
Abstract
Genetic and molecular mechanisms that play a causal role in mental illnesses are challenging to elucidate, particularly as there is a lack of relevant in vitro and in vivo models. However, the advent of induced pluripotent stem cell (iPSC) technology has provided researchers with a novel toolbox. We conducted a systematic review using the PRISMA statement. A PubMed and Web of Science online search was performed (studies published between 2006–2020) using the following search strategy: hiPSC OR iPSC OR iPS OR stem cells AND schizophrenia disorder OR personality disorder OR antisocial personality disorder OR psychopathy OR bipolar disorder OR major depressive disorder OR obsessive compulsive disorder OR anxiety disorder OR substance use disorder OR alcohol use disorder OR nicotine use disorder OR opioid use disorder OR eating disorder OR anorexia nervosa OR attention-deficit/hyperactivity disorder OR gaming disorder. Using the above search criteria, a total of 3515 studies were found. After screening, a final total of 56 studies were deemed eligible for inclusion in our study. Using iPSC technology, psychiatric disease can be studied in the context of a patient’s own unique genetic background. This has allowed great strides to be made into uncovering the etiology of psychiatric disease, as well as providing a unique paradigm for drug testing. However, there is a lack of data for certain psychiatric disorders and several limitations to present iPSC-based studies, leading us to discuss how this field may progress in the next years to increase its utility in the battle to understand psychiatric disease.
Similar content being viewed by others
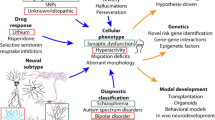

Introduction
Psychiatric disorders are currently one of the most challenging diseases to treat. Somatic disorders such as cancer have shown significant progress in the hunt for effective therapeutics, whereas the failure rate for novel psychopharmacological agents in drug development and clinical trials remains high (Arnerić et al. 2018; Cummings 2018; Kinch 2015). This is partly due to a lack of suitable in vitro and in vivo models for drug testing, as central nervous system tissue is difficult to access (Cummings 2018). Even when precious tissue can be accessed (e.g. post-mortem brain tissue), its use is limited by factors such as terminal cellular differentiation (no propagation possible), end-stage pathology, lack of adjustable experimental conditions, and the confounding effects of long-term medication and patient lifestyle (Silva and Haggarty 2019). Research has therefore relied heavily on animal models, only introducing the human context later at the clinical trial phase, which presents a number of inherent problems. Psychiatric disorders are by nature highly complex with multiple subjective symptoms, making the extrapolation of human phenotypes highly difficult (Nestler and Hyman 2010), and large neuroanatomical differences exist between humans and animals (Seok et al. 2013). This was highlighted in a recent review by Logan et al. (2019), who observed that in multiple studies of genetically engineered mice expressing human microcephaly-related gene mutations, the reduced brain size observed in humans was not able to be recapitulated (Logan et al. 2019).
In 2006, Takahashi and Yamanaka revolutionized the study of psychiatric disease by introducing pluripotent stem cell technology, which helped solve many of the problems associated with the use of animal models (Takahashi and Yamanaka 2006). Using a combination of only four transcription factors they demonstrated that human fibroblast cells could be transduced to form induced pluripotent stem cells (iPSCs), which were capable of forming all 3 germ layers and therefore had the potential to differentiate into any cell type of the body (Takahashi and Yamanaka 2006). Since then, it has been demonstrated that iPSCs can form neural progenitor cells (NPCs) using defined growth factors, resulting in homogenous, expandable and self-renewable cultures (Chambers et al. 2009; Cheng et al. 2017; Koch et al. 2009). Moreover, it was found that these cultures could be further stimulated to differentiate into various CNS cell types, including astrocytes and functionally active neurons (Muratore et al. 2014; Silva et al. 2016).
