Genesis of Complex Polyiodide Networks: Insights on the Blue Box/I−/I2 Ternary System


Department of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia, 3-13, 50019 Sesto Fiorentino, Italy
*
Authors to whom correspondence should be addressed.
Crystals 2020, 10(5), 387; https://doi.org/10.3390/cryst10050387
Submission received: 20 April 2020
/
Revised: 6 May 2020
/
Accepted: 7 May 2020
/
Published: 9 May 2020
(This article belongs to the Section Crystal Engineering)
Abstract
:The Stoddart’s blue box (BB) (cyclobis(paraquat-p-phenylene))/iodide binary system was recently demonstrated to give rise to porous three-dimensional networks which can hardly be classified as common XOF-type materials (X = M, C, S, i.e., metal, covalent, or supramolecular organic frameworks), leading to the definition of permutable organized frameworks (POFs). The ternary BB/iodide/iodine system was reported to generate pentaiodide-based structures constituted by the most complex interlocked polyiodides so far isolated (up to an infinite supramolecular pseudopolyrotaxane with a poly[3]catenane axle). The missing link, i.e., the XRD structure of the BB/triiodide complex, is herein reported: structural similarities and novel Raman evidence, opening perspectives in the genesis of solid-state BB-based complex polyiodide networks from solution.

1. Introduction
Polyiodide chemistry is a long-lasting research field which, these days, still possesses a theoretical and structural connotation related to bonding theory and hypervalency of polyiodide species [1,2]. At the same time, pragmatic interest in iodine-containing solids, both as crystalline phases and iodine-doped polymers [3,4], has recently re-flourished, especially in view of the potential use of these materials as solid-state electrical conductors [5,6,7]. Beyond pragmatic interest, studies aimed at selective synthesis of polyiodides [8,9,10,11] or at disentangling different stabilizing contributions for these systems [12,13], in view of the recent interest for halogen bonding [14], are also appearing. In this sense, the “feeble force”, with which iodides retain molecular iodine in Gay Lussac’s writings, appears to have somewhat anticipated whole branches of modern chemistry [1].
Among our recent contributions, we demonstrated the use of tetrazine-based ligands for the coordination of inorganic [15,16] and organic anions [17], emphasizing coordination of halide [18] and pseudohalide anions [19]. Stabilization of anion coordination networks [20], including polyiodide-dense ones [21], has been achieved with s-tetrazine-based ligands through anion-π interactions [22], building on previous evidences of the usefulness of anion-π forces for polyiodide stabilization in perfluoroarene-based systems [23]. We re-evaluated azacyclophanes as templating agents to orchestrate the synthesis of polyiodides: while Ilioudis and Steed [24] reported formation of highly charged polyhalides (e.g., I42−) with cyclophanes matching the size of iodide anions, we demonstrated that size mismatch can be used to some extent to make the organic ligand act as a mold for the template self-assembly of larger, low-valency polyiodides (e.g., I7−), leading to extremely iodine-dense crystalline phases [25]; contextually, to benchmark our work with reported literature systems, a novel macroscopic descriptor, the iodine number IN, has been introduced to allow easy comparison of different crystalline materials in terms of iodine density [25].
Our pursuit of highly dense polyiodide-based frameworks and consolidated interest for anion-π interactions eventually led us to consider the organic tetracation cyclobis(paraquat-p-phenylene) (Scheme 1) known as Stoddart’s blue box (BB) [26], which is the object of present study. This ligand has initially drawn our attention, as it offers a highly charged scaffold, whose charge is delocalized over a rather large surface: this fixes the number of I− anions required for charge neutrality to four, without imposing limits on the formal number of I2 molecules that can be co-crystallized; assembly of polyiodides of varying stoichiometries is thus expected, with BB geometry and molecular cavity playing an orchestrating role.
The BB/I− binary system has been recently investigated, leading to interesting findings [27].
Robustness, due to charge-charge attraction, and flexibility, due to reduced directionality of BB…I− interactions, led to the preparation of a novel type of porous networks named permutable organized frameworks (POFs) [27]. POFs feature a 3D hollow architecture held together by supramolecular ball joints, which allow for different mutual orientation of constituents in the solid, granting the network a flexibility which is generally made impossible by design for the rigid jointed metal organic frameworks (MOFs), covalent organic frameworks (COFs), and supramolecular organic frameworks (SOFs) (generally based around strictly directional H-bonds) [27].
