
Abstract
Autoimmune encephalitis (AE) is a group of disorders causing synaptic receptor dysfunction with a broad range of neurological symptoms that has been historically difficult to differentiate clinically. Today, AE represents an excellent example of the rapid determination of the cause of a disease and the ability to identify potential treatments using relatively simple basic science techniques of investigation. Of the number of autoimmune encephalitides identified thus far, one of the best examples of the impact of basic science studies on disease management is NMDA receptor mediated autoimmune encephalitis (NMDAr-AE). In this review, we will provide an overview of the epidemiology of NMDAr-AE, clinical features and treatments, and the basic science tools and techniques that were used to identify the cause, correlate symptoms to underlying pathophysiology, and to understand the mechanism of disease pathology.
Similar content being viewed by others


Introduction
Encephalitis is a debilitating condition that includes a spectrum of CNS disorders resulting in parenchymal inflammation. Acute or subacute progression is common and often necessitates admission into the neurointensive care unit. The causes can be broadly categorized as infectious, paraneoplastic, and autoimmune etiologies, with the later attributed to auto-antibody formation to parenchymal epitopes. The scope of clinical presentations of autoimmune encephalitis is broad, including seizures, movement disorders, cranial neuropathies, dysautonomias, and often psychiatric manifestations. The spectrum of recognizable syndromes suggests immunosensitive structures localized to limbic, lenticulostriate, cerebellar, and brainstem anatomy.
Based on a consensus definition of autoimmune encephalitis [1], it represents a subacute progression of neuropsychiatric or cognitive deficits with the presence of focal deficits, seizures, or objective evidence of CNS inflammation (MRI or CSF studies), which form the minimum criteria for a large group of disorders, including several well-known clinical syndromes. For example, limbic encephalitis, first described in the 1960s [2], is a subacute encephalopathy with symptoms (e.g., seizures, memory loss, and hallucinations) associated with lesions involving the medial temporal lobe and hippocampus, and is the most predominant presentation within the autoimmune encephalitis spectrum. Other well-described entities, such as Bickerstaff’s encephalitis, neuromyelitis optica, Hashimoto’s encephalopathy, and Morvan syndrome have also been included in the spectrum of autoimmune encephalitis, each with a unique antibody discovered to be relatively specific to the disease [1, 3,4,5]. Over the last three decades, modern techniques of cell-based assays and immunocytochemistry have demonstrated multiple antibodies responsible for limbic encephalitis. Most were found to be triggered by a malignancy or infection, but over the last decade, several auto-antibodies have been discovered that appear to be primarily autoimmune in nature [6,7,8,9,10,11,12,13]. The distinction of intracellular versus extracellular antigen pathogenesis is becoming increasingly relevant given the diagnostic and therapeutic implications. Along this vein, the ability to identify pathophysiological antigens has improved over the years which aids in diagnostic and treatment strategies. In this review, we will refer to autoimmune encephalitis (AE) as those caused by extracellular or neuronal surface antibodies. NMDA receptor autoimmune encephalitis (NMDA-AE) has become the prototypical example, providing a great deal of clinical data and models for us to guide our therapies and diagnostic approaches. In this review, we provide a brief overview of the epidemiology of autoimmune encephalitis, the clinical aspects of NMDAr-AE specifically, and the bench science techniques used to define and better understand the clinical features and response to therapy of NMDAr-AE.
Epidemiology
Encephalitis has an overall prevalence worldwide ranging from 0.07 to 12.6/100 k persons and is cryptogenic at least 40% of the time [14, 15]. The age distribution is typically bimodal with a predilection for infants and adults > 65 years of age [14]. There are several subtypes of encephalitis, which are largely dependent on their anatomical predilection to different CNS structures based on their pathogenesis. They all tend to present with alteration of consciousness, progressive, and insidious course and often focal neurological features which suggest CNS involvement. Known entities can range from limbic encephalitis, encephalomyelitis, and brainstem encephalitis, and arise from a multitude of causes such as autoimmune processes, infectious/peri-infectious causes, paraneoplastic syndromes, or idiopathic processes in which the trigger or causative agent is poorly understood or not yet classified. In the United States, encephalitis had an estimated prevalence of 7/100 k persons and accounted for nearly $2 billion dollars in hospital-based charges in 2010 [16]. This burden is amplified when the etiology is autoimmune. A single institution retrospective analysis revealed autoimmune encephalitis patients to have significantly longer length of stay (LOS), higher utilization of ICU admission, and increased hospital charges when compared with those with herpes simplex encephalitis [17]. In addition, autoimmune encephalitis patients admitted to the ICU had a LOS four times greater and a median hospital charge three times greater than those not admitted to the ICU [16, 17]. Historically, the most common identified causes were infectious, however, paraneoplastic and autoimmune etiologies have been recognized more frequently as testing has become commercially available. Given the high burden of disease, investigations into the etiology of encephalitis have discovered that the incidence of autoimmune encephalitis to be at least as common as infectious causes based on two different population based studies, the California Encephalitis Project and Olmsted County, Minnesota [18, 19].