Several previous reviews have been published regarding iPSC-derived models of neuropsychiatric disorders, with early reviews including only a few published original studies and giving more an outlook on the promises and pitfalls of this new technology (for examples see (Brennand and Gage 2012; Dolmetsch and Geschwind 2011; Tobe et al. 2011; Vaccarino et al. 2011). As more research was published using iPSC-derived neuronal cells from patients with neuropsychiatric disorders, more specific reviews emerged. Initial research and reviews focused on neurodegenerative disorders such as Alzheimer’s disease (Essayan-Perez et al. 2019; Goldstein et al. 2015; Ooi et al. 2013), Parkinson’s disease (Parmar et al. 2020; Schwamborn 2018) and Huntington’s disease (Benraiss and Goldman 2011; Golas and Sander 2016; Wu et al. 2019). The rationale was that stem cell-based therapies might be easier to implement in these more defined diseases than in complex mental illnesses such as schizophrenia or major depression. However, there were several early studies and reviews on autism spectrum disorder (Aigner et al. 2014; Freitas et al. 2014; Russo et al. 2019) and schizophrenia (Brennand and Gage 2011; Brennand et al. 2014; Hoffman et al. 2019; Watmuff et al. 2016), as well as rare research on syndromic, monogenetic and/or chromosomal aberration disorders that can cause psychiatric phenotypes such as 22q11.2 microdeletion syndrome which is strongly associated with autism spectrum disorder, schizophrenia but also ADHD and affective disorders (Drew et al. 2011), Down syndrome (Faundez et al. 2018), Fragile X Syndrome (Faundez et al. 2018) and Timothy’s syndrome (Coskun and Lombardo 2016).
As iPSC technology developed and became more accessible, researchers began to focus on less well-defined psychiatric diseases, such as bipolar disorder (Hoffmann et al. 2018; Miller and Kelsoe 2017; O'Shea and McInnis 2015; Viswanath et al. 2015; Watmuff et al. 2016), alcohol use disorder (Prytkova et al. 2018) and anorexia nervosa (Maussion et al. 2019). There has also been a great interest in the use of 3D brain organoids instead of 2D neuronal cells, and the use of CRISPR/cas9 to genetically edit iPSC-derived models, which have been extensively reviewed elsewhere (Korhonen et al. 2018; Lee et al. 2017; Rehbach et al. 2020; Tian et al. 2020). In this systematic review, we aim to present an update of iPSC-based research into already relatively well-studied polygenic psychiatric disorders, less focusing on the syndromic and monogenetic forms, and review recent novel data on less well-studied disorders. Lastly, we will describe the present limitations of iPSC-based studies, and discuss how researchers can work to overcome them.
Methods
Our systematic review was performed according to the Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) statement (Moher et al. 2009). We conducted an online PubMed (on the 25/3/2020) and Web of Science (19/4/2020) search using the following search strategy: hiPSC [ti] OR iPSC [ti] OR iPS [ti] OR stem cells [ti] AND schizophrenia disorder OR personality disorder OR antisocial personality disorder OR psychopathy OR bipolar disorder OR major depressive disorder OR obsessive compulsive disorder OR anxiety disorder OR substance use disorder OR alcohol use disorder OR nicotine use disorder OR opioid use disorder OR eating disorder OR anorexia nervosa OR attention-deficit/hyperactivity disorder OR gaming disorder. Additional search criterion was studies must have been published between 2006 and 2020. Papers were initially screened by title only, and excluded if deemed not relevant because the study was not on iPSCs, not written in the English language, or no abstract was available. In a second round of screening, full-text papers were assessed for eligibility, and excluded if not relevant to the topic. We excluded reviews and meta-analysis and meeting abstracts as well as stem cell studies derived from animal models, studies on rare monogenetic disorders/syndromes and articles reporting protocols of hiPSC differentiation without including the generation of a disorder- and patient-specific iPSC cell line as well as study protocols of planned studies without actual results. This was firstly done subdivided into the disorder entities by the co-authors (GZ, FR, MN, KPL, SKS) and then again independently for all topics from one co-author (RVMcN) as well as the Web of Science search of another co-author (SKS).
