.ajhg.2013.03.007
Riley LG, Rudinger-Thirion J, Schmitz-Abe K, Thorburn DR, Davis RL, Teo J, Arbuckle S, Cooper ST, Campagna DR, Frugier M, Markianos K, Sue CM, Fleming MD, Christodoulou J (2016) LARS2 variants associated with hydrops, lactic acidosis, sideroblastic anemia, and multisystem failure. JIMD Rep 28:49–57. https://doi.org/10.1007/8904_2015_515
Roca-Ayats N, Balcells S, Garcia-Giralt N, Falcó-Mascaró M, Martínez-Gil N, Abril JF, Urreizti R, Dopazo J, Quesada-Gómez JM, Nogués X, Mellibovsky L, Prieto-Alhambra D, Dunford JE, Javaid MK, Russell RG, Grinberg D, Díez-Pérez A (2017) GGPS1 mutation and atypical femoral fractures with bisphosphonates. N Engl J Med 376:1794–1795. https://doi.org/10.1056/NEJMc1612804
Sadedin SP, Dashnow H, James PA, Bahlo M, Bauer DC, Lonie A, Lunke S, Macciocca I, Ross JP, Siemering KR, Stark Z, White SM, Melbourne Genomics Health A, Taylor G, Gaff C, Oshlack A, Thorne NP (2015) Cpipe: a shared variant detection pipeline designed for diagnostic settings. Genome Med 7:68. https://doi.org/10.1186/s13073-015-0191-x
Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnstrom K, Mallick S, Kirby A, Wall DP, MacArthur DG, Gabriel SB, DePristo M, Purcell SM, Palotie A, Boerwinkle E, Buxbaum JD, Cook EH Jr, Gibbs RA, Schellenberg GD, Sutcliffe JS, Devlin B, Roeder K, Neale BM, Daly MJ (2014) A framework for the interpretation of de novo mutation in human disease. Nat Genet 46:944–950. https://doi.org/10.1038/ng.3050
Sobreira N, Schiettecatte F, Valle D, Hamosh A (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 36:928–930. https://doi.org/10.1002/humu.22844
Stiles AR, Simon MT, Stover A, Eftekharian S, Khanlou N, Wang HL, Magaki S, Lee H, Partynski K, Dorrani N, Chang R, Martinez-Agosto JA, Abdenur JE (2016) Mutations in TFAM, encoding mitochondrial transcription factor A, cause neonatal liver failure associated with mtDNA depletion. Mol Genet Metab 119:91–99. https://doi.org/10.1016/j.ymgme.2016.07.001
Theunissen TE, Szklarczyk R, Gerards M, Hellebrekers DM, Mulder-Den Hartog EN, Vanoevelen J, Kamps R, de Koning B, Rutledge SL, Schmitt-Mechelke T, van Berkel CG, van der Knaap MS, de Coo IF, Smeets HJ (2016) Specific MRI abnormalities reveal severe Perrault syndrome due to CLPP defects. Front Neurol 7:203. https://doi.org/10.3389/fneur.2016.00203
Tran C, Hewson S, Steinberg SJ, Mercimek-Mahmutoglu S (2014) Late-onset Zellweger spectrum disorder caused by PEX6 mutations mimicking X-linked adrenoleukodystrophy. Pediatr Neurol 51:262–265. https://doi.org/10.1016/j.pediatrneurol.2014.03.020
Tucker EJ, Compton AG, Calvo SE, Thorburn DR (2011) The molecular basis of human complex I deficiency. IUBMB Life 63:669–677. https://doi.org/10.1002/iub.495
Tucker EJ, Grover SR, Bachelot A, Touraine P, Sinclair AH (2016) Premature ovarian insufficiency: new perspectives on genetic cause and phenotypic spectrum. Endocr Rev 37:609–635. https://doi.org/10.1210/er.2016-1047
van der Knaap MS, Bugiani M, Mendes MI, Riley LG, Smith DEC, Rudinger-Thirion J, Frugier M, Breur M, Crawford J, van Gaalen J, Schouten M, Willems M, Waisfisz Q, Mau-Them FT, Rodenburg RJ, Taft RJ, Keren B, Christodoulou J, Depienne C, Simons C, Salomons GS, Mochel F (2019) Biallelic variants in LARS2 and KARS cause deafness and (ovario)leukodystrophy. Neurology 92:e1225–e1237. https://doi.org/10.1212/WNL.0000000000007098
Xu N, Shen N, Wang X, Jiang S, Xue B, Li C (2015) Protein prenylation and human diseases: a balance of protein farnesylation and geranylgeranylation. Sci China Life Sci 58:328–335. https://doi.org/10.1007/s11427-015-4836-1
Yu HL, Shen Y, Sun YM, Zhang Y (2019) Two novel mutations of PEX6 in one Chinese Zellweger spectrum disorder and their clinical characteristics. Ann Transl Med 7:368. https://doi.org/10.21037/atm.2019.06.42
Yubero D, Montero R, Santos-Ocana C, Salviati L, Navas P, Artuch R (2018) Molecular diagnosis of coenzyme Q10 deficiency: an update. Expert Rev Mol Diagn 18:491–498. https://doi.org/10.1080/14737159.2018.1478290
Acknowledgements
WGS was performed as part of the Australian Genomics Health Alliance (Australian Genomics) project, funded by a National Health and Medical Research Council (NHMRC) Targeted Call for Research Grant (1113531). This work was supported by an NHMRC program Grant (1074258, to A.H.S.), and NHMRC fellowships (1054432 to E.J.T., 1062854 to A.H.S., 1155244 to D.R.T), and a CONACYT Postgraduate Research Scholarship (R.R). The research conducted at the Murdoch Children’s Research Institute was supported by the Victorian Government's Operational Infrastructure Support Program.
Funding
WGS was performed as part of the Australian Genomics Health Alliance (Australian Genomics) project, funded by a National Health and Medical Research Council (NHMRC) Targeted Call for Research Grant (1113531). This work was supported by an NHMRC program Grant (1074258, to A.H.S.), and NHMRC fellowships (1054432 to E.J.T., 1062854 to A.H.S., 1155244 to D.R.T), and a CONACYT Postgraduate Research Scholarship (R.R). The research conducted at the Murdoch Children’s Research Institute was supported by the Victorian Government's Operational Infrastructure Support Program.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethics approval
Procedures were in accordance with the ethical standards of the Human Research Ethics Committee of the Royal Children’s Hospital, Melbourne (HREC/22073). WGS was performed as part of the Mitochondrial Flagship study of the Australian Genomics Health Alliance research project, which also has Human Research Ethics Committee approval (HREC/16/MH251).
Informed consent
Written informed consent was obtained from all participants.
Consent for publication
Participants consented to publication of non-identifiable data.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tucker, E.J., Rius, R., Jaillard, S. et al. Genomic sequencing highlights the diverse molecular causes of Perrault syndrome: a peroxisomal disorder (PEX6), metabolic disorders (CLPP, GGPS1), and mtDNA maintenance/translation disorders (LARS2, TFAM). Hum Genet 139, 1325–1343 (2020). https://doi.org/10.1007/s00439-020-02176-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-020-02176-w