The most common risk factors for patients with autoimmune encephalitis are what are thought to be the most likely triggers: malignancy and infections. Most autoimmune encephalitides will occur without an identified trigger, but each has several well-recognized associations with specific malignancies (Table 1). For example, NMDA-AE is well known to have a strong association with ovarian teratomas. In adult females, teratoma is found in about 60% of patients; however, it also occurs less commonly in males and those without any identified malignancy [20]. In addition, anti-NMDA antibodies have been known to form in the setting herpes encephalitis in up to 20% of patients [21], suggesting viral causes may also be the underlying trigger. Evidence of antibody production on both sides of the blood brain barrier supports a model for antibody triggers proposed by Dalmau et al. [20]. The lack of consistent identification of an immune trigger suggests that there may be genetic predispositions to these conditions as well. Multiple studies have confirmed HLA class II genes associations with ant-LGI1 and anti-CASPR2 encephalitis patients in different populations (e.g., HLA-DR7 (B1*07) in LGI-1 and HLA-DRB1*11:01 in CASPR-2) [22,23,24], but no specific associations were seen with NMDA-AE.
Based on retrospective reviews, NMDAr-AE is the most commonly identified cause of AE [19, 25]. It has a high frequency in the young and may represent 1% of all young adults admitted to the ICU [25]. In one study, the prevalence of specific antibody associated AE ranged from 0.6 to 0.7 per 100,000 persons [18]. However, this is subject to debate as the incidence of AE is increasing rapidly, most likely due to more awareness and testing. The next most frequent AE is often considered to be leucine-rich, glioma-inactivated 1 (LGI-1) antibody related, which is responsible for faciobrachial dystonic seizures, and then CASPR-2 antibodies. One limitation to these epidemiological studies is that patients may not be captured as they may be detected and treated prior to formal testing and diagnosis of autoimmune encephalitis. For example, LGI-1 Ab positive patients may only present with seizures and never be classified as the classic autoimmune limbic encephalitis. More rare causes include gamma-amino-butyric acid receptor B (GABA(B)r) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAr) antibodies, which are also described as causes of limbic encephalitis [10, 13, 26, 27]. One must consider that this will likely change over the years as more antibody epitopes are discovered and testing specificity increases (Table 1).
Clinical Correlates for NMDA Receptor Interactions
The NMDA receptor (NMDAr) is one of the better studied and understood receptors in the CNS. The excessive activation of the receptor is implicated in many disease processes of the CNS including status epilepticus and hypoxic ischemic damage [28]. In animal models, the NMDAr in the hippocampi is strongly tied to learning and memory; thus, with inhibition of the receptor with NMDAr antagonists, there was an expected effect on the ability to learn and perform memory-based tasks, particularly in acquisition of new memory [28].
Excessive glutamate, a major excitatory neurotransmitter in the CNS, is implicated in dysfunction and or destruction of neurons due to a toxic effect on the receptors. Interestingly, in cases of NMDAr-AE, there is a decrease of NMDAr on the cell surface, and increase in inflammatory B cells and plasma cells and even immunoglobulin G deposits, though no complement fixation [29]. These observations would suggest that the clinical features of the disease are a result of alteration of the synapse from a pathophysiologic standpoint [30]. Based on postmortem studies, it has been observed that the presence of NMDAr antibodies leads to internalization of the receptor which leads to a decrease in the number of receptors in the synapse [31].
In humans, the best studied effects of NMDAr dysfunction and excitotoxicity exist in use of ketamine and phencyclidine (PCP). Both NMDAr antagonists result in reduced NMDAr activity similar to NMDAr antibodies. However, the antagonists act directly on the receptors to antagonize NMDAr function whereas the pathological antibodies decrease the actual number of receptors on the cell membrane [28]. Phencyclidine, a powerful recreational hallucinogenic drug, is known to cause significant behavioral activation, psychosis, hallucination, agitation, and hyperthermia. Long-term use of PCP is known to have effects on memory, cognitive function, and poor reaction time.