Results
A total of 3515 records were retrieved using our search criteria, and split based on disorder/search term (see Fig. 1). After initial screening of titles and removing of duplicates, 836 papers were excluded. 2267 papers were excluded due to unrelated articles, not written in English, no abstract available. A further 356 papers were excluded due to unrelated articles, repeated publications, no abstract available, reviews, meta-analysis, meeting abstracts, animal models, studies on rare monogenetic disorders/syndromes, protocols of hiPSC differentiation without including the generation of a disorder- and patient-specific iPSC cell line, study protocols of planned studies without actual results. The final number of papers included in our review was 56, which again was split based on disorder/search term. Six disorder/search terms did not yield any papers after eligibility assessment; personality disorder, antisocial personality disorder, anxiety disorder, substance use disorder, eating disorder and gaming disorder. Synthesised results from each of the other disorders/search terms are described below (see Fig. 1 and Table 1).

PRISMA flow diagram
Autism-spectrum disorders
Individuals diagnosed with autism spectrum disorder (ASD) show characteristic deficits in social communication and interaction, and often demonstrate restricted, repetitive behaviors and interests (Ivanov et al. 2015). The disorder is highly heritable, with concordance rates in monozygotic twins of 70–90%. A monogenetic cause has been defined for some forms of ASD, however the exact genetic cause is known in only 20–25% of cases (Ivanov et al. 2015). Studies investigating hiPSC models of rare, monogenetic forms and/or syndromic of ASD have previously been reviewed elsewhere (Brito et al. 2018). In non-syndromic cases of autism, there has been little research performed using iPSC-based models. Transcriptome and gene network analyses of iPSC-derived organoids revealed that gene expression involved in cell proliferation, neuronal differentiation, and synaptic assembly was increased in ASD compared to controls (Mariani et al. 2015). Furthermore, the organoids showed abnormally high expression of inhibitory GABAergic interneurons, as well as an accelerated cell cycle. Consistent with these results, increased proliferation of neural progenitor cells has been found in iPSC-derived 2D cultures from ASD patients, along with reduced synaptogenesis leading to abnormal function of neural networks (Marchetto et al. 2017). Corroborating these results, investigation of neuronal activity in iPSC-derived neurons from ASD patients revealed bursting and spike interval parameters that pointed to a significant reduction in dynamical complexity (Amatya et al. 2019). Furthermore, an impairment in iPSC-derived neuronal differentiation has been observed and linked to micro RNA function; specifically the downregulation of miR-1290, which is involved in neural proliferation and differentiation (Moore et al. 2019). More recently, iPSC-derived neural cell models have helped to further characterize newly identified genetic risk genes for ASD, such as PTCHD-1-AS (Ross et al. 2019). Alterations in this gene were found to cause synaptic dysfunction, identified using electrophysiological experiments. Additionally, in models using iPSC-derived neural cells with disruptions in the TRPC6 gene, it was found that altered neuronal differentiation, morphology, and function was associated with dysregulation of CREB phosphorylation (Griesi-Oliveira et al. 2015).
Bipolar disorder
Bipolar disorder is a primarily episodic disorder which is characterised by alternating manic episodes, and depressed episodes. In between, there are so called euthymic phases with no or only little affective symptoms (Shastry 2005). Bipolar disorder affects 1 to 2% of the world’s population, showing similar incidence rates in both males and females (Hirschfeld et al. 2003). It is a highly familial disorder, with heritability estimates of up to 80% (Craddock and Jones 1999). Recent genome-wide association studies revealed several common risk gene variants to be associated with bipolar disorder (Stahl et al. 2019), and rare genetic variants do not appear to play an important role in the complex genetic architecture of bipolar disorder (Georgieva et al. 2014). The first studies investigating iPSC-derived neuronal cells from bipolar disorder patients were published in 2014. Chen and colleagues reported differential gene expression in iPSC-derived neuronal cells from bipolar patients in comparison to healthy controls (n = 3 biological replicates each). Specifically, expression of genes encoding membrane-bound receptors, ion channels and telencephalic neuronal differentiation was