
Abstract
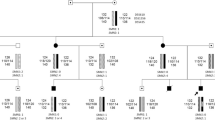
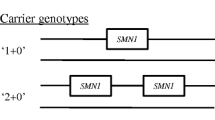
Proximal spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease caused by homozygous deletion in the seventh exon of the SMN1 gene. The aim of this work is to analyze the association of the allelic polymorphism of telomeric genes SMN1 and NAIP and the centromeric gene SMN2 of the 5q13 region with the clinical phenotype of SMA. It was shown that the homozygous genotype, which contains a telomeric deletion, covering both SMN1 and NAIP, is significantly more often observed in patients with the most severe type of SMA. Three or more copies of SMN2 are associated with a milder phenotype; the number of SMN2 copies affects the SMA phenotype more heavily than the length of the telomeric deletion. It was shown that one SMN2 copy is significantly more frequent than three or more copies of this gene in SMA-patients with homozygous deletion of SMN1 and NAIP. This fact may indicate the presence of a large deletion of all the three studied genes in SMA genotypes associated with the most severe type of SMA. It is noted that congenital SMA (type 0) is significantly less common in female patients, which may indicate the presence of SMA modifier genes on the X-chromosome.


Similar content being viewed by others

REFERENCES
Ogino, S. and Wilson, R., Spinal muscular atrophy: molecular genetics and diagnostics, Expert.Rev., 2004, vol. 4, no. 1, pp. 15–29. https://doi.org/10.1586/14737159.4.1.15
Mesfin, A., Sponseller, P.D., and Leet, A.I., Spinal muscular atrophy: manifestations and management, J. Am. Acad. Orthop. Surg., 2012, vol. 20, no. 6, pp. 393–401. https://doi.org/10.5435/JAAOS-20-06-393
Grotto, S., Cuisset, J.M., Marret, S., Drunat, S., Faure, P., Audebert-Bellanger, S., Desguerre, I., Flurin, V., Grebille, A.G., Guerrot, A.M., Journel, H., Morin, G., Plessis, G., Renolleau, S., Roume, J., Simon-Bouy, B., Touraine, R., Willems, M., Frébourg, T., Verspyck, E., and Saugier-Veber, P., Type 0 spinal muscular atrophy: further delineation of prenatal and postnatal features in 16 patients, J. Neuromuscul. Dis., 2016, vol. 3, no. 4, pp. 487–95. https://doi.org/10.3233/JND-160177
Butchbach, M.E., Copy number variations in the survival motor neuron genes: implications for spinal muscular atrophy and other neurodegenerative diseases, Front. Mol. Biosci., 2016, vol. 10, no. 3, pp. 7. https://doi.org/10.3389/fmolb.2016.00007
Jedrzejowska, M., Milewski, M., and Zimowski, J., Phenotype modifiers of spinal muscular atrophy: the number of SMN2 gene copies, deletion in the NAIP gene and probably gender influence the course of the disease, Acta Biochim. Pol., 2009, vol. 56, no. 1, pp. 103–111.