Ketamine, which is a commonly used anesthetic, has similar properties to PCP but at a lower intensity and with less predilection to NMDA receptors than PCP, thus less NMDA excitotoxicity, but still with clinical effects which we often harness clinically to help treat patients. Low doses of ketamine produce a dissociative amnesia due to the drug’s effects on inhibiting memory acquisition [28]. Larger doses can impair consciousness and have analgesic effects which are helpful for ketamine’s role in procedural sedation and pain control. As a caution, a small cohort study from China found that chronic ketamine use left structural damage particularly in the limbic system and striatum [28]. The toxicity of ketamine is thought to be due to activation of an apoptotic cascade, in addition to possibly influencing glial cells to produce inflammatory responses [32]. Although the number of patients studied was small (n = 21), the structural effects of long-term ketamine use were similar among the patients [33]. Ketamine toxicity lead to radiographic changes in the limbic system, internal capsule, white matter degeneration, and cortical atrophy, and many of these areas are also involved in NMDAr-AE suggesting a link in pathological activities in these areas anatomically.
Clinical Overview of NMDAr Autoimmune Encephalitis
The multiphase or staged clinical deterioration seen in NMDAr-AE is the result of a progressive decrease in NMDAr function due to loss of the receptors from the neuronal cell surface. Expectedly, as antibodies are removed with treatment, the surface expression of NMDAr can reverse and increase back to near normal levels [34]. A decrease in surface NMDAr in GABA-ergic neurons can contribute to excitotoxicity from multiple mechanisms. Disinhibition of excitatory paths can lead to hyperexcitation and more sustained excitatory activity. This higher activity creates robust metabolic demand and stress on neurons, which increases extracellular glutamate in the synapse and also activates excitatory pathways, resulting in neuronal damage if continued for long periods of time [28]. Initial stages of this NMDAr dysfunction can result in loss of executive function and paranoid behaviors manifested as neuropsychiatric symptoms that are often new in onset for the patient, which should trigger clinical concern [35]. If this autoimmune NMDAr antagonism goes untreated or unrecognized, patients often go on to develop more significant cognitive impairment, agitation, and seizures. High levels of antagonism manifest as motor stereotypies (dyskinesia, oculogyric crisis, chorea, dystonia), psychosis, mutism, and alteration in consciousness, which are manifestation of pathological frontostriatal syndromes [30, 35].
Treatment and Outcomes
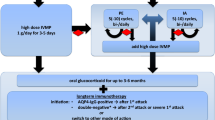
There is no current standard treatment pathway for NMDAr-AE, but many reasonable approaches have been proposed and reported to be effective. First and foremost, evaluation of possible tumor or viral illness (i.e., herpes simplex virus) should be completed. Generally, for autoimmune encephalitis, if there is no cell surface antibody nor tumor identified, first-line therapy will consist of a trial of high dose steroids (e.g., 1 g/day of IV methylprednisolone for 5 days) in conjunction with intravenous immunoglobulin (IVIg) or plasmapheresis (PLEX) [36]. If patients are minimally responsive or have relapses, second-line therapy will consist of immunotherapy with Rituxan or cyclophosphamide, and usually the patient is started on additional maintenance therapy with agents Azathioprine or Mycophenolate mofetil. Recently, alternative therapies (possibly third line) such as Bortezomib or Tocilizumab have also been used with patient’s refractory to Rituxan with similar success rates. A brief summary of the treatment options, mechanisms, and side effects is seen in Table 2.
A course of IVIg, 2 g/kg dosing over 5 days, is typically used to attempt to inhibit the pathological antibodies effects, but no large clinical cohort has been successfully used to evaluate the proper dosing or timing of immunoglobulin; much of its use is based on what is known on its effect on other neuroimmune disorders [37]. One large retrospective database in the UK was created to evaluate the efficacy of IVIg, but the study was limited in its conclusions due to limited outcome data availability [38].
Plasmapheresis or selective immunoadsorption (sIA) are treatment options that remove pathologic substances from plasma, alter the immune system, and have a possible synergistic effect with steroids [36]. Plasmapharesis (plasma exchange or PLEX) has been suggested to rapidly reduce the antibody titer in the serum [39,40,41,42]. If PLEX or sIA is used, it is important to use it before, or at least not immediately after IVIG has been administered, because this will theoretically remove IVIg from the circulation and limit its clinical effectiveness. No single approach has been proven to be superior to the other in disease modification or outcome response. No current clinical trials have compared the first-line therapeutic agents (corticosteroids, IVIg, or PLEX) against each other individually. A retrospective review (after steroids and IVIg) compared the efficacy of PLEX in multiple sclerosis and autoimmune encephalitis, with 75% of the AE group showing mild or good clinical improvement [43]. Another retrospective review with sIA showed an early clearance (average 5 days) of median antibody titers in serum (97%) and CSF (64%), and significant clinical improvement with 86% having at least 1 point decrease in mRS at 4 months [41]. One prospective clinical trial [44] is comparing IVIg versus placebo specifically in children for all causes of encephalitis.