significantly increased in the neurons generated from bipolar patients compared to controls. Additionally, in vitro lithium treatment had a significant effect on calcium signaling and electrophysiological properties in bipolar neurons but not in controls (Chen et al. 2014). Bavamian and colleagues compared miR-34a expression in post-mortem brain tissue from medication-naïve bipolar patients and healthy controls with miR-34a levels from directly induced neuronal cells (iNs) and iPSC-derived neuronal cells, from a bipolar son and his unaffected father. In post-mortem cerebellum tissue, iNs and iPSC-derived neuronal cells, miR-34a expression was increased in the bipolar patients in comparison to healthy controls. The effect of miR-34a on previously identified bipolar risk genes was also investigated, and ANK3, DDN, and CACNB3 were shown to be targeted and potentially silenced by miR-34a. In a later study, overexpression of miR-34a in vitro was reported to decrease CACNB3 and ANK3 gene expression, and alter the neuronal differentiation and morphology of human iPSC-derived neurons from healthy controls (Bavamian et al. 2015). Madison and colleagues derived iPSCs from the fibroblasts of the unaffected father and mother from a bipolar patient and two brothers with bipolar disorder type I. This family was also genotyped for the previously published risk SNPs (single nucleotide polymorphisms) associated with bipolar disorder. All four family members were either homozygous or heterozygous for the bipolar risk genotypes in the SYNE1, ANK3, CACNA1C, ODZ4 (TNM4) and ZNF804A genes, of which ZNF804A is also a schizophrenia risk allele. The iPSCs were differentiated into CXCR4+ NPCs, and abnormalities in the early steps of NPC formation in the bipolar siblings were reported. Additionally, alterations in WNT/GSK3 signaling were observed, suggesting dysregulated ion channel expression in NPCs and neuronal cells (Madison et al. 2015). Mertens and colleagues showed differential responses to in vitro lithium treatment regarding hyperexcitability in iPSC-derived hippocampal dentate gyrus-like neuronal cells from bipolar patients (n = 6), generated from clinical lithium responders vs. bipolar lithium non-responders and healthy controls (n = 4). Evidence for mitochondrial abnormalities in neurons from bipolar patients was also shown (Mertens et al. 2015, 2016). The finding of hyperexcitability in iPSC-derived hippocampal dentate gyrus granule-cell neurons could be replicated in another study using Epstein-Barr immortalized lymphoblast cells as primary cells from bipolar patients and healthy controls (three lithium-responders, three lithium non-responders and four healthy controls). The iPSC-derived neuronal cells were treated with lithium and the electrophysiological data was used to train a Naïve Bayes (NB) classifier that could predict if a novel bipolar patient was lithium-responsive or non-responsive with an accuracy of up to 98% (Stern et al. 2018). Proteomic differences between cells of lithium responders and non-responders have been investigated in iPSC-derived dorsal anterior forebrain cortical neurons generated from bipolar patients, unaffected family members, a family member affected with major depression and a cell line from a Parkinson’s patient. The results suggested that the molecular lithium-response pathway in bipolar patients may function via CRMP2 (collapsin response mediator protein-2), which acts to modify neuronal cytoskeletal dynamics (specifically dendrite and dendritic spine formation) (Tobe et al. 2017). Kim et al. reported the generation of iPSC lines from 4 bipolar disorder affected and 4 unaffected relatives in an Old Order Amish pedigree that were differentiated into NPCs and further differentiated into cortical neurons in vitro. They found several genes differentially expressed between bipolar patients and healthy controls using microarray analysis, but only in the more mature neuronal cells. The most significantly differentially regulated pathways included RNA metabolic processes, protein trafficking and receptor-mediated signaling (Kim et al. 2015). Vizlin-Hodzic and colleagues used primary adipocytes for generating iPSCs and differentiated them into cortical neuronal cells from six bipolar I patients and three healthy controls. Using RNA-sequencing, the expression of the inflammasome NLRP2 gene (NLR family, pyrin domain containing 2) was the most significantly differentially regulated gene between BD I and control lines in both iPSC and cortical neurons. Several cytoskeleton- and inflammatory-associated genes, and GABA and dopamine receptor signaling pathways, were also significantly dysregulated in bipolar neuronal cells compared to the control cells (Vizlin-Hodzic et al. 2017).