The ternary BB/I−/I2 system led instead to the formation of some of the more intricate, and arguably charming, polyiodide architectures of all time [28].
In fact, I5@(BB)(I3)3 crystal structure features a peculiar infinite supramolecular [3]catenane constituted by 14-membered I5− rings interlocked with two BB molecules each. By tuning crystallization conditions, i.e., fostering the conversion of a further I3− anion into a second I5−, the I5@BB@(I5)(I3)2 crystal structure is obtained, where the infinite [3]catenane is converted into the axle of an infinite supramolecular pseudopolyrotaxane, where the “macrocyclic” components are (I5)2(I3)4 22-membered polyiodide rings [28]. It is noteworthy that, while being both mixed I3−/I5− complexes, the core (I5@BB)∞ [3]catenane is the cornerstone of these structures and passes unmodified from one structure to the other.
Here, we present the missing link between the five reported BB/I− crystal structures and the two mixed BB/I3−/I5− crystals, i.e., a structure featuring only triiodide as anion. As we show, this structure features analogies with both families of I−- and I5−-based crystal structures reported so far, and it is especially relevant for the understanding of the genesis of these crystalline phases.
2. Materials and Methods
I3@BB(I3)3·2DMSO crystals were obtained by leaving exposed to the air a DMSO solution of BB(PF6)4 containing 1:2 mol:mol NaI and I2. Alternatively, I3@BB(I3)3·2DMSO were obtained by dissolving I5@BB(I3)3 crystals [28] in DMSO and waiting for subsequent spontaneous crystallization (see Section 3.4). Both methods are quite slow and required several (7–10) weeks. All employed materials were high purity commercial products.
Black crystals of I3@BB(I3)3·2DMSO were used for X-ray diffraction analysis. A summary of the crystallographic data is reported in Table S1. The integrated intensities were corrected for Lorentz and polarization effects, and an empirical absorption correction was applied [29]. The structure was solved by SHELXS-97 [30]. Refinements were performed by means of full-matrix least-squares, using SHELXL Version 2014/7 [30].
Raman spectra of I5@BB(I3)3 and I−/I2 DMSO solutions were recorded with a Brucker FT-Raman Multiram spectrometer equipped with a Nd-YAG laser emitting at 1064 nm as excitation source. The spectra were recorded on solid DMSO-wetted I5@BB(I3)3 as it gradually dissolved, giving a DMSO solution of BB-polyiodide complexes. Acquisition settings were: 4 cm−1 resolution, 150 mW incident power, 50 acquisition scans. Recrystallization of I3@BB(I3)3·2DMSO crystals happened spontaneously over time from these solutions.
3. Results and Discussion
3.1. I3@BB(I3)3·2DMSO
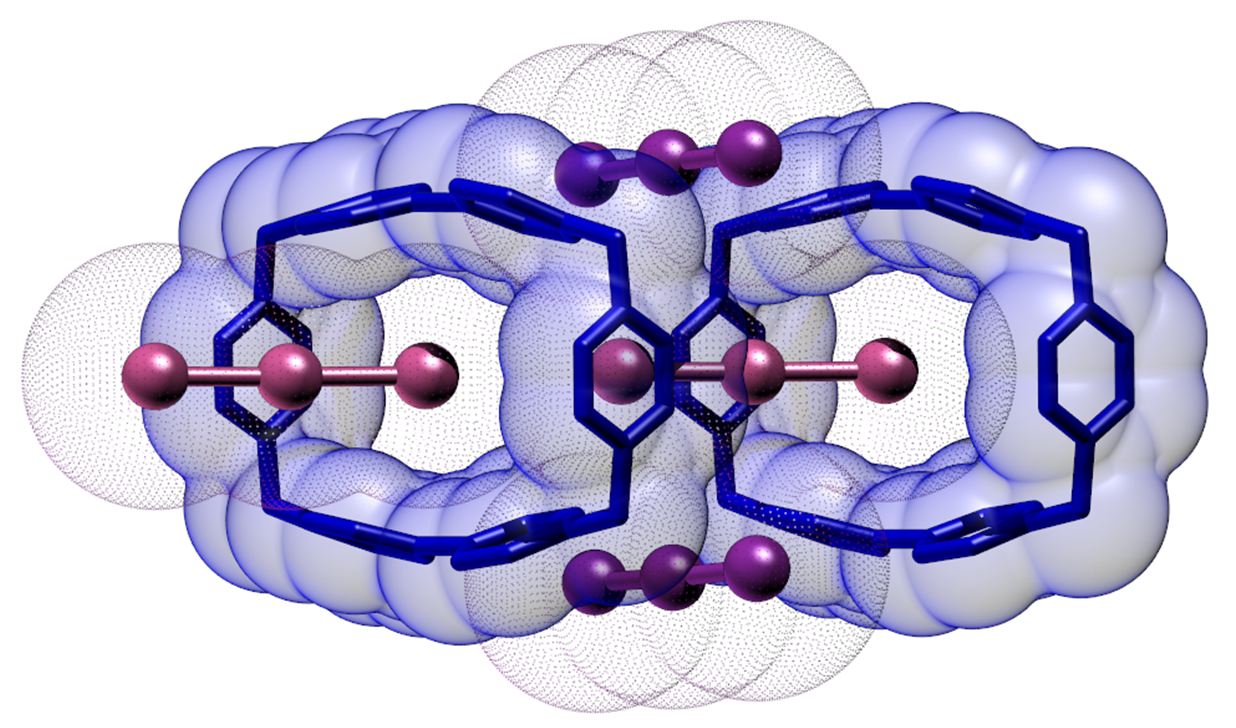
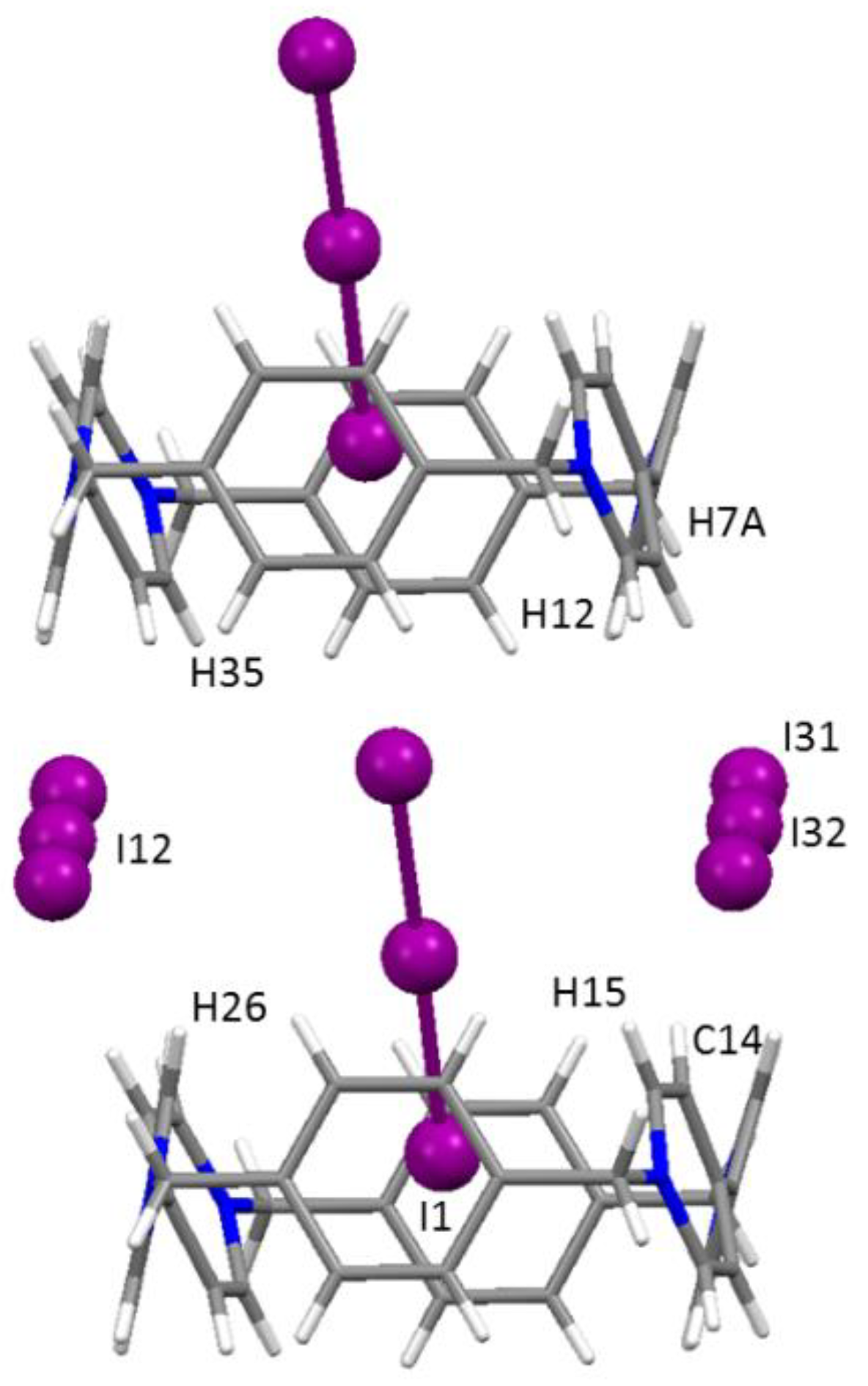
Evaluated in isolation, the I3@BB(I3)3·2DMSO crystal structure can be described as a piling of BB molecules in pillars along the a direction. Pillars are held together by two of the four non-equivalent I3− anions located along the widest (paraquat) BB side (Figure 1).
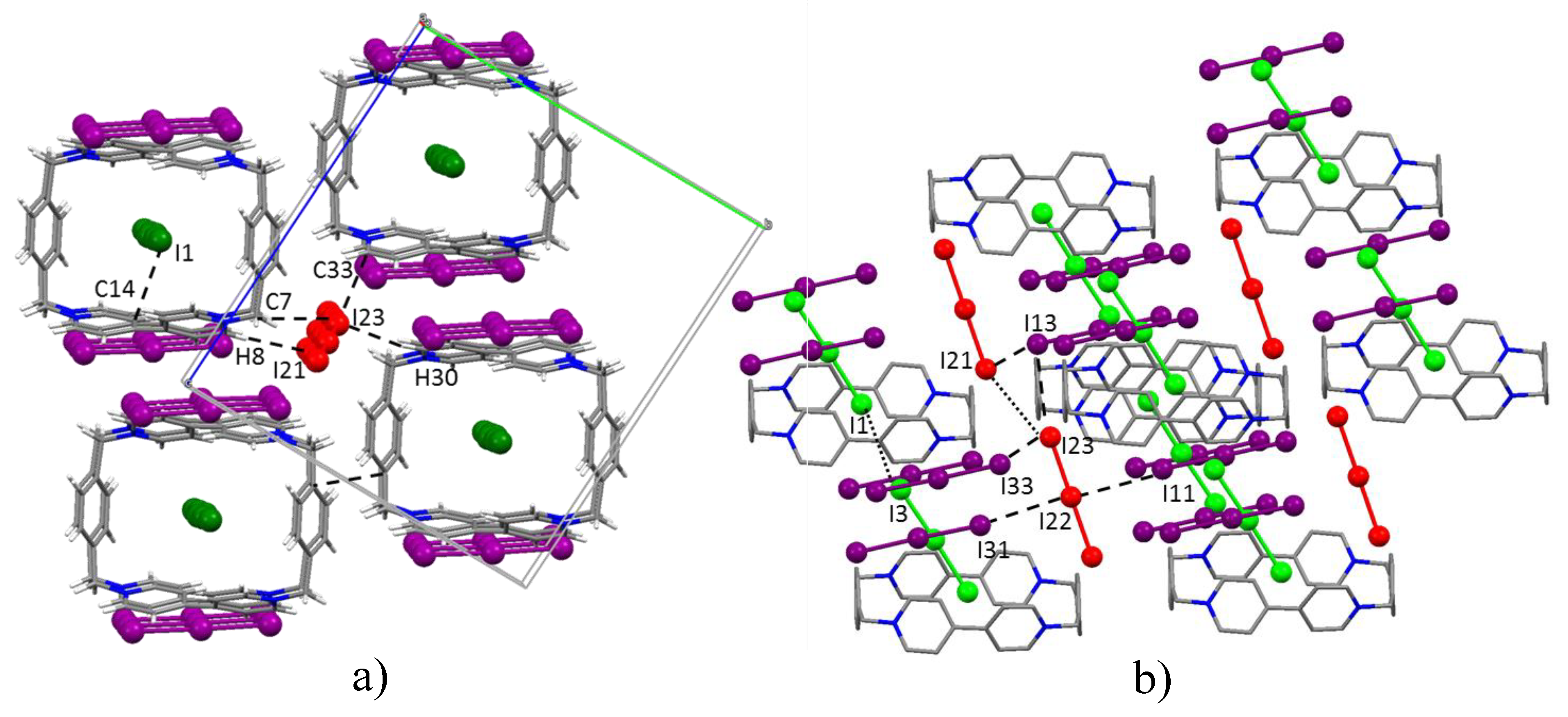
A third I3− is found to occupy the pillar cavity, hosting one of its I atoms within the BB frame. As a result, looking along the a direction, a wire of triiodides passing through BB molecules spans the whole crystal. The fourth non-equivalent I3− anion is also organized in wires passing in between adjacent BB pillars. Figure 2 offers a complete view of the crystal packing. Contact distances according to the labelling scheme of Figure 1 and Figure 2 are reported in the Supplementary Materials (Table S2).
The result is that of a highly dense polyiodide network (IN = 0.354, i.e., ≈ ⅓ of the iodine density of crystalline I2 [25]). Differently from I5−-based crystal structures, however, we observe the lack of short I…I supramolecular contacts among individual I3−: the shortest one being 4.2834(5) Å (I13…I21). This implies the lack of maintenance of orbital contact and the consequent impossibility of the I3@BB(I3)3·2DMSO structure to exhibit any conductive behavior [28].
Arguably, however, the most interesting approach for examining this structure includes its comparison with I−- and I5−-based ones.
3.2. Structural Comparison
The family of I−-based structures, obtained from acetonitrile (ACN) solvent, was found to exhibit a ball-and-socket joint architecture, where spherical iodide anions occupy preferentially BB aromatic corners, the charge-assisted CH…I contacts are more favorable, and maintain a degree of angular freedom in the direction of pillar growth [27]. A major exception to this is constituted by the (BB)I4·8H2O structure, where, due to the higher polarity of the solvent (ACN/MeOH, Scheme 1), burrowing of I− anions inside the BB was observed [27].
According to these observations, the prototypical (ACN)2@(BB)I4 crystal structure, and the abovementioned (BB)I4·8H2O [27], result as the most relevant I−-based structures to compare with I3@BB(I3)3·2DMSO.