For more chronic therapy, regular immunosuppression with IV rituximab or cyclophosphamide has also been suggested with supporting data, particularly in those patients with relapse of symptoms and recurrent antibody titers [31, 45]. In small prospective cohorts, treatment with rituximab has been shown to significantly improve seizure frequency, and improved functional outcomes as assessed by the modified Rankin Score (mRS) at 6–36 months [46,47,48]. Most current clinical trials are being done in small cohorts and focus on novel immunotherapies or adjunct treatment refractory to second-line therapy. Tocilizumab has been used in a small prospective, randomized trial for patients refractory to rituximab, showing significant improvement in mRS at 1 month (53.3% vs 12.9%, p < 0.002) [49]. In addition, there are two phase II trials, the GENERATE-BOOST trial in Germany and UT Southwestern in Dallas, TX, evaluating Bortelezomib [50] and Ocrelizumab [51] as second-line therapy versus placebo, respectively.
In immunotherapy refractory cases, it is important to consider the continued evaluation and prompt treatment of potential sources. If there is an identified malignancy, such as a teratoma, this should be surgically addressed to remove the source of pathological antibodies. Those without contributory tumor or who are diagnosed with NMDAr-AE late in its course tend to require more aggressive treatment courses such as aggressive immunosuppression with chemotherapeutics [31].
Bench to Bedside
Immunohistochemistry (IHC) was the pivotal technique that led to our current understanding of NDMAr AE. The basis for IHC is founded in the discovery and characterization of antibodies in the late 1890s and early 1900s, and the development of antibody labeling in the 1960s. Although this history is outside the scope of this review, the interested reader is referred to the article by Childs in which the work of several Nobel Laureates is summarized as it pertains to the understanding of antibody mediated immunity and the development of IHC methodology [52]. Here, we review the features of antibody-antigen interactions and the approaches used to characterize AE, and NMDAr-AE in particular. The fundamental aspects of the antibody and two forms of labeling are summarized in Fig. 1A. Antibodies have two heavy chains that carry species-specific sequences and two light chains that combined with the heavy chains to form two antigen binding domains (Fab - Fig. 1A, yellow highlights) with identical antigen binding regions and a crystallization fragment (Fc – red highlight in Fig. 1A) domain [53]. The antibody that contains the binding region for the specific antigen of interest is referred to as the primary antibody. To visualize the binding reaction location, the heavy chain is commonly labeled in one of two ways. Direct labeling is the attachment of an enzyme or chromophore to the heavy chain of the primary antibody (Fig. 1B). Indirect labeling is the use of an antibody that recognizes the species-specific regions (epitopes) in the heavy chain of the primary antibody and is labeled with an enzyme or chromophore on its heavy chain (Fig. 1C). The preferred chromophores today are fluorescent and can allow multiple antigens to be studied simultaneously in the same samples by using labels that fluoresce in different wavelength bands. Both labeling approaches were used extensively in the study and characterization of the causative antibodies associated with NMDAr-AE.

Antibody structure and approaches to labeling for immunostaining. (A) Typical antibody structure with two heavy chains and two light chains in a complex that forms two identical antigen binding regions (green sections in the yellow highlight, Fab portion). The dimer portion between heavy chains contain species-specific regions that provide epitopes for antibodies to bind and label indirectly (red FC portion). (B) Example of direct labeling of a primary antibody. A chromophore or enzyme can be attached to the heavy chain dimer portion of the primary antibody that recognizes the target antigen and provides a signal to localize the site of antigen binding interactions. (C) Example of indirect primary antibody labeling. Antibodies directed at the species-specific epitope on the heavy chain dimers can be labeled with a chromophore or enzyme and added after a primary antibody has been applied. This allows for localization of the primary antibodies and by extension the location of their binding interactions
The application of IHC as a diagnostic tool relied on using the serum or CSF from affected patients as the primary antibody in the assay. Then, either the patient antibodies were labeled directly, or anti-human antibodies raised in another species (such as goat or rabbit) and tagged with a fluorophore were applied as secondary antibodies to visualize the binding of the antibodies from the patient samples. Application of the patient samples to sections of rat brain followed by one of the labeling techniques allowed for the determination of the distribution of target antigens in neural tissue [9, 54]. Critical assumptions were that rat brain slices would contain the same antigenic proteins as human brains, that the proteins would be expressed in the same brain regions as in humans, and that the proteins would contain the same epitope found in the patients’ brains. This was a reasonably safe set of assumptions given the severity of symptoms produced in humans suggested a critical role for the target protein, making it highly likely to be conserved across species. Although the process has been optimized since it was introduced [55], the fundamental aspects of IHC have remained the basis for both diagnosis and characterizing AE antibodies from patient samples [5, 35].