Groen, E.J.N., Perenthaler, E., Courtney, N.L., Jordan, C.Y., Shorrock, H.K., van der Hoorn, D., Huang, Y.-T., Murray, L.M., Viero, G., and Gillingwater, T.H., Temporal and tissue-specific variability of SMN protein levels in mouse models of spinal muscular atrophy, Hum. Mol. Genet., 2018, vol. 27, no. 16, pp. 2851–2862. https://doi.org/10.1093/hmg/ddy195
Alrafiah, A., Alghanmi, M., Almashhadi, S., Aqeel, A., and Awaji, A., The expression of SMN1, MART3, GLE1 and FUS genes in spinal muscular atrophy, Folia Histochem. Cytobiol., 2018, vol. 56, no. 4, pp. 215–221. https://doi.org/10.5603/FHC.a2018.0022
Aquilina, B. and Cauchi, R.J., Genetic screen identifies a requirement for SMN in mRNA localisation within the Drosophila oocyte, BMC Res. Notes, 2018, vol. 11, no. 1, p. 378. https://doi.org/10.1186/s13104-018-3496-1
Beattie, C.E. and Kolb, S.J., Spinal muscular atrophy: selective motor neuron loss and global defect in the assembly of ribonucleoproteins, Brain. Res., 2018, vol. 1693 (Pt. A), pp. 92–97. https://doi.org/10.1016/j.brainres.2018.02.022
Mattis, V.B., Butchbach, M.E., and Lorson, C.L., Detection of human survival motor neuron (SMN) protein in mice containing the SMN2 transgene: applicability to preclinical therapy development for spinal muscular atrophy, J. Neurosci. Methods, 2008, vol. 175, no. 1, pp. 36–43. https://doi.org/10.1016/j.jneumeth.2008.07.024
Butchbach, M.E., Rose, F.F., Jr., Rhoades, S., Marston, J., McCrone, J.T., Sinnott, R., and Lorson, C.L., Effect of diet on the survival and phenotype of a mouse model for spinal muscular atrophy, Biochem. Biophys. Res. Commun., 2010, vol. 391, no. 1, pp. 835–40. https://doi.org/10.1016/j.bbrc.2009.11.148
Rouault, F., Christie-Brown, V., and Broekgaarden, R., Disease impact on general well-being and therapeutic expectations of European type II and type III spinal muscular atrophy patients, Neuromuscul. Disord., 2017, vol. 27, no. 5, pp. 428–438. https://doi.org/10.1016/j.nmd.2017.01.018
Gidaro, T. and Servais, L., Nusinersen treatment of spinal muscular atrophy: current knowledge and existing gaps, Dev. Med. Child. Neurol., 2019, vol. 61, no. 1, pp. 19–24. https://doi.org/10.1111/dmcn.14027
Maniatis, T., Fritsch, E.E., and Sambrook, J., Molecular Cloning: A Laboratory Manual, 4th ed., Cold Spring Harbor Laboratory, 2012.
Stabley, D.L., Harris, A.W., Holbrook, J., Chubbs, N.J., Lozo, K.W., Crawford, T.O., Swoboda, K.J., Funanage, V.L., Wang, W., Mackenzie, W., Scavina, M., Sol-Church, K., and Matthew, E.R., Butchbach SMN1 and SMN2 copy numbers in cell lines derived from patients with spinal muscular atrophy as measured by array digital PCR, Mol. Genet. Genom. Med., 2015, vol. 3, no. 4, pp. 248–257.https://doi.org/10.1002/mgg3.141
Feldkötter, M., Schwarzer, V., Wirth, R., Wienker, T.F., and Wirth, B., Quantitative analyses of SMN1 and SMN2 based on real-time LightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy, Am. J. Hum. Genet., 2002, vol. 70, pp. 358–368.https://doi.org/10.1086/338627
Anhuf, D., Eggermann, T., Rudnik-Shöneborn, S., and Zerres, K., Determination of SMN1 and SMN2 copy number using TaqMan technology, Hum. Mutat., 2003, vol. 22, pp. 74–78. https://doi.org/10.1002/humu.10221
Soloviov, O.O., Livshits, G.B., Podlesnaya, S.S., and Livshits, L.A., Implementation of the quantitative Real-Time PCR for the molecular-genetic diagnostics of spinal muscular atrophy, Biopolym. Cell, 2010, vol. 26, no. 1, pp. 51–55. https://doi.org/10.7124/bc.000144
Solov’ev, A.A., Grishchenko, N.V., and Livshits, L.A., Spinal muscular atrophy carrier frequency in Ukraine, Genetika, 2013, vol. 49, no. 9, pp. 1126–1133. https://doi.org/10.1134/S1022795413080140