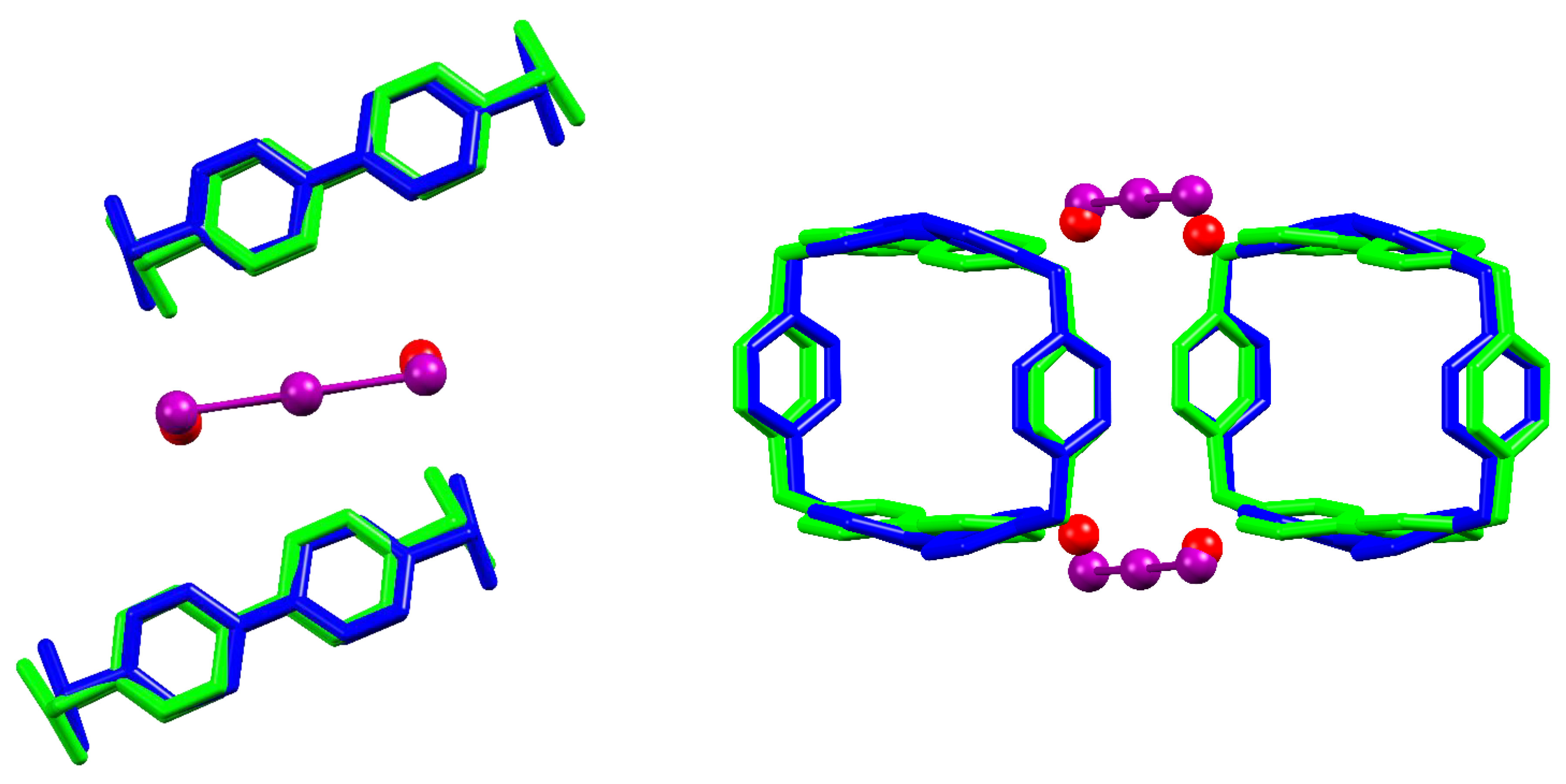
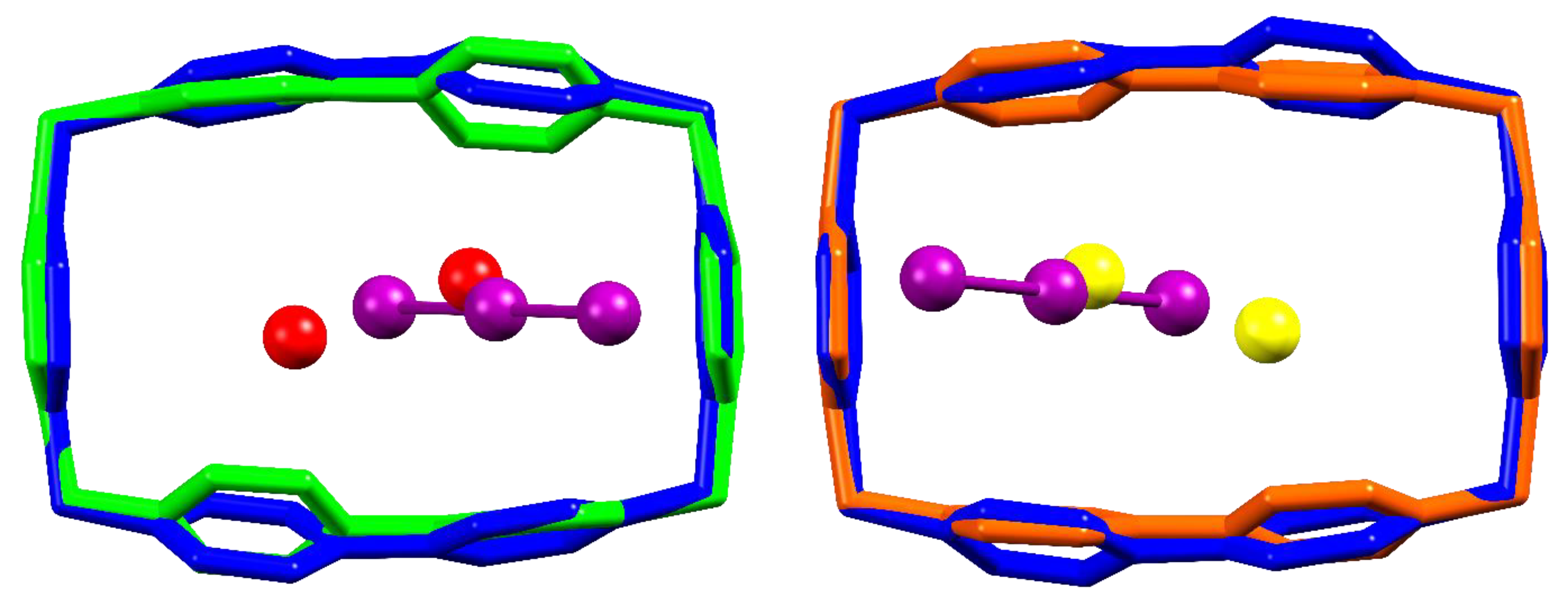
As can be seen from Figure 3, the terminal atoms of the two I3− anions responsible for intra-pillar BB…BB bridging in the I3@BB(I3)3·2DMSO structure occupy almost the same spots of simple I− anions in the (ACN)2@(BB)I4 crystal, most likely due to a fortunate coincidence between the length of I3− anion and the spacing of BB CH binding sites. Triiodides are found slightly more detached from BB than iodide anions, however this could be expected due to change in the anion charge density affecting the strength of charge-charge interactions. As shown in Figure 3, the pillar step, in terms of BB…BB distance, is basically unaffected, while a certain adjustment of the pillar growth angle is observed: this type of flexibility is a leitmotiv of I−-based structures [27].
The (BB)I4.8H2O crystal structure allows instead for comparison of the mutual positions of BB and hosted I− or I3− anion. As shown in Figure 4 for both non-equivalent BB molecules found in (BB)I4·8H2O (termed molecule A and molecule B) [27], it is observed that the position occupied by the iodide anion is almost coincident with the position of the central atom of the triiodide. This does not appear fortuitous, as this atom is formally the charge-bearing one, indicating that the charge-charge component of the piridinium-I3− anion-π contact is the most significant for the host-guest interaction.
As can be concluded from the above examination, the main differences between I3−- and I−-based structures are not to be ascribed to significant differences in the BB pillar motif or in the host-guest interactions. The main factor in play appears to be tied to charge balance and anion size reasons. Four I− per BB were required to hold together the BB pillars in the prototypical (ACN)2@(BB)I4 crystal structure, yet the same structural role can be fulfilled by only two I3− anions (as displayed in Figure 3), thus leaving two BB positive charges to be counterbalanced. One of these charges is provided by the encapsulated I3− anion; this again shows similarity with BB-I− host-guest complex, as found in (BB)I4·8H2O (Figure 4). In the case of (BB)I4·8H2O, two I− anions could be accommodated within the cavity, yet, as manifest from Figure 4, maintenance of the position of the negative charge inside the cavity, and the I3− increased size, prevent accommodating two negative charges within each BB in the case of I3@BB(I3)3·2DMSO. This leaves a last +1 charge to be compensated. The line of triiodides passing in between adjacent BB pillars (Figure 2, red) is thus the most noticeable distinguishing element between the I3@BB(I3)3·2DMSO crystal structure and I−-based ones, and it is brought about not by a difference in interactions subtending to I3− and I− BB complexes, but by the necessity of accommodating a fourth negative charge per BB molecule.
This same argument can be raised also in the comparison with I5−-based structures.
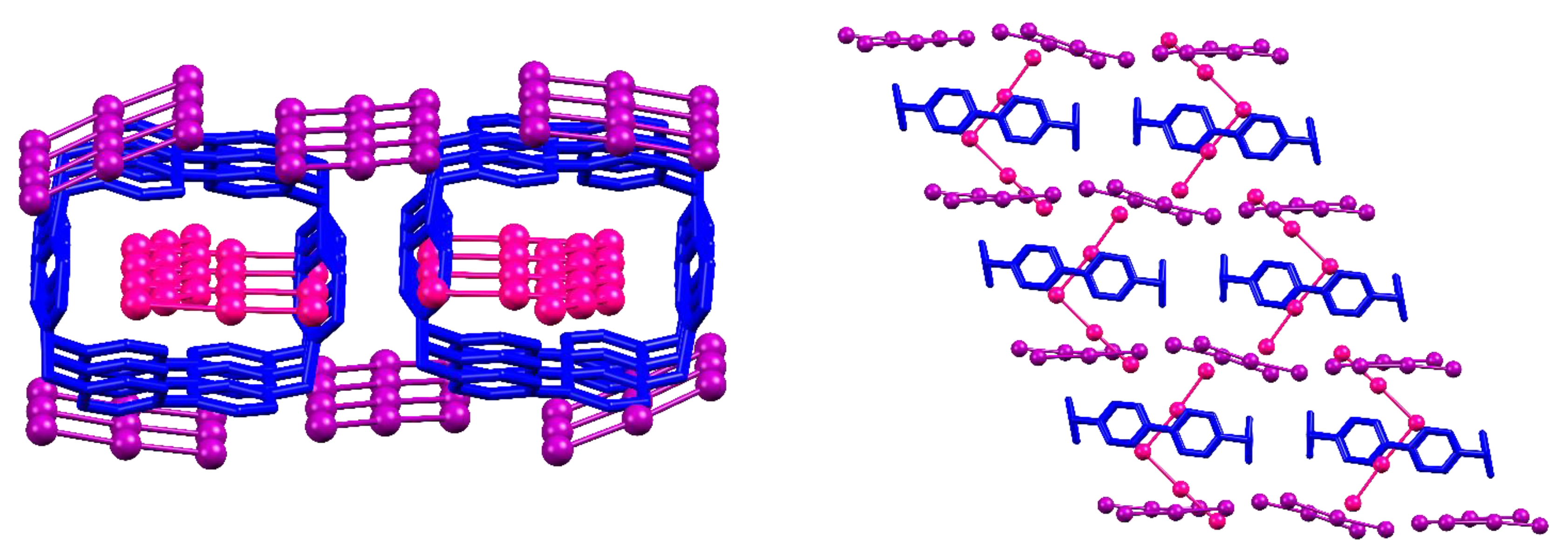
In fact, as displayed in Figure 5, these structures feature coupled BB pillars held together by I5…I5 interactions and π-π stacking forces [28].