In Vitro Immortalized Cell Lines
Another important tool used in the characterization of NMDAr-AE was the use of the HEK-293 cells (HEK) in vitro. The HEK cell line was derived from human fetal kidney cells in 1973. The origin of the cells is traced to a legal abortion in the Netherlands [56]. This method was developed by using adenovirus to transfect human cells so they could survive in culture indefinitely. Although the original cell cultures were thought derived from kidney tissue samples, transcriptome studies suggest they were likely transfected adrenal cells, as the closest pattern of gene expression is in adrenal cells [57, 58]. This makes sense given the proximity of these cells to renal tissue and the low transfection rate of adenovirus in epithelial cells compared to neuronal cell lines [57, 58]. HEK cells are now commonly used for gene expression studies by transfecting the cells with DNA plasmids containing genes of interest resulting in the expression of the proteins in mammalian cells in vitro [59, 60]. Using this technology, different combinations of NMDAr subunits could be studied in isolation so that the combination best recognized by patient antibodies could be determined. Identifying the target epitope, NMDAr1, using this approach has led to a better understanding of the physiologic basis for the clinical symptoms and presentation [9, 34].
Discovering the Cause of NMDAr-AE
The discovery and characterization of voltage-gated potassium channel antibodies predated the work on NMDAr-AE and provided a foundation for the rapid application of the same assays to the new group of patients, with a slightly different clinical picture and negative evaluation for known antibodies [54, 61]. Antibodies were suspected based on the clinical observations that immunomodulatory therapy was an effective treatment [54, 61]. Using optimized screening assays, the first step was to determine if antibodies were present that recognized antigens in neural tissue. In the first few NMDAr-AE patients to be tested and characterized, patients’ serum and spinal fluid was applied to fixed sections of rat brain [54, 61]. The patterns of staining that resulted could be compared to prior known AEs, and in particular the voltage-gated potassium channel (VGKC) associated AE [61]. These initial studies demonstrated significant staining in the hippocampus, and to a much lesser extent cerebellum, and were distinct from the pattern of VGKC antibodies [61]. To further confirm this observation and to determine if more than one epitope was responsible for the new patient samples, competition assays were used. The amount of fluorescent signal was measured as increasing amounts of unlabeled antibody samples from patients was mixed with samples containing a fixed concentration of labeled antibodies. As the unlabeled antibody concentration is increased the fluorescent signal was reduced. The reduction in signal suggests a competition for common epitope binding sites and a common antigen. The new samples did not compete for binding with known VGKC patient samples, and only 2 patient samples directly competed with each other, though both partially reduced signals in other parings from similar patients, indicating the antigen target was distinct from VGKC and there was likely more than one epitope on the target antigen [61].
From the rat sections, it appeared the new antigen was densely expressed in the hippocampus, though was not clearly co-localizing with synaptic markers [54, 61]. To further explore this, primary neuronal cultures from rat pups were used to allow better assessment of co-localization [9]. By using primary antibodies to known cell surface antigens and patient samples as primary antibody, detailed confocal microscopy in these more isolated cells allowed for clear demonstration of co-localization of known NMDAr subunits in the postsynaptic dendrite membranes, as well as distinct patterns of cell surface staining with absence of intracellular labeling and extra-synaptic membrane labeling seen with VGKC [9, 35, 62].
To further confirm the identity of the antigen, co-localization studies were performed between patient samples and antibodies to known surface proteins in synaptic neuropil [9, 35, 62]. In these assays, it was noted that antibodies to NR2B subunits of the NMDAr were significantly co-localized with the patient samples. This relationship was then further explored using HEK 293 cells. HEK cells are an immortalized line of neuroepithelial cells that can be easily grown in large numbers [56, 57, 59, 60]. They are commonly used as a mammalian cell line that can be can exploited to express specific proteins in high concentration to allow study [59, 60]. In the study of NMDAr-AE, IHC was applied to HEK cells that were fixed and either left intact or prorated to determine what side of the cell membrane the NMDAr antigen resided. The ability to transfect these cells, which do not normally have NMDAr expression, allowed for expression of specific subsets of NMDAr subunits and the establishment of which subunits or combinations of subunits resulted in antigen epitope formation and binding of the patient antibodies [34, 35, 63]. Based on these studies, it was determined that the NMDAr1 subunit is the primary target for the antibodies [34, 35, 63]. These experimental tools remain clinically important and provide the foundation for the clinical assay for diagnosis [35, 62, 63].