Boratyn, G.M., Camacho, C., and Cooper, P.S., BLAST: a more efficient report with usability improvements, Nucleic Acids Res., 2013, vol. 41, pp. W29–W33. https://doi.org/10.1093/nar/gkt282
Wangkumhang, P. and Chaichoompu, K., WASP: a Web-based Allele-Specific PCR assay designing tool for detecting SNPs and mutations, BMC Genomics, 2007, vol. 14, no. 8, pp. 275. https://doi.org/10.1186/1471-2164-8-275
Casper, J. and Zweig, A.S., The UCSC Genome Browser database: 2018 update, Nucleic Acids Res., 2018, vol. 46 (database issue), pp. D762–D769. https://doi.org/10.1093/nar/gkx1020
Livak, K.J. and Schmittgen, T.D., Analysis of relative gene expression data using real-time quantitative PCR and the 2 CT method, Methods, 2001, vol. 25, pp. 402–408. https://doi.org/10.1006/meth.2001.1262
Cusco, I. and Barcelo, M., Characterisation of SMN hybrid genes in Spanish SMA patients: de novo, homozygous and compound heterozygous cases, Hum. Genet., 2001, vol. 108, pp. 222–229. https://doi.org/10.1007/s004390000452
Ping, F. and Liang, L., Molecular characterization and copy number of SMN1, SMN2 and NAIP in Chinese patients with spinal muscular atrophy ànd unrelated healthy controls, BMC Musculoskeletal Disord., 2015, vol. 16, pp. 11–15. https://doi.org/10.1186/s12891-015-0457-x
Crawford, T.O., Paushkin, S.V., and Kobayashi, D.T., Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study, PLoS One, 2012, vol. 7, no. 4. e33 572. https://doi.org/10.1371/journal.pone.0033572
Ogino, S., Gao, S., Leonard, D.G., Paessler, M., and Wilson, R.B., Inverse correlation between SMN1 and SMN2 copy numbers: evidence for gene conversion from SMN2 to SMN1, Eur. J. Hum. Genet., 2003, vol. 11, no. 3, pp. 275–281. https://doi.org/10.1038/sj.ejhg.5200957
Chen, T.H. and Tzeng, C.C., Identification of bidirectional gene conversion between SMN1 and SMN2 by simultaneous analysis of SMN dosage and hybrid genes in a Chinese population, J. Neurol. Sci., 2011, vol. 308, no. 1–2, pp. 83–89. https://doi.org/10.1016/j.jns.2011.06.002
ACKNOWLEDGMENTS
We thank the Kharkiv Charity Foundation Children with Spinal Muscular Atrophy headed by V.N. Matyushenko for many years of comprehensive assistance in the research of this disease and the popularization of knowledge about SMA among the population of Ukraine. We also thank the doctors of the medical-genetic centers of Ukraine for collecting and analyzing clinical data of patients. We thank the staff of the Human Genomics Department of the IMBG NAS of Ukraine, primarily A.Yu. Ekshiyan, A.B. Livshits, A.A. Solov’ev, and S.S. Podlesnaya, who actively participated in the formation of the collection of DNA samples from patients with SMA and in the genotyping of patients by SMN1 and NAIP in the period of 1995—2016.
Funding
This work was carried out in the framework of the budget topics of the IMGB NAS of Ukraine 2011–2015 (topic code 2.2.4.13, state registration number 0105U005341) and 2016–2020 (topic code 2.2.4.13, state registration number 0115U003747).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest. The authors declare that they have no conflict of interest.
Statement of compliance with standards of research involving humans as subjects. All procedures carried out in the studies involving people were consistent with international and national standards, the Helsinki Declaration of 1964, and its later amendments, and approved by the Bioethics Commission of the IMBG NAS of Ukraine.
Additional information
Translated by K. Lazarev
About this article

Cite this article
Hryshchenko, N.V., Yurchenko, A.A., Karaman, H.S. et al. Genetic Modifiers of the Spinal Muscular Atrophy Phenotype. Cytol. Genet. 54, 130–136 (2020). https://doi.org/10.3103/S0095452720020073
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3103/S0095452720020073