If, as shown in Figure 3, pillars formed by piling of one BB above another can be stabilized either by two triiodides or four iodides acting as bridging elements, coupled BB pillars as found in I5−-based structures are too large for that, requiring a total of six triiodides to bridge each coupled BB complex with the next one. In terms of charges, for each two BB molecules (+8), a total of six triiodides is required (−6), leaving a +2 charge to be counterbalanced, which is easily achieved by the presence of the I5− anions within the BB cavity (one for each) (which in turn is the very reason for coupling of BB pillars).
Again, the presence of a second line of polyiodides spanning the crystal beyond the one passing through BB cavities is entirely unique of I3@BB(I3)3·2DMSO, and due to charge balance reasons. All the other architectures use as many iodide or triiodide anions as required to stabilize intra-pillar piling of BB molecules and place the remaining anions within the ligand cavity. Only for I3@BB(I3)3·2DMSO, when this process is repeated, are additional anions required for charge neutrality, leading to a train of polyiodides passing in between different BB pillars.
Conversely, the core architecture of iodide/polyiodide-stabilized BB pillars, together with main supramolecular interactions subtending to it, is essentially preserved from one structure to another. Despite different space groups and contents of the asymmetric unit, all systems present a lattice vector directed along the BB pillars’ growth direction of invariant modulus ((ACN)2@(BB)I4, b, 10.5041(2) Å; I3@BB(I3)3·2DMSO, a, 10.3559(6) Å; I5@(BB)(I3)3, a, 10.3325(2) Å), signifying the presence of a characteristic interaction pattern beyond the specificity of each crystal packing.
Comparison with I5−-based structures, specifically with prototypical I5@(BB)(I3)3, can also be undertaken in terms of host-guest complex structural features.
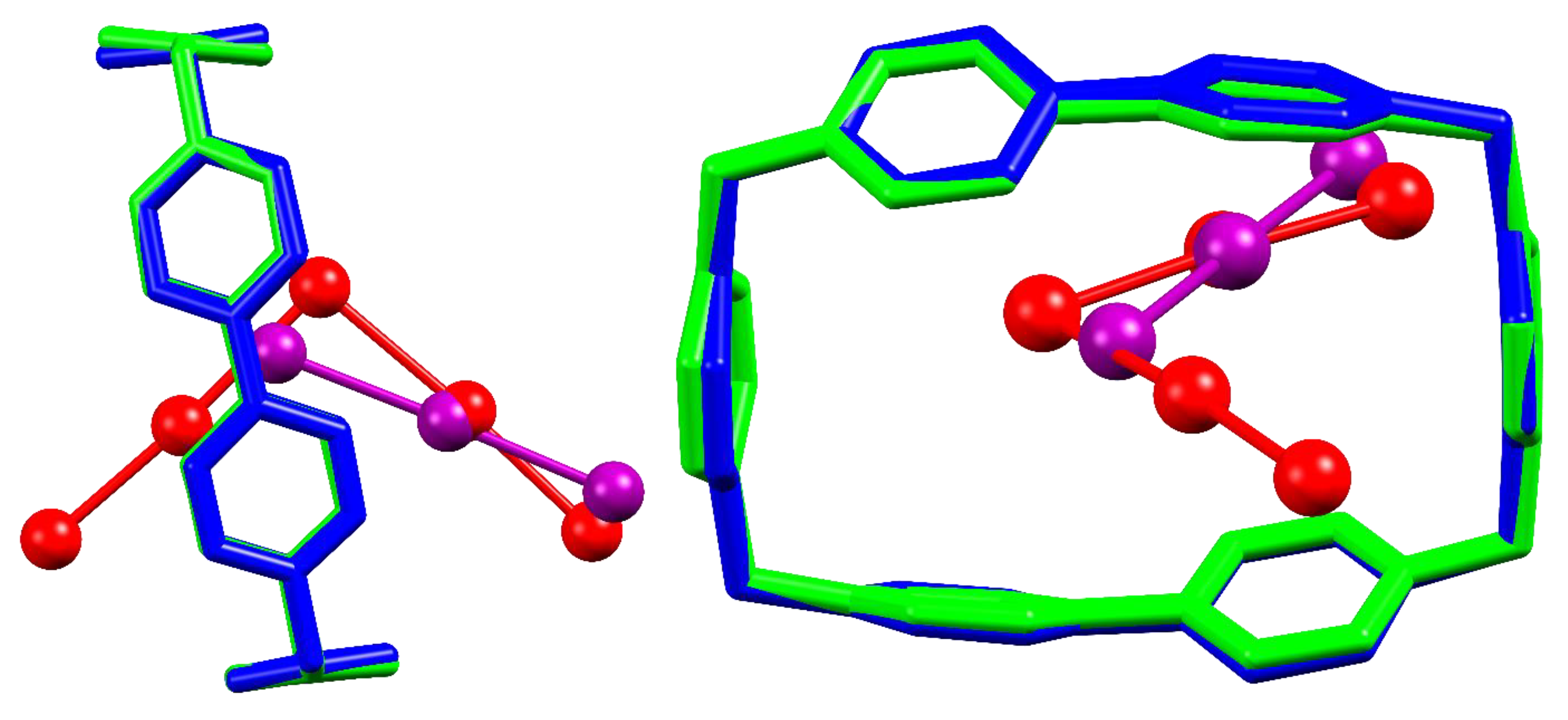
This time, the BB-anion’s mutual disposition between I3− and I5− are different, and tellingly similar at the same time (Figure 6).
As is well-known, I5− can exist in a symmetric V-shaped form, rationalized as an [(I2)·(I−)·(I2)] complex formally charged on the central atom, or in a L-shaped form, schematized as an [(I3−)·(I2)] complex with the charge localized on one of the proximal I atoms [2,21]. Both forms are frequently occurring in the solid state, and proper assignment is generally a focal point for case-by-case assessment of I5− electronic structure [2]. This point has been previously investigated for I5@(BB)(I3)3; as demonstrated by reported Raman spectra of DMSO-soaked I5@(BB)(I3)3 (solvent is necessary to avoid immediate radiation damage on the crystals preventing recording of any spectrum [28], cf. Section 3.4), in this case I5− is observed to be in the fully symmetric V-form.
What we are now able to notice from crystal structure superimposition is that, if on one hand there is no atom by atom coincidence, still the position of the central formally charge-bearing atom of I3− is relatively close not to the central I5− atom, as would be expected if it were charge-bearing, but to one of the proximal I atoms. Since the position of BB encapsulated charge-bearing I atoms was found not to differ much also in the I−-based structures, where charge localization is straightforward, it could be argued whether observed V-symmetry for I5− in I5@(BB)(I3)3 is tied to intrinsic preferences of the BB-I5− host-guest complex, or rather due to the tight network of I5−-I5− interactions within the crystal (Figure 5). As a matter of fact, this led us to seek further Raman evidence for I5− complexation in solution, where it could be possible to obtain information on the isolated (solvated) I5−@BB complex and assess the V- or L-shaped nature of the anion (see Section 3.4).
3.3. Hirshfeld Surface Analysis
Hirshfeld surface can be defined in plain words as the region of space where the electron distribution of a theoretical collection of spherical atoms, representing the molecule under discussion (the promolecule), dominates the corresponding summation over the whole crystal depicted in the same manner (the procrystal): mathematical definition, properties, and usefulness of Hirshfeld surface are found in dedicated literature [33,34]. This allows for a comprehensive and “non-biased” (in the sense that it does not involve choices concerning what is deemed important and worth discussing and what is not) evaluation of all contacts within a crystal structure. Moreover, the same treatment allows for the visualization of a crystal structure through the lens of so-called fingerprint plots [35]. These are complete maps of external distance (de) (i.e., the point-by-point distance of the nearest atom belonging to another molecule to the Hirschfeld surface of considered species) vs. internal distance (di) (i.e., the point-by-point distance from a molecule’s Hirshfeld surface and the nearest atom belonging to the molecule itself) from the Hirshfeld surface under consideration, color-coded to show relative abundance (from blue: few contacts, to red: many contacts) of intermolecular contacts occupying di × de square bins of 0.01 × 0.01 Å2.
These tools were developed to obtain a comprehensive view of a crystal and assist in formalizing and quantifying concepts like similarity among different crystal structures. In a previous study, we demonstrated how such an approach is of particular significance in our case, as crystal structures generated by the BB/I−/I2 ternary system all develop about a single invariant molecular component (BB), around which the whole crystal architecture unfolds [27]. This allows us to directly compare these structures through this methodology.
Results of the analysis for (ACN)2@(BB)I4, I3@(BB)(I3)3·2DMSO and I5@(BB)(I3)3 crystal structures are reported below in Figure 7 and Figure 8.