Having proven that antibodies against neural tissue were present and that they bound to NMDAr subunits, the final step was to prove that the antibodies identified in patient samples were causative of the symptoms. This determination of causality has been approached in several ways. First, other agents that interact with NMDAr and impair its function such as PCP and Ketamine result in similar subsets of symptoms to those seen in NMDAr-AE [28], for example aberrant behavioral changes and memory disturbances, and chronic use results in permanent damage to brain regions that are commonly affected in NMDAr-AE [28, 32, 33]. The second line of evidence is based on the expectation that clinical symptoms should be related to the concentration of antibodies present. It was known that immunotherapy to reduce antibody titers resulted in improvement of symptoms [61]. In several cases, the relationship of symptoms to titer was carefully tracked and showed a strong correlation of Ab titers to clinical states during the course of treatment and recurrence [34, 62]. Finally, exposure to the antibodies derived from patients should result in the disease. Infusion of human CSF samples from affected patients into mice in vivo resulted in the development of behavioral symptoms such as progressive memory deficits, and anhedonic and depressive-like behaviors [64, 65]. After chronic infusion, histologic evaluation showed a reduction of NMDAr staining in the hippocampus, increased glutamate release and impaired regulation of glutamate [64, 66]. This observation, in combination with the demonstration in primary neuronal cultures and HEK cells that NMDAr expression was reduced with exposure to patient samples, has proven that the antibodies are causative and work by reducing the pool of available surface NMDAr subunits through internalization and loss from the cell surface.
Conclusion
In summary, the autoimmune encephalitides, and NMDAr-AE in particular, represent great examples of true translational neuroscience. The use of established bench technologies to directly address a clinical question using patient samples was an important aspect of this work and led to the rapid identification of the causative agent, an understanding of treatment effects, and the characterization of the underlying pathophysiology in a remarkably short period of time of less than 10 years. The diagnostic tools used clinically are based on the research tools used to characterize the disease [35]. Fortunately for patients, an effective treatment exists that reverses the neurologic impairment when applied early in the disease. The combination of clinical observations, application of disease modifying therapy, and an underlying pathophysiology that has been well studied and understood for many years were key contributors to the rapid discovery and characterization of this class of diseases.
References
Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016;15:391-404.
Brierley JB, Corsellis JAN, Hierons R. Subacute encephalitis of later adult life - mainly affecting the limbic areas. Brain. 1960(83):357-70.
Irani SR, Pettingill P, Kleopa KA, Schiza N, Waters P, Mazia C, et al. Morvan syndrome: clinical and serological observations in 29 cases, Ann Neurol 2012;72:241-55.
Varley J, Taylor J, Irani SR. Autoantibody-mediated diseases of the CNS: Structure, dysfunction and therapy. Neuropharmacology. 2018;132:71-82.
Younger DS. Autoimmune Encephalitides. Neurol Clin 2019;37(2):359-81.
Irani SR, Michell AW, Lang B, Pettingill P, Waters P, Johnson MR, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. 2011;69(5):892-900.
Lai M, Huijbers MG, Lancaster E. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776-85.
Ohkawa T, Fukata Y, Yamasaki M. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci 2013;33:18161-74.
Dalmau J, Tuzun E, Wu HY, Masjuan J, Rossi JE, Voloschin A, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007;61(1):25-36.
Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, et al. Antibodies to the GABAB receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010;9:67-76.
Dogan Onugoren M. Limbic encephalitis due to GABAB and AMPA receptor antibodies: a case series. J Neurol Neurosurg Psychiatry 2015;86(9):965-72.
Boronat A, Gelfand JM, Gresa-Arribas N. Encephalitis and antibodies to dipeptidylpeptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol 2013;73:120-8.
Höftberger R, van Sonderen A, Leypoldt F. Encephalitis and AMPA receptor antibodies: novel findings in a case series of 22 patients. Neurology. 2015;84:2403-12.
Granerod J, Ambrose HE, Davies NW, Clewley JP, Walsh AL, Morgan D, et al. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis 2010;10(12):835-44.
Granerod J, Tam CC, Crowcroft NS, Davies NW, Borchert M, Thomas SL. Challenge of the unknown. A systematic review of acute encephalitis in non-outbreak situations. Neurology. 2010;75(10):924-32.
Vora NM, Holman RC, Mehal JM, Steiner CA, Blanton J, Sejvar J. Burden of encephalitis-associated hospitalizations in the United States, 1998-2010. Neurology. 2014;82(5):443-51.
Cohen J, Sotoca J, Gandhi S, Yeshokumar AK, Gordon-Lipkin E, Geocadin RG, et al. Autoimmune encephalitis: A costly condition. Neurology. 2019;92(9):e964-e72.