There have been several studies investigating specific risk gene variants in iPSC-derived neuronal cells from bipolar patients. Ishii et al. compared glutamatergic and GABAergic neurons derived from two bipolar patients carrying a PCDH15 deletion with seven healthy control cell lines and isogenic controls. In bipolar neurons, decreased MAP2+ dendrite length and synapse number were shown (Ishii et al. 2019). Additionally, the generation of a bona fide iPSC cell line of a Chinese bipolar patient carrying the potential risk variants SLC1A3 rs117588697 A-G/, MAPT rs577501443 G-A, SIRT1 rs12415800 G-A was recently reported (Wang et al. 2018a). In our own lab, we have generated one bone fide iPSC cell lines from a bipolar patient who is a carrier of a risk haplotype in the diacylglycerol kinase eta gene (DGKH; rs994856/rs9525580/rs9525584 GAT) and from another bipolar patient who is a carrier of the wildtype haplotype (Palladino et al. 2018). This DGKH risk haplotype was previously shown to be associated with bipolar disorder, but also with major depression and adult ADHD, and was observed to have functional effects on brain volume and peripheral gene expression (Kittel-Schneider et al. 2014, 2016; Weber et al. 2011).
Schizophrenia
Schizophrenia (SCZ) is a highly heritable, severe, and complex psychiatric disorder, that affects ~ 1% of the general population (Owen et al. 2016). Despite high heritability estimates (Hilker et al. 2018; Sullivan et al. 2003) and a century of biological investigation (Ahmad et al. 2018), the genetics underling SCZ remain unclear and no conclusive disease mechanisms have been identified. It is thought that the disorder is highly polygenic (Giegling et al. 2017; Gratten et al. 2014), and GWAS have detected multiple common genetic variants of small effect size that explain up to 50% of variability in genetic susceptibility (Purcell et al. 2014). The first study using an iPSC-based model to investigate SCZ was conducted in 2011 by Brennand et al. who reported decreased connectivity, neurite outgrowth and expression of post-synaptic density protein 95 (PSD95) in SCZ iPSC-derived NPCs (SCZ iPSC-NPCs) (Brennand et al. 2011). Significant alterations were also observed in gene expression relating to glutamate and Wnt signaling. Interestingly, treatment with the antipsychotic loxapine appeared to ameliorate these effects, enhancing neuronal connectivity and rescuing gene expression. However, it should be noted that other structurally related antipsychotic drugs failed to rescue these deficits. Despite this, changes in Wnt signaling in SCZ iPSC-derived NPCs was later replicated in other studies (Topol et al. 2015), as was inhibited NPC growth and migration (Brennand et al. 2015; Lee et al. 2015). Collectively, the studies to date suggest that early neurodevelopmental pathways may be perturbed in SCZ. Mitochondrial (Mt) dysfunction has been consistently implicated in SCZ. Increased extra-Mt oxygen consumption (Paulsen et al. 2012) and reactive oxygen species (ROS) levels in SCZ iPSC-derived NPCs have been observed (Brennand et al. 2015; Paulsen et al. 2012; Robicsek et al. 2013), which could be reversed by treatment with valproic acid. Altered Mt protein expression has additionally been found directly in SCZ iPSCs, which correlated with decreased membrane potential and an uneven cellular distribution of Mt, which is characteristic of impaired function (Robicsek et al. 2013). Perturbed Mt morphology was further observed by other groups, with SCZ iPSC-NPCs containing Mt that were decreased in size, less connected and not as densely placed around the nucleus (Brennand et al. 2015). As proof-of-principle, Robicsek et al. (2018) recently transferred healthy Mt to SCZ iPSCs using isolated active normal mitochondria (IAN-MIT) methodology, which improved membrane potential and Mt distribution (Robicsek et al. 2018). These studies imply that perturbed Mt respiration and morphology combined with increased oxidative stress may play a key role in SCZ, and moreover, that Mt transfer may be a feasible treatment option for reversing these deficits.