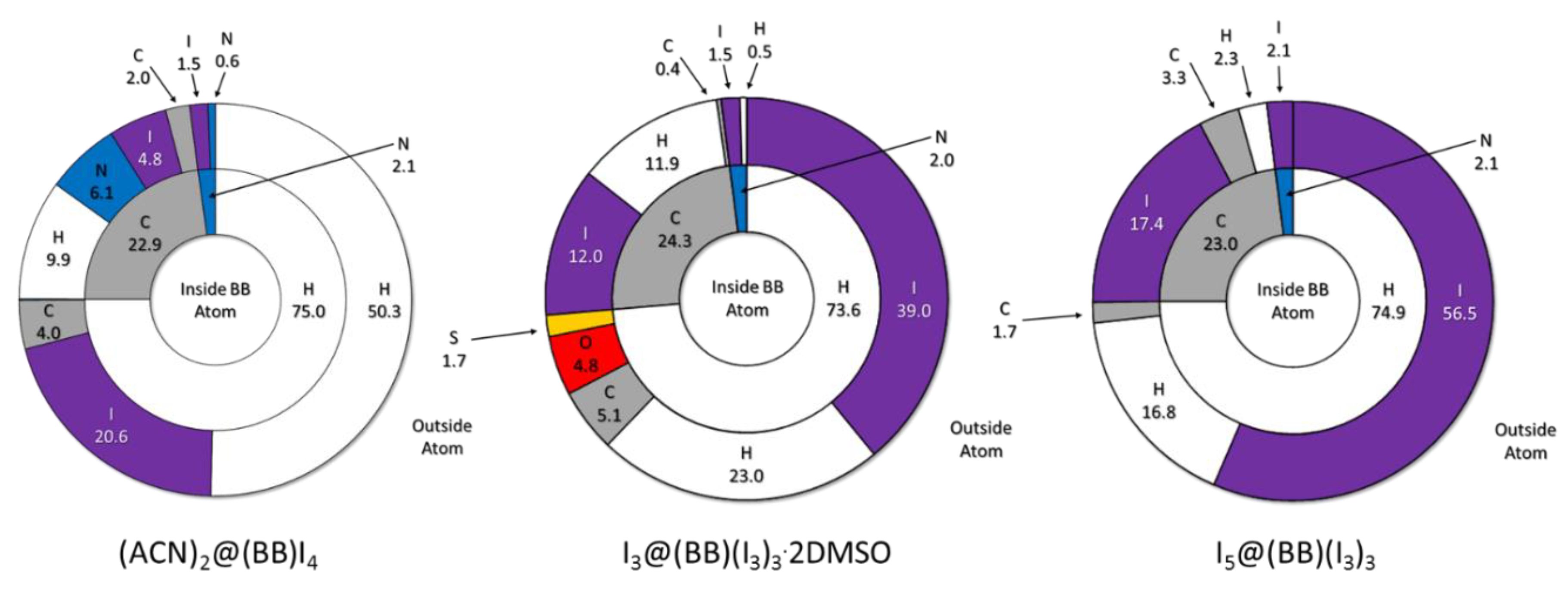
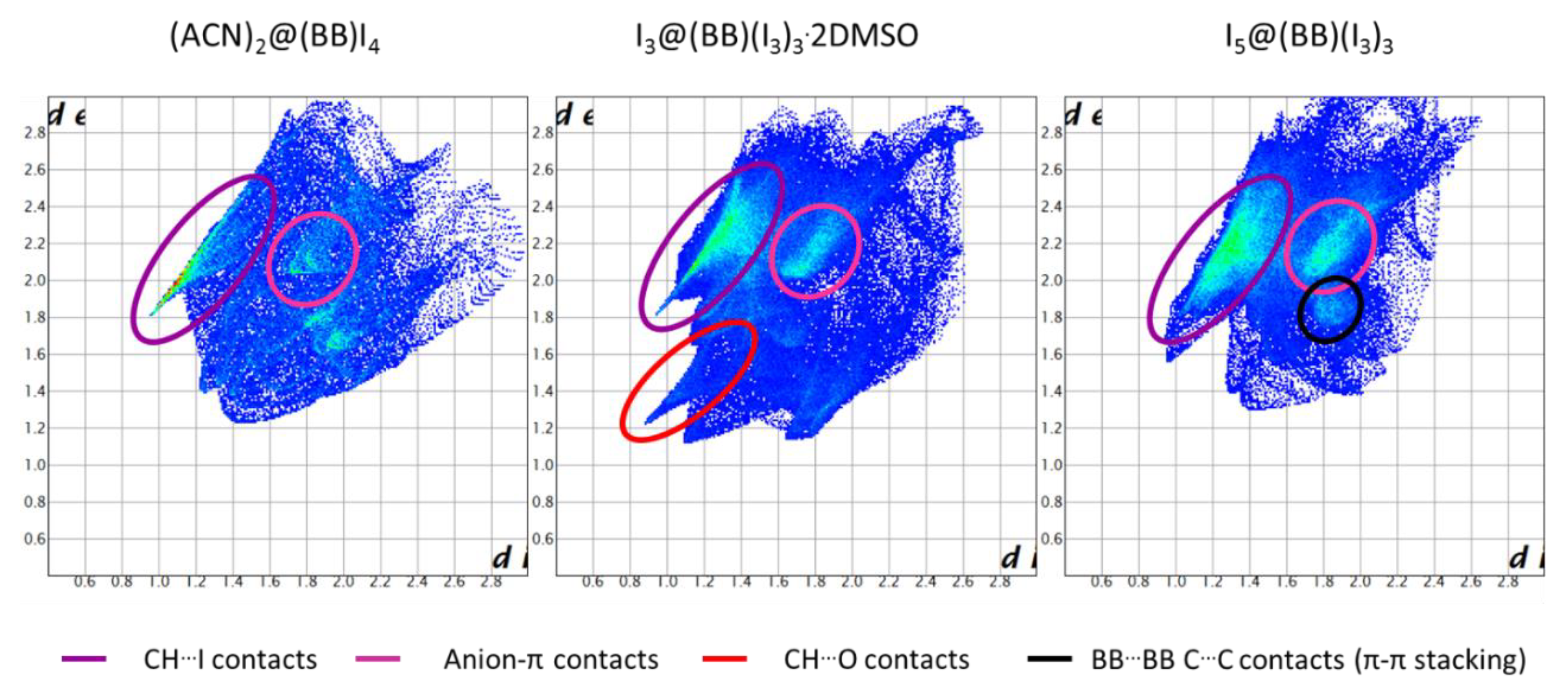
The main observation from Hirshfeld surface composition (Figure 7) is the progressive increase of BB…I contacts with increasing I content (or iodine number, IN = 0.154, 0.354, and 0.447, for considered I−, I3−, and I5− based structures, respectively). This might seem unsurprising, yet fingerprint plots allow evidence for a certain change in the type of BB…I main interactions.
As shown in Figure 8, and better detailed in Figure S1, in the case of (ACN)2@(BB)I4, main BB…I contacts are of the H…I type, giving rise to the classic tip-like feature expected for H-bond type interactions. A minor contact-dense zone related to C…I, i.e., pyridinium-I− anion-π interaction, is also manifest [27]. As we move towards polyiodide-based structures, the predominance of CH…I contacts and H-bonds is gradually reduced, together with the anion charge density. Polarizability and size of the anion increases, and so does the relevance of stabilizing contributions that can take advantage of it; in this case, anion-π interactions. The defined sharp H-bond tip observed for H…I contacts in (ACN)2@(BB)I4 gets smeared and blurred the more we move towards I5@(BB)(I3)3, while at the same time, the relevance of anion-π contacts, just glimmering in the (ACN)2@(BB)I4 fingerprint, become a major feature. I3@BB(I3)3·2DMSO indeed appears as a bridging instance between these two different trends, showing similarities with both I− and I5−-based structures.
Another point worth mentioning is the extent of BB…BB face-to-face π-π stacking. This is absent in the case of the (ACN)2@(BB)I4 structure (C…C contacts in Figure S1 are entirely due to host-guest C…C contacts between ACN and hosting BB molecule), barely present in I3@BB(I3)3·2DMSO, and relevant for I5@(BB)(I3)3. This is due to the abovementioned coupling of BB pillars existing in I5− structures (both I5@(BB)(I3)3 and I5@BB@(I5)(I3)2) and, judging from the invariance of the core (I5@BB)∞ [3]catenane from one structure to another, most likely points out the stability of such an arrangement (also consolidated by I5−…I5− interactions, cf. Figure 5). This view is experimentally supported by TGA analysis of I5@(BB)(I3)3 crystals, showing loss of the first I2 molecule only beyond 110 °C [28].
3.4. Raman Evidence
Direct Raman spectroscopic investigation of I3@BB(I3)3·2DMSO crystals was not possible, due to the same issues affecting I5@(BB)(I3)3 crystals (strong radiation absorption resulting in crystal damage).
The lack of coincidence between charge-bearing atoms for BB-hosted I3− and I5− in the crystal structure (cf. Figure 6 and related discussion) sheds doubt over pentaiodide geometry/electronic structure. V-shaped symmetry substantiated for pentaiodide in I5@(BB)(I3)3 could be due to packing features (cf. Figure 5) [28] and is not conclusive for the isolated I5−@BB host-guest complex; therefore, further Raman investigation in solution was performed.
This was done starting from I5@(BB)(I3)3 crystals because they are DMSO soluble (differently from I3@BB(I3)3·2DMSO crystals, which moreover are obtained by letting these solutions crystallize).
In reconsidering the matter, interesting pieces of evidence emerge.
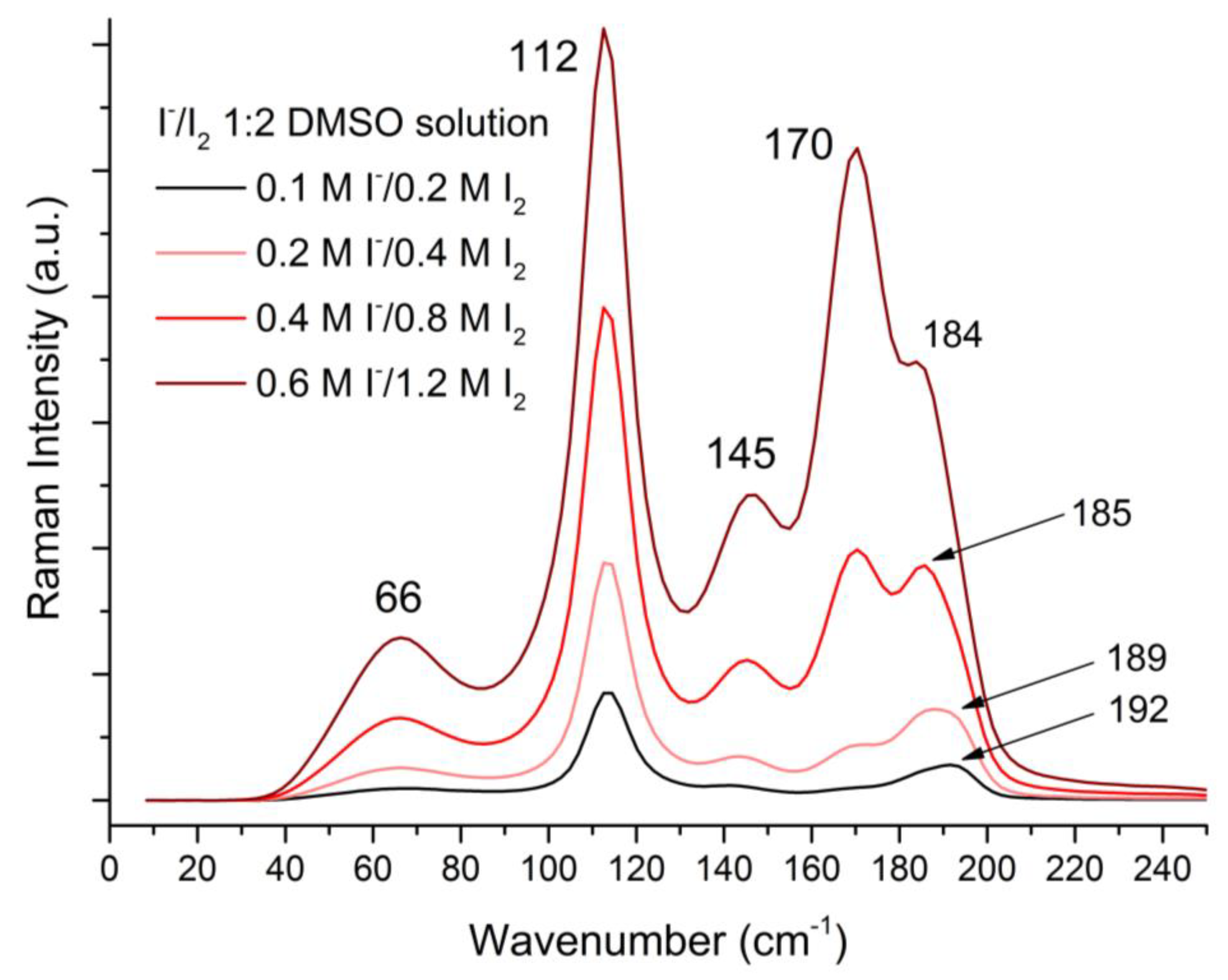
Despite a long literature tradition of dismissing the matter by saying that I5− is never present in solution [36], we, as others before us [37], were able to show that free I5− exists in solution. This was done by preparing 1:2 NaI:I2 DMSO solutions and recording their Raman spectra. As it can be seen in Figure 9, a clear I5− band centered at 170 cm−1 becomes more and more manifest the more I5− formation in solution is forced by mass action (minor shift of neighboring bands is also observed); assignment of observed bands is reported in Table 1.