Dubey D, Pittock SJ, Kelly CR, McKeon A, Lopez-Chiriboga AS, Lennon VA, et al. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann Neurol. 2018;83(1):166-77.
Gable MS, Sheriff H, Dalmau J, Tilley DH, Glaser CA. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis 2012;54:899-904.
Dalmau J, Geis C, Graus F. Autoantibodies to synaptic receptors and neuronal cell surface proteins in autoimmune diseases of the central nervous system. Physiol Rev 2017;97:839-87.
Armangue T, Moris G, Cantarín-Extremera V. Autoimmune postherpes simplex encephalitis of adults and teenagers. Neurology. 2015;85:1736-43.
van Sonderen A, Dl R, Ja S. Anti-LGI1 encephalitis is strongly associated with HLA-DR7 and HLA-DRB4. Ann Neurol 2017;81:193-8.
Kim TJ, Lee ST, Moon J. Anti-LGI1 encephalitis is associated with unique HLA subtypes. Ann Neurol 2017;81:183-92.
Binks S, Varley J, Lee W, Makuch M, Elliott K, Gelfand JM, et al. Distinct HLA associations of LGI1 and CASPR2-antibody diseases. Brain. 2018;141(8):2263-71.
Prüss H, Dalmau J, Harms L. Retrospective analysis of NMDA receptor antibodies in encephalitis of unknown origin. Neurology. 2010;75:1735-9.
Hoftberger R, Titulaer MJ, Sabater L. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology. 2013;81:1500-6.
Lai M, Hughes EG, Peng X. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009;65:424-34.
Newcomer JW, Farber NB, Olney JW. NMDA receptor function, memory, and brain aging. Dialogues Clin Neurosci 2000;2(3):219-32.
Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, Sangha N, Martinez-Lage M, Dalmau J. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology. 2011;77(6):589-93.
Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology. 2011;77(2):179-89.
Rosenfeld MR, Dalmau JO. Paraneoplastic disorders of the CNS and autoimmune synaptic encephalitis. Continuum (Minneap Minn) 2012;18(2):366-83.
Mak YT, Lam WP, Lü L, Wong YW, Yew DT. The toxic effect of ketamine on SH-SY5Y neuroblastoma cell line and human neuron. Microsc Res Tech 2010;73(3):195-201.
Wang C, Zheng D, Xu J, Lam W, Yew DT. Brain damages in ketamine addicts as revealed by magnetic resonance imaging. Front Neuroanat 2013;7:23.
Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008;7(12):1091-8.
Dalmau J. NMDA receptor encephalitis and other antibody-mediated disorders of the synapse: The 2016 Cotzias Lecture. Neurology. 2016;87(23):2471-82.
Shin Y-W, Lee S-T, Park K-I, Jung K-H, Jung K-Y, Lee SK, et al. Treatment strategies for autoimmune encephalitis. Ther Adv Neurol Disord 2018;11:1756285617722347.
Jacob S, Rajabally Y. Current proposed mechanisms of action of intravenous immunoglobulins in inflammatory neuropathies. Curr Neuropharmacol 2009;7:337-42.
Kinsella JA, Irani SR, Hollingsworth R, O'Shaughnessy D, Kane P, Foster M, et al. Use of intravenous immunoglobulin for the treatment of autoimmune encephalitis: audit of the NHS experience. JRSM Open 2018;9(9):2054270418793021.
Klingel R, A H, Fassbender C. Plasma exchange and immunoadsorption for autoimmune neurologic diseases - current guidelines and future perspectives. Atheroscler Suppl 2009;10:129-32.
Kohler W, Ehrlich S, Dohmen C. Tryptophan immunoadsorption for the treatment of autoimmune encephalitis. Eur J Neurol 2015;22:203-6.
Dogan Onugoren M, Golombeck KS, Bien C, Abu-Tair M, Brand M, Bulla-Hellwig M, et al. Immunoadsorption therapy in autoimmune encephalitides. Neurol Neuroimmunol Neuroinflamm. 2016;3(2):e207.
Heine J, Ly LT, Lieker I. Immunoadsorption or plasma exchange in the treatment of autoimmune encephalitis: a pilot study. J Neurol 2016;263:2395-402.
Moser T, Harutyunyan G, Karamyan A, Otto F, Bacher C, Chroust V, et al. Therapeutic Plasma Exchange in Multiple Sclerosis and Autoimmune Encephalitis: a Comparative Study of Indication, Efficacy and Safety. Brain Sci. 2019;9(10).