Altered micro RNA (miRNA) expression and/or function has been demonstrated in case/control in SCZ-iPSC studies, affecting both individual genes and complex networks. A GWAS-predicted functional SCZ risk SNP in the MIR137 gene was demonstrated to reduce miR-137 expression during iPSC-derived glutamatergic forebrain neuron differentiation, using isogenic cell lines generated using CRISR-Cas9 technology (Forrest et al. 2017). Decreased miR-9 activity has also been observed in SCZ iPSC-NPCs, concomitant with inhibited radial migration of neurospheres, which could be rescued by subsequent overexpression of miR-9 (Topol et al. 2016). miR-219 is brain-specific and has additionally been reported dysregulated in SCZ, resulting in abnormal neural stem cell proliferation and depletion of the neural stem cell pool (Murai et al. 2016). These effects could be rescued by treatment with a miR-219 inhibitor, suggesting that the miR-219 pathway plays a key role in neural stem cell proliferation and could be a potential therapeutic target. Other miRNAs found to be differentially expressed in SCZ iPSC-based models include miR-34, miR-4449, miR-146b-3p, and miR-23a-5p (Zhao et al. 2015). 22q11.2 deletion is a well-recognized risk factor for the development of SCZ (Marshall et al. 2017), and has been associated with both altered miRNA (Zhao et al. 2015) and gene expression (Lin et al. 2016) in SCZ iPSC-derived neurons. Moreover, 22q11.2 deletion SCZ-iPSCs were observed to have impaired differentiation capacity, smaller neurosphere size and inhibited neurite outgrowth/migration (Pedrosa et al. 2011; Toyoshima et al. 2016). However, a lack of genetically engineered cell models that can rescue these effects means that a candidate causal gene in this deleted chromosome region has yet to be identified. In contrast, a putative causal gene has been identified in 15q11.2 deletion, another risk factor for SCZ (Stefansson et al. 2008). SCZ iPSC-NPCs were found haploinsufficient for the cytoplasmic FMR1 interacting protein 1 (CYFIP1) gene, which is located in this deleted region (Yoon et al. 2014). The cells also demonstrated deficiency in apical polarity and adherens junctions, implying dysregulated neuronal maturation. Subsequent studies supported these findings, with decreased CYFIP1 and altered morphology additionally observed in 15q11.2 deletion iPSC-derived neurons (Das et al. 2015). The rare DISC1 mutation is another well-replicated risk gene for SCZ (Millar et al. 2000) that has been investigated using iPSC-based cell models. iPSC-derived neurons containing mutated DICS1 were found to have impaired synaptic vesicle release, which could be induced in control cells and reversed in patient cell lines using TALENs to specifically edit the DISC1 gene, strongly implicating a role of this gene in synaptic dysfunction (Wen et al. 2014). TALEN and CRISPR-Cas9 editing was further used to mutate DISC1 directly in iPSCs, resulting in abnormal neural differentiation and dysregulated Wnt signaling, consistent with previous findings in SCZ (Srikanth et al. 2015). Moreover, in the previously described study by Murai et al. (2016) which showed increased miR-219 expression and altered proliferation, DISC1 mutations were also found to be present in SCZ iPSC-derived neural stem cells (Murai et al. 2016). Other studies have since replicated this result (Ye et al. 2017), suggesting a role for DISC1 in the regulation of proliferation and providing a potential pathomechanism for SCZ development. Reelin (RELN) is another gene involved in brain developmental processes that has been associated with SCZ, with exonic deletions specifically reported (Costain et al. 2013; Ishii et al. 2016). A recent study also employed the use of genomic editing, comparing glutamatergic and GABAergic iPSC-derived neurons from a SCZ patient with RELN deletion to those from isogenic controls genetically engineered to possess a RELN deletion (Ishii et al. 2019). The resulting phenotypes were similar, with dendrite-shortening and decreased synapse numbers observed, further implicating this gene in SCZ development.