The observed I5− symmetric stretch vibration frequency perfectly corresponds to literature values for symmetric V-shaped I5− [2], indicating that I5− solvation in DMSO can be considered isotropic, or at least isotropic enough not to cause a significant distortion of the anion converting it in its L shape. Proper determination of an equilibrium constant for the I3− + I2 = I5− was not possible, due to deviation from ideality and non-linear response of the Raman signal in solutions of such high concentration; however, observation of spectral changes only at sub-molar concentration are in agreement with reported very small K values (log K ≈ 1 in water [37]).
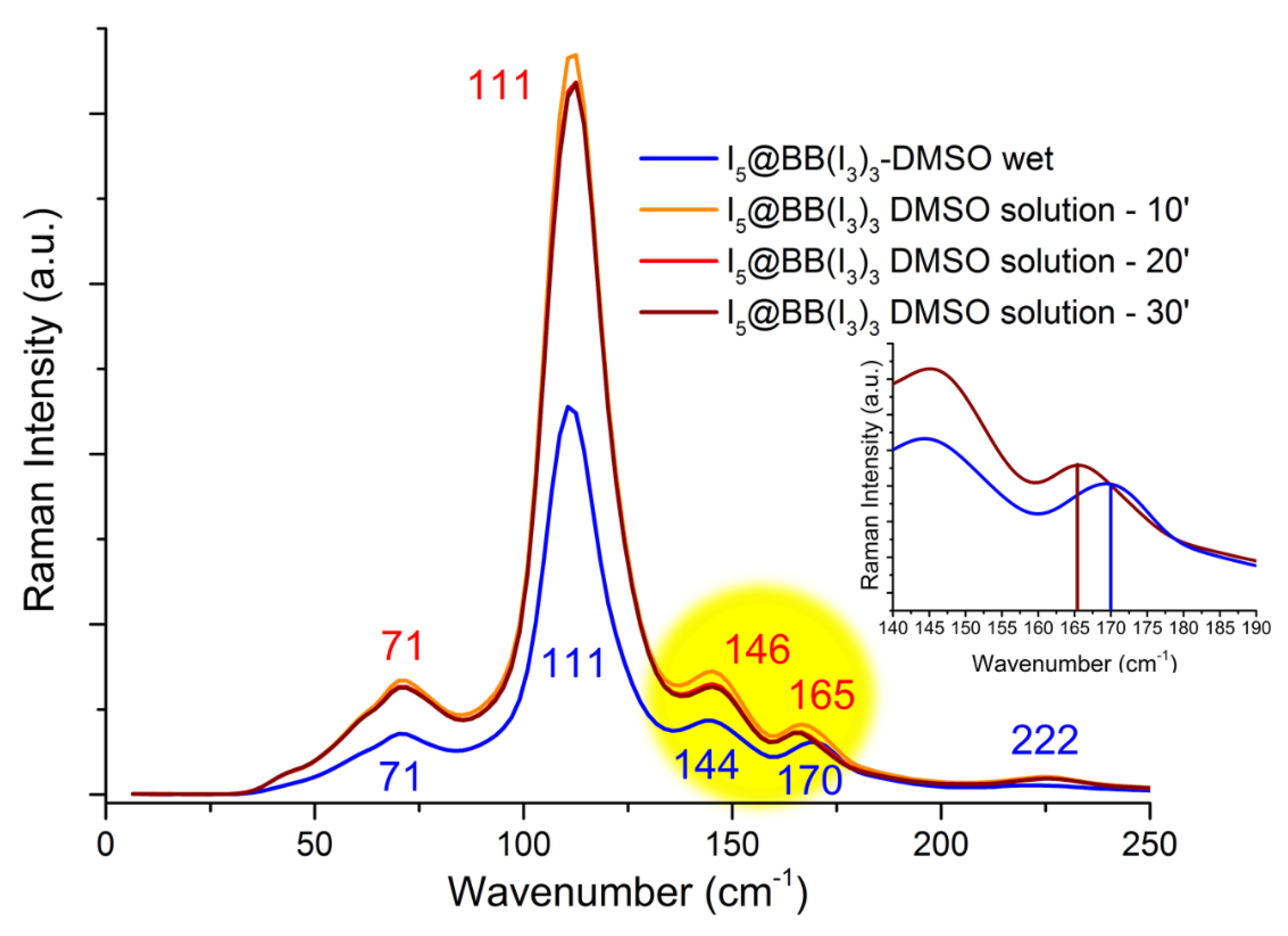
Raman spectra of the DMSO-soaked I5@(BB)(I3)3 were re-recorded in the very same manner as in [26] and showed analogous results. This time, however, spectra were recorded from the initial wetting of the crystals for up to 30 min (after which no further changes were observed) while crystal dissolution in DMSO proceeded; data are reported in Figure 10.
Beyond a difference in intensity caused by the dissolution process, all bands remained at the same frequencies except the one related to I5− stretching. For tiny the shift of I5− vibration frequency may seem, it cannot be overlooked, as it actually covers the whole expected literature range of variability from a perfectly V-shaped (170 cm−1) to a markedly L-shaped (165 cm−1) pentaiodide [2].
Since we demonstrated that pentaiodide is V-shape symmetrical in DMSO in the absence of ligand, as well as in the I5@(BB)(I3)3 crystal (both from Raman spectra, Figure 10 blue line, and crystal data: central I-I 3.076(1)-3.139(1) Å, distal I-I 2.805(2)–2.785(1) Å, angle 94.54(4)°), its observed transition to L-shaped form must be assigned to discrete [(I3−)·(I2)]@BB host-guest complexes, which exist as isolated entities in solution.
It becomes clear that parameters affecting polyiodide formation equilibria (I− + I2 = I3−, I3− + I2 = I5−, etc.), among which the choice of the solvent plays a fundamental role, have a deep influence on the resulting crystal packing for the BB/I−/I2 ternary system.
It can be concluded that a fine game of equilibria, perhaps not fully understood due to the limits imposed by solubility (all I− and I5− structures are obtained from acetonitrile, a solvent eluding direct investigation due to complexes extreme insolubility), subtends to the formation of these systems.
Both I3− and I5−, as demonstrated by crystal structures, can be hosted within BB cavities. Such host-guest interaction exists also in DMSO solution and causes a reduction of symmetry of I5− anions. Due to the stability of the (I5@BB)∞, it is possible for discrete I5−@BB complexes to associate, giving a crystalline solid, where I5− regains its V-shaped symmetry due to additional I5−…I5− contacts. The solvent of choice must implicitly play a major role by affecting the position of I3− + I2 = I5− equilibrium, this being most likely the reason for the impossibility to isolate I5−-based crystals from DMSO, while their formation is commonplace in ACN.
4. Conclusions
The I3@BB(I3)3·2DMSO crystal structure represents an important conceptual link between reported I−- and I5−-BB-based architectures. Accordingly, it allows for a broad rationalization of the most important phenomena governing the formation of these systems.
In the first place, the original idea of having a large charged scaffold imposing limits on the number of required iodide anions, but not on the number of formal additional I2 molecules, indeed resulted in systems which are interesting and intricate at the same time. In particular, charge balance is at the core of some of the major differences observed among these structures.
For the BB/I− family of structures, as manifest from Figure 7, the role of charge-charge interactions is fundamental and leads to four multiple CH…I contacts, exploiting BB corners, which are responsible for holding together BB pillars and balancing out the BB charge.
The BB/I5− family of structures seems to be dominated by the stability of the (I5−@BB)∞ [3]catenate, that passes unmodified from one structure to the other. The main type of forces in play in this type of structure are of the anion-π type, according to the polarizability of both BB and larger polyiodides. The other three required charges are provided by three almost aligned I3− anions, which are just big enough to bridge the BB coupled pillars brought about by the core [3]catenane architecture.
The I3@BB(I3)3·2DMSO is a mix of both trends. It maintains BB…I corner contacts as in I−-based structure, though requiring only two I3− to bridge a pair of adjacent BB molecules, rather than four I−. At the same time, I3−, due to its large surface allowing for multiple contacts, is more easily stabilized than I− within the BB cavity (yet not as much as I5−). One last charge remains to be counterbalanced, giving rise to a line of triiodides passing in between pillars: a feature unique to this structure.
The second focal point regards the role of the solvent and its importance for the position of the I3− + I2 = I5− equilibrium in solution, which in turn is crucial to determine which polyiodide architecture is formed.