Iro MA, Sadarangani M, Absoud M. ImmunoglobuliN in the treatment of encephalitis (IgNiTE): protocol for a multicentre randomised controlled trial. BMJ Open 2016;6:012356.
VanHaerents S, Gerard EE. Epilepsy Emergencies: Status Epilepticus, Acute Repetitive Seizures, and Autoimmune Encephalitis. Continuum (Minneap Minn). 2019;25(2):454-76.
Byun J-I, Lee S-T, Jung K-H, Sunwoo J-S, Moon J, Lim J-A, et al. Effect of Immunotherapy on Seizure Outcome in Patients with Autoimmune Encephalitis: A Prospective Observational Registry Study. PLoS One 2016;11(1):e0146455.
Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12(2):157-65.
Lee WJ, Lee ST, Byun JI. Rituximab treatment for autoimmune limbic encephalitis in an institutional cohort. Neurology. 2016;86:1683-91.
Lee W-J, Lee S-T, Moon J, Sunwoo J-S, Byun J-I, Lim J-A, et al. Tocilizumab in Autoimmune Encephalitis Refractory to Rituximab: An Institutional Cohort Study. Neurotherapeutics 2016;13(4):824-32.
Trial to Evaluate Efficacy and Safety of Bortezomib in Patients With Severe Autoimmune Encephalitis - Full Text View - ClinicalTrials.gov.
Efficacy of Ocrelizumab in Autoimmune Encephalitis - Full Text View - ClinicalTrials.gov.
Childs GV. History of Immunohistochemistry. In: McManus LM, Mitchell RN editors. Pathobiology of Human Disease: A Dynamic Encyclopedia of Disease Mechanisms. ACADEMIC PRESS LTD-ELSEVIER SCIENCE LTD 2014:3775-96. https://doi.org/10.1016/B978-0-12-386456-7.07401-3.
Crisp SJ, Dm K, Vincent A. Autoimmune synaptopathies. Nat Rev Neurosci 2016;17:103-17.
Bataller L, Kleopa KA, Wu GF, Rossi JE, Rosenfeld MR, Dalmau J. Autoimmune limbic encephalitis in 39 patients: immunophenotypes and outcomes. J Neurol Neurosurg Psychiatry 2007;78(4):381-5.
Chiu NC, Lin YJ, Tzang RF, Li YS, Lin HJ, Das S, et al. Optimization of an Anti-NMDA Receptor Autoantibody Diagnostic Bioassay. Front Neurol 2018;9:661.
Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 1977;36(1):59-74.
Louis N, Evelegh C, Graham FL. Cloning and sequencing of the cellular-viral junctions from the human adenovirus type 5 transformed 293 cell line. Virology. 1997;233(2):423-9.
Shaw G, Morse S, Ararat M, Graham FL. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J 2002;16(8):869-71.
Stepanenko AA, Dmitrenko VV. HEK293 in cell biology and cancer research: phenotype, karyotype, tumorigenicity, and stress-induced genome-phenotype evolution. Gene. 2015;569(2):182-90.
Lin YC, Boone M, Meuris L, Lemmens I, Van Roy N, Soete A, et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat Commun 2014;5:4767.
Ances BM, Vitaliani R, Taylor RA, Liebeskind DS, Voloschin A, Houghton DJ, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain. 2005;128(Pt 8):1764-77.
Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011;10(1):63-74.
Lancaster E, Dalmau J. Neuronal autoantigens--pathogenesis, associated disorders and antibody testing. Nat Rev Neurol 2012;8(7):380-90.
Planaguma J, Leypoldt F, Mannara F, Gutierrez-Cuesta J, Martin-Garcia E, Aguilar E, et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain. 2015;138(Pt 1):94-109.
Wright S, Hashemi K, Stasiak L. Epileptogenic effects of NMDAR antibodies in a passive transfer mouse model. Brain. 2015;138:3159-67.
Manto M, Dalmau J, Didelot A, Rogemond V, Honnorat J. In vivo effects of antibodies from patients with anti-NMDA receptor encephalitis: further evidence of synaptic glutamatergic dysfunction. Orphanet J Rare Dis. 2010;5:31. Published 2010 Nov 26. doi:https://doi.org/10.1186/1750-1172-5-31.
Acknowledgments
The authors would like to thank Stan Coffman of MedmediaSolutions.com for creating the artwork presented in Fig. 1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 498 kb).
Rights and permissions
About this article

Cite this article
Kolls, B.J., O’Keefe, Y.A. & Sahgal, A.K. Autoimmune Encephalitis: NMDA Receptor Encephalitis as an Example of Translational Neuroscience. Neurotherapeutics 17, 404–413 (2020). https://doi.org/10.1007/s13311-020-00861-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00861-2