There are several environmental risk factors for SCZ, such as prenatal starvation (St Clair et al. 2005; Susser et al. 1996; Susser and Lin 1992) and infection-induced maternal immune activation (MIA) (Brown and Derkits 2010; Estes and McAllister 2016), that are starting to be investigated using iPSC-based models. SCZ iPSC-NPCs were observed to differ in their sensitivity to subthreshold environmental stressors such as alcohol and mercury, as inferred by differential heat shock factor 1 (HSF1) activation (Hashimoto-Torii et al. 2014). However, the mechanisms underlying this different susceptibility were not further investigated. Increased copy number of LINE-1 retrotransposons has been observed as a consequence of MIA in animal models, and was also observed in SCZ iPSC-derived neurons from 22q.11 deletions patients, suggesting prior infection (Bundo et al. 2014). SCZ iPSC-derived neurons have been directly infected with the herpes simplex virus type 1 (HSV-1), and alterations in expression of genes involved in glutamatergic signaling and Mt function were consequently observed (D'Aiuto et al. 2015). Despite these initial studies, comprehensive iPSC-based studies investigating the effect of MIA on SCZ-relevant molecular mechanisms are still lacking (Balan et al. 2019). Moreover, the iPSC-based models used so far are simplistic and not reflective of the in vivo scenario, where viral response requires the concerted effort of neuronal, immune and glial cells (D'Aiuto et al. 2015). To overcome this, future studies should aim to use co-culture systems, which utilize a number of cell types and permit cell–cell interaction (Balan et al. 2019).
There are several limitations to using iPSC-based models for SCZ. Even after prolonged cell culture, iPSC-derived neurons lack the myelination required for maturation and still resemble fetal stage neurons, therefore caution is needed when extrapolating results from iPSC-based models to adolescent/adult SCZ patients (Ahmad et al. 2018; Balan et al. 2019). Additionally, SCZ generally has its onset in adolescence (Häfner et al. 1994), further questioning the validity of using immature neurons to investigate its etiology. A further problem is that of sample heterogeneity, with iPSCs generated from different subtypes of SCZ shown to demonstrate differential results, for example in undifferentiated (Brennand et al. 2011) versus paranoid SCZ (Robicsek et al. 2013). This has also been observed in SCZ iPSCs derived from treatment-responsive versus treatment-resistant patients (Grunwald et al. 2019; Nakazawa et al. 2017; Paulsen et al. 2012). Moreover, patient genetic background has been found to have opposing effects on miRNA expression (Ahmad et al. 2018). In the future, sample heterogeneity may be counteracted by selecting ‘general’ SCZ samples based on phenotype and/or polygenic risk score, and utilizing familial samples to reduce the confounding effect of differential genetic background (Balan et al. 2019).
Attention-deficit/hyperactivity disorder
Attention-deficit/hyperactivity disorder (ADHD) is a neurodevelopmental disorder characterised by inappropriate and persistently high levels of inattention, locomotor hyperactivity, and impulsivity (American Psychiatric Association 2013). ADHD is one of the most common neurodevelopmental disorders of childhood, and shows a high persistence of symptoms into adulthood (approximately two-thirds) (Faraone et al. 2006). Impairment by ADHD symptoms entails a considerable socioeconomic burden for affected individuals (Gjervan et al. 2012). It is well established that heritability rates for ADHD rank amongst the highest of all mental disorders at ~ 75% (Faraone and Larsson 2019), similar to schizophrenia (Sullivan et al. 2003), bipolar disorder (McGuffin et al. 2003), and autism spectrum disorders (Sandin et al. 2017). As with most common psychiatric disorders, ADHD demonstrates a polygenic inheritance pattern with a multiplicity of genes involved, most of them contributing only a small proportion to the overall genetic variance of the disorder (Faraone and Larsson 2019). Early genetic studies focused on the investigation of monoaminergic candidate genes (Cook et al. 1995; Kustanovich et al. 2004; Swanson et al. 1998). Several disease-associated candidate genes have also been identified, such as ADGRL3 (= LPHN3) (Arcos-Burgos et al. 2010), GRM5 (Elia et al. 2012), and NOS1 (Weber et al. 2015), which play fundamental roles in excitatory glutamatergic neurotransmission (Lesch et al. 2012). Furthermore, 12 significant loci revealed in a recent ADHD GWAS were found to contain several promising genes, such as ST3GAL3, SORCS3, and FOXP2, all of which show strong expression in the central nervous system (CNS) (Demontis 2019). These genes are additionally involved in neurodevelopmental processes, further supporting a possible role in ADHD