As a matter of fact, from ACN solvent it is possible to obtain an I−-based structure in absence of I2 or I5− based structures in the presence of I2, while stopping at phases containing only I3− is not observed. The fact that I3@BB(I3)3·2DMSO is obtained from the same solvent (DMSO) that was used for Raman investigation of I5@(BB)(I3)3 prompted further study. It was found that the symmetric V-shaped I5−, as found in I5@(BB)(I3)3, shifts to an L-shaped I5− in DMSO solution, due to host-guest interactions with BB which reduce its symmetry.
This indicates that when the choice of solvent is such to allow compresence of I5− and BB in solution over a certain threshold, monomeric I5−@BB host-guest complexes present in solution start interacting and form inevitably the very stable (I5−@BB)∞ [3]catenate, leading to I5−-based architectures. Conversely, the more the solvent favors I5− dissociation, the more the process becomes unlikely, allowing for the formation of I3−-based phases. In the case of DMSO, even starting from 1:2 I−:I2 solutions does not lead to I5−-containing structures; this appears indicative of the ability of DMSO to dissociate I5− more effectively in comparison to ACN; in turn this being probably the reason underlying to the solubility of I5−-based crystals in DMSO.
In the end, given on one hand the interest for I2-dense solids for solid-state conductor applications [5,6,7], and on the other one, the recent demonstration of the usage of I− anions as supramolecular ball joints, leading to a new class of porous materials, POFs, whose structure is inherently dependent on crystallization conditions [27], we can anticipate that fine understanding of the “homeostasis” of polyiodides in the BB/I−/I2 system will be a guiding light in the design of related materials intended for applications.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/10/5/387/s1, Table S1: Crystallographic information; Table S2: Selected contacts for the I3@BB(I3)3·2DMSO crystal structure; Table S3: Overview of BB contacts in the examined crystal structures according to Hirshfeld surface analysis; Figure S1: Synopsis of fingerprint plots. CCDC deposition number 1996957 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
Author Contributions
Conceptualization: A.B.; methodology: all authors; validation: C.B. and C.G.; formal analysis: all authors; investigation: C.B., C.G., and M.S.; resources: A.B., C.B., and C.G.; writing—original draft preparation: M.S.; writing—review and editing: all authors; visualization: M.S.; supervision: A.B. and C.B.; project administration: A.B.; funding acquisition: A.B. and C.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
CRIST, Crystallography Centre, University of Florence, is gratefully acknowledged for technical support.
Conflicts of Interest
The authors declare no conflicts of interest.
References and Note
- Gay-Lussac, J.L. Mémoire sur l’iode. Ann. Chimie 1814, 91, 5–160. [Google Scholar]
- Svensson, P.H.; Kloo, L. Synthesis, structure, and bonding in polyiodide and metal iodide-iodine systems. Chem. Rev. 2003, 103, 1649–1684. [Google Scholar] [CrossRef] [PubMed]
- Reiss, G.J.; van Megen, M. Two new polyiodides in the 4,4′-bipyridinium diiodide/iodine system. Zeitschrift Für Naturforschung B 2012, 67, 5–10. [Google Scholar]
- Van Megen, M.; Reiss, G.J. The pseudosymmetric structure of bis(pentane-1,5-diaminium) iodide tris(triiodide). Acta Cryst. E 2012, 68, o1331–o1332. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Z.-S. Lithium-coordinating ionic conductor for solid-state dye-sensitized solar cells. RSC Adv. 2015, 5, 56967–56973. [Google Scholar] [CrossRef]
- Wang, H.; Li, J.; Gong, F.; Zhou, G.; Wang, Z.-S. Ionic Conductor with High Conductivity as Single-Component Electrolyte for Efficient Solid-State Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2013, 135, 12627–12633. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, H.; Xue, B.; Wang, Z.; Meng, Q.; Chen, L. Solid-State Composite Electrolyte LiI/3-Hydroxypropionitrile/SiO2 for Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2005, 127, 6394–6401. [Google Scholar] [CrossRef]
- Svensson, P.H.; Gorlov, M.; Kloo, L. Dimensional caging of polyiodides. Inorg. Chem. 2008, 47, 11464–11466. [Google Scholar] [CrossRef]
- Abate, A.; Brischetto, M.; Cavallo, G.; Lahtinen, M.; Metrangolo, P.; Pilati, T.; Radice, S.; Resnati, G.; Rissanen, K.; Terraneo, G. Dimensional encapsulation of I−···I2···I− in an organic salt crystal matrix. Chem. Commun. 2010, 46, 2724–2726. [Google Scholar] [CrossRef]
- García, M.D.; Martí-Rujas, J.; Metrangolo, P.; Peinador, C.; Pilati, T.; Resnati, G.; Terraneo, G.; Ursini, M. Dimensional caging of polyiodides: Cation-templated synthesis using bipyridinium salts. CrystEngComm 2011, 13, 4411–4416. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Martí-Rujas, J.; Metrangolo, P.; Pilati, T.; Radice, S.; Resnati, G.; Terraneo, G. Solution and Solid State Synthesis of the Discrete Polyiodide I73– under Modular Cation Templation. Cryst. Growth Des. 2012, 12, 5757–5762. [Google Scholar] [CrossRef]
- Peuronen, A.; Rinta, H.; Lahtinen, M. N⋯I Halogen Bonding Supported Stabilization of a Discrete Pseudo-Linear [I12]2− Polyiodide. CrystEngComm 2015, 17, 1736–1740. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Van Megen, M.; Reiss, G.J. I62− Anion Composed of Two Asymmetric Triiodide Moieties: A Competition between Halogen and Hydrogen Bond. Inorganics 2013, 1, 3–13. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; García-Gallarín, C.; López de la Torre, M.D.; Pichierri, F.; Bianchi, A.; Melguizo, M. Anion Complexes with Tetrazine-Based Ligands: Formation of Strong Anion−π Interactions in Solution and in the Solid State. Inorg. Chem. 2016, 55, 8013–8024. [Google Scholar] [CrossRef]
- Savastano, M.; García-Gallarín, C.; Giorgi, C.; Gratteri, P.; López de la Torre, M.D.; Bazzicalupi, C.; Bianchi, A.; Melguizo, M. Solid State and Solution Study on the Formation of Inorganic Anion Complexes with a Series of Tetrazine-Based Ligands. Molecules 2019, 24, 2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savastano, M.; Bazzicalupi, C.; García-Gallarín, C.; López de la Torre, M.D.; Bianchi, A.; Melguizo, M. Supramolecular forces and their interplay in stabilizing complexes of organic anions: Tuning binding selectivity in water. Org. Chem. Front. 2019, 6, 75–86. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; García-Gallarín, C.; Giorgi, C.; López de la Torre, M.D.; Pichierri, F.; Bianchi, A.; Melguizo, M. Halide and hydroxide anion binding in water. Dalton Trans. 2018, 47, 3329–3338. [Google Scholar] [CrossRef]
- Savastano, M.; García-Gallarín, C.; López de la Torre, M.D.; Pichierri, F.; Bazzicalupi, C.; Bianchi, A.; Melguizo, M. Interplay between salt bridge, hydrogen bond and anion-π interactions in thiocyanate binding. Inorg. Chim. Acta 2018, 470, 133–138. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; Mariani, P.; Bianchi, A. Network Formation via Anion Coordination: Crystal Structures Based on the Interplay of Non-Covalent Interactions. Molecules 2018, 23, 572. [Google Scholar] [CrossRef] [Green Version]
- Savastano, M.; Bazzicalupi, C.; García-Gallarín, C.; Gellini, C.; López de la Torre, M.D.; Mariani, P.; Pichierri, F.; Bianchi, A.; Melguizo, M. Iodide and triiodide anion complexes involving anion–π interactions with a tetrazine-based receptor. Dalton Trans. 2017, 46, 4518–4529. [Google Scholar] [CrossRef] [PubMed]
- Savastano, M.; García-Gallarín, C.; López de la Torre, M.D.; Bazzicalupi, C.; Bianchi, A.; Melguizo, M. Anion-π and lone pair-π interactions with s-tetrazine-based ligands. Coord. Chem. Rev. 2019, 397, 112–137. [Google Scholar] [CrossRef]
- Müller, M.; Albrecht, M.; Gossen, V.; Peters, T.; Hoffmann, A.; Raabe, G.; Valkonen, A.; Rissanen, K. Anion–π interactions in salts with polyhalide anions: Trapping of I42−. Chem. Eur. J. 2010, 16, 12446–12453. [Google Scholar] [CrossRef] [PubMed]
- Ilioudis, C.A.; Steed, J.W. Complexation of I42− and I82− by protonated azacyclophanes. CrystEngComm 2004, 6, 239–242. [Google Scholar] [CrossRef]
- Savastano, M.; Martínez-Camarena, A.; Bazzicalupi, C.; Delgado-Pinar, E.; Llinares, J.M.; Mariani, P.; Verdejo, B.; García-España, E.; Bianchi, A. Stabilization of Supramolecular Networks of Polyiodides with Protonated Small Tetra-azacyclophanes. Inorganics 2019, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Odell, B.; Reddington, M.V.; Slawin, A.M.Z.; Spencer, N.; Fraser Stoddart, J.; Williams, D.J. Cyclobis(paraquat-p-phenylene). A Tetracationic Multipurpose Receptor. Angew. Chem. Int. Ed. Engl. 1988, 27, 1547–1550. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; Bianchi, A. Porous Frameworks Based on Supramolecular Ball Joints: Bringing Flexibility to Ordered 3D Lattices. Chem. Eur. J. 2020. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; Gellini, C.; Bianchi, A. Infinite supramolecular pseudo-polyrotaxane with poly[3]catenane axle: Assembling nanosized rings from mono- and diatomic I− and I2 tectons. Chem. Commun. 2020, 56, 551–554. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17. Available online: http://hirshfeldsurface.net (accessed on 8 May 2020).
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermol-ecular interactions in molecular crystals. Acta Cryst. B 2004, 60, 627–668. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Which is correct under commonly employed and relatively diluted concentration ranges, but not an absolute truth.