etiology (Hu et al. 2011) (Breiderhoff et al. 2013) (Yoo et al. 2015) (Chen et al. 2016). Several groups have reported the generation of iPSCs derived from ADHD patients (Jansch et al. 2018; Re et al. 2018; Sochacki et al. 2016b; Tong et al. 2019). However, functional analyses of these new cell lines were not conducted, and therefore form the next logical step of investigation. These studies should integrate hypothesis-driven and hypothesis-free gene expression experiments, under both basal and drug-treatment conditions, to assess possible modes of drug action. A number of studies have already addressed the effects of psychostimulants in cell culture, however these studies relied on animal cell culture models, non-neuronal human cell models and/or transfection systems for overexpression of the dopamine (DAT) or norepinephrine transporter (Grünblatt et al. 2013; Schwarz et al. 2014). For example, in a murine stem cell model, it was shown that methylphenidate treatment in vitro could enhance neuronal differentiation (Bartl et al. 2013). Furthermore, differences in axonal outgrowth and synaptic connectivity between patient and healthy control iPSC-derived neurons could be assessed with microfluidic chips in future studies. In summary, ADHD iPSC-based models offer the unique potential to close the gap between basic genetic and neural network associations, by identifying potential disease-modifying cellular pathways and adding valuable information to the endophenotypic finger print of ADHD (Castellanos and Tannock 2002). However, research has been inhibited by the lack of functional characterisation of these models, which should now be the priority for researchers working in this field.
Eating disorders
To date, there has only been one study conducted using iPSCs to investigate the etiology of an eating disorder (ED). The authors focused on anorexia nervosa (AN), which is a multifactorial neurodevelopmental disorder that affects ~ 1% of the population (Smink et al. 2012). The disorder presents with distorted body image and severe caloric restriction, usually as a result of a highly anxiogenic response to food, which ultimately leads to emaciation and/or death (Sharan and Sundar 2015). AN has the highest mortality rate of all psychiatric diseases (Arcelus et al. 2011), and there are currently no highly efficient treatments (Bulik et al. 2007; Watson and Bulik 2013). This is largely due to a lack of understanding of disease etiology, and research lags behind other psychiatric diseases. This is likely due to the traditional view that EDs are non-biologically based problems that are caused by vanity and poor parenting, and only occur in specific groups of people (O'Hara and Smith 2007). However, studies have suggested that 50–75% AN risk is due to genetics (Bulik et al. 2005), although no genome-wide association study has so far been powered enough to identify specific genes significantly associated with the disease (Brandys et al. 2015; Wang et al. 2011). Negraes et al. (2017) generated iPSCs from 4 AN patients and 4 healthy controls, expanding two clones from each sample (Negraes et al. 2017). To reduce sample heterogeneity, all AN patients included had similar severity of symptoms and medical/behavioral consequences from AN symptoms. However, some patients also used compensatory behavior such as purging. AN binge-eating/purging has been recognized as a clinically relevant subtype, demonstrating higher levels of core ED psychopathology and more severe cognitive impairments, and therefore may cause significant sample heterogeneity (Reas and Rø 2018; Tamiya et al. 2018). The authors were able to successfully generate iPSC-derived cortical neurons, with no differences in differentiation capability observed. This was supported by RNA sequencing data (RNASeq), which showed no changes in genes related to neural development/differentiation, suggesting that developmental anomalies may not be present in AN patient brains. RNASeq also revealed that in contrast to previous genetic studies (Gervasini et al. 2013; Kaye et al. 1988, 2005; Levine et al. 2000; Phillipou et al. 2014; Rask-Andersen et al. 2010), there were no changes in serotonin or dopamine-related genes in AN patient neurons. However, 361 differentially expressed genes were observed. 13 candidate genes that showed highly different expression were selected and confirmed by RT-qPCR (Negraes et al. 2017). Tachykinin receptor 1 (TACR1) was the mostly significantly upregulated gene in AN patient neurons, and increased protein expression was also observed. This gene has already been associated with psychiatric disorders such as anxiety (Muñoz and Coveñas 2014) and addiction (Schank 2014). However, in animal models this gene increases the risk of high BMI, which is at odds with AN presentation (Pillidge et al. 2016). Therefore it remains unclear as to what