- Ramette, R.W.; Sandford, R.W., Jr. Thermodynamics of Iodine Solubility and Triiodide Ion Formation in Water and in Deuterium Oxide. J. Am. Chem. Soc. 1965, 87, 5001–5005. [Google Scholar] [CrossRef]
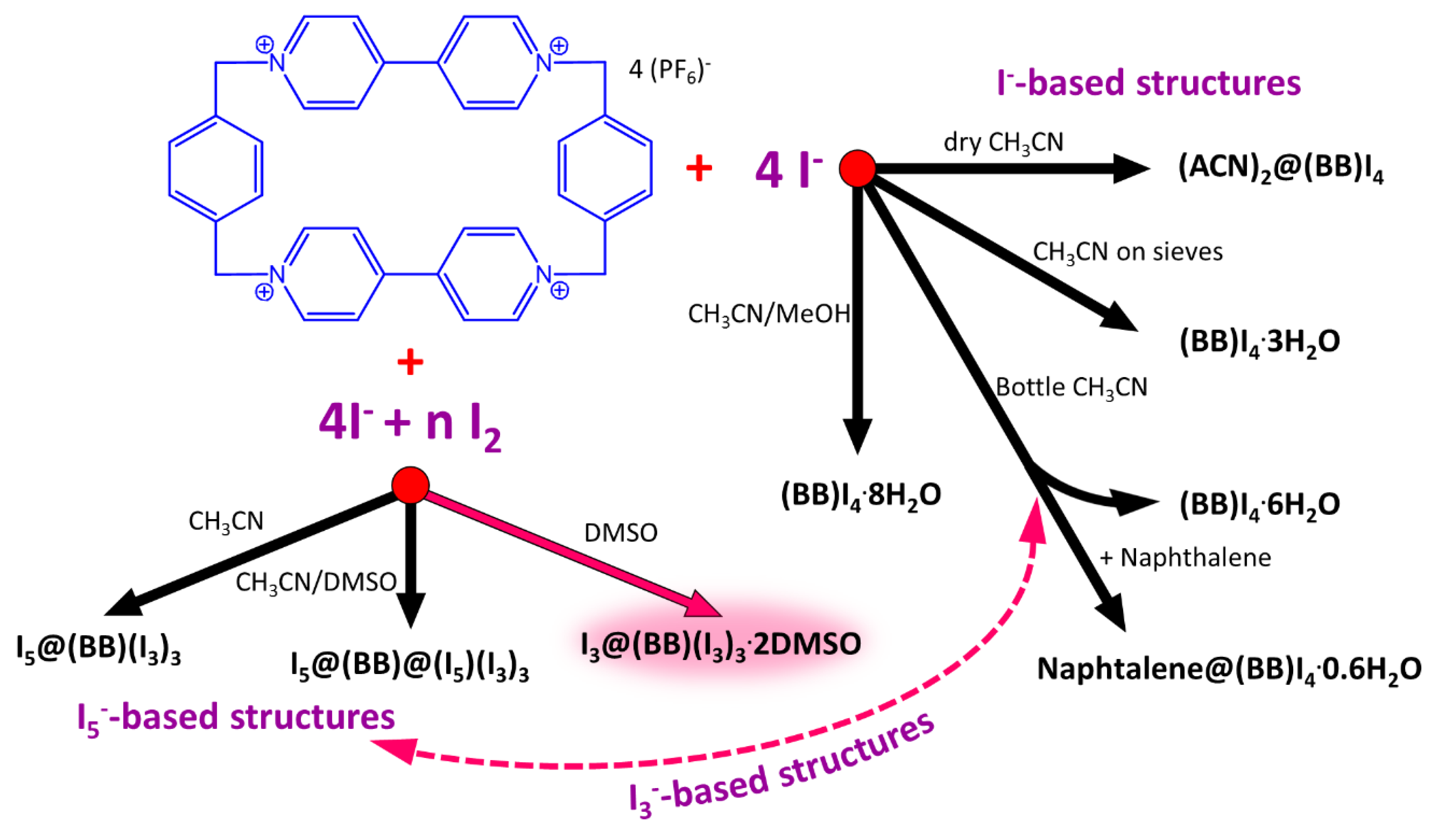
Scheme 1.
Overview of reported crystalline phase for the binary blue box (BB)/I− [27] and ternary BB/I−/I2 [28] systems. The two subgroups of I−- and I5−-based structures were hardly relatable, due to the lack of an intermediate I3−-based architecture. The I3@(BB)(I3)3·2DMSO crystal structure herein reported fills the gap allowing substantial comparison of packing features. Adapted from [27] with permission from John Wiley and Sons.
Scheme 1.
Overview of reported crystalline phase for the binary blue box (BB)/I− [27] and ternary BB/I−/I2 [28] systems. The two subgroups of I−- and I5−-based structures were hardly relatable, due to the lack of an intermediate I3−-based architecture. The I3@(BB)(I3)3·2DMSO crystal structure herein reported fills the gap allowing substantial comparison of packing features. Adapted from [27] with permission from John Wiley and Sons.

Figure 1.
Column of macrocycles and triiodides growing in the a crystallographic direction in the I3@BB(I3)3·2DMSO crystal structure.
Figure 1.
Column of macrocycles and triiodides growing in the a crystallographic direction in the I3@BB(I3)3·2DMSO crystal structure.

Figure 2.
I3@BB(I3)3·2DMSO crystal structure. Top view (a) and lateral view (b) of the BB/I3− pillars growing along the a axis. Color code: purple: triiodides bridging piled BB; green: triiodides inside the BB cavity; red: triiodides outside the BB pillars. DMSO molecules omitted for clarity. I…I, C…I, and H…I contacts as dashed lines.
Figure 2.
I3@BB(I3)3·2DMSO crystal structure. Top view (a) and lateral view (b) of the BB/I3− pillars growing along the a axis. Color code: purple: triiodides bridging piled BB; green: triiodides inside the BB cavity; red: triiodides outside the BB pillars. DMSO molecules omitted for clarity. I…I, C…I, and H…I contacts as dashed lines.

Figure 3.
Side and top view of I3@BB(I3)3·2DMSO structure (in blue and violet (I3−)) superimposed to (ACN)2@(BB)I4 (in green and red (I−)). Superimposition obtained by overlap of the eight C atoms of BB methylenic corners (RMS = 0.383).
Figure 3.
Side and top view of I3@BB(I3)3·2DMSO structure (in blue and violet (I3−)) superimposed to (ACN)2@(BB)I4 (in green and red (I−)). Superimposition obtained by overlap of the eight C atoms of BB methylenic corners (RMS = 0.383).

Figure 4.
Comparison of host-guest interaction for BB…I3 in I3@BB(I3)3·2DMSO (blue and violet) and BB…I− in (BB)I4·8H2O (green and red: molecule A; orange and yellow: molecule B). Superimposition obtained by overlap of the four C atoms of BB methylenic corners (RMS = 0.110 and 0.057 for molecule A and B, respectively).
Figure 4.
Comparison of host-guest interaction for BB…I3 in I3@BB(I3)3·2DMSO (blue and violet) and BB…I− in (BB)I4·8H2O (green and red: molecule A; orange and yellow: molecule B). Superimposition obtained by overlap of the four C atoms of BB methylenic corners (RMS = 0.110 and 0.057 for molecule A and B, respectively).

Figure 5.
Front and top view of coupled BB pillars as found in the I5@(BB)(I3)3 crystal structure.

Figure 6.
Side and front comparisons of host-guest interaction for BB…I3 in I3@BB(I3)3·2DMSO (blue and violet) and BB…I5− in I5@(BB)(I3)3 (green and red). Superimposition is obtained by overlap of the four C atoms of BB methylenic corners (RMS = 0.0617).
Figure 6.
Side and front comparisons of host-guest interaction for BB…I3 in I3@BB(I3)3·2DMSO (blue and violet) and BB…I5− in I5@(BB)(I3)3 (green and red). Superimposition is obtained by overlap of the four C atoms of BB methylenic corners (RMS = 0.0617).

Figure 7.
Overview of BB Hirshfeld surface composition for all discussed crystal structures (same data in tabular form are found in Table S3). Inner ring: internal composition of Hirshfeld surface, i.e., a depiction of BB molecular surface; outer ring: closest outer atom, i.e., atom in contact with BB Hirshfeld surface. Color code: white: H; grey: C; blue: N; red: O; yellow: S; violet: I. Data from (ACN)2@(BB)I4 crystal structure taken from [27]. Partly adapted from [27] with permission from John Wiley and Sons.
Figure 7.
Overview of BB Hirshfeld surface composition for all discussed crystal structures (same data in tabular form are found in Table S3). Inner ring: internal composition of Hirshfeld surface, i.e., a depiction of BB molecular surface; outer ring: closest outer atom, i.e., atom in contact with BB Hirshfeld surface. Color code: white: H; grey: C; blue: N; red: O; yellow: S; violet: I. Data from (ACN)2@(BB)I4 crystal structure taken from [27]. Partly adapted from [27] with permission from John Wiley and Sons.

Figure 8.
Overview of BB fingerprint plots for all discussed crystal structures. A breakdown of assignment of recognizable features is presented in Figure S1. Partly adapted from [27] with permission from John Wiley and Sons.

Figure 9.
Raman spectra of DMSO I−/I2 1:2 solutions at different concentrations showing the concentration-dependent presence of a strong band at 170 cm−1, diagnostic of V-shaped I5−.
Figure 9.
Raman spectra of DMSO I−/I2 1:2 solutions at different concentrations showing the concentration-dependent presence of a strong band at 170 cm−1, diagnostic of V-shaped I5−.

Figure 10.
Variation of Raman spectra as I5@(BB)(I3)3 slowly dissolves in DMSO.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Breakdown and assignment of Raman bands observed in Figure 9.
Table 1.
Breakdown and assignment of Raman bands observed in Figure 9.
| I2 | I3− | I5− | Assignment |
|---|---|---|---|
| 66 m | bending v2 | ||
| 112 vs | symmetric stretch v1 | ||
| 145 m | asymmetric stretch v3 | ||
| 170 s | symmetric stretch V-shaped | ||
| 192 m | Symmetric stretch v1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Savastano, M.; Bazzicalupi, C.; Gellini, C.; Bianchi, A. Genesis of Complex Polyiodide Networks: Insights on the Blue Box/I−/I2 Ternary System. Crystals 2020, 10, 387. https://doi.org/10.3390/cryst10050387
AMA Style
Savastano M, Bazzicalupi C, Gellini C, Bianchi A. Genesis of Complex Polyiodide Networks: Insights on the Blue Box/I−/I2 Ternary System. Crystals. 2020; 10(5):387. https://doi.org/10.3390/cryst10050387
Chicago/Turabian StyleSavastano, Matteo, Carla Bazzicalupi, Cristina Gellini, and Antonio Bianchi. 2020. "Genesis of Complex Polyiodide Networks: Insights on the Blue Box/I−/I2 Ternary System" Crystals 10, no. 5: 387. https://doi.org/10.3390/cryst10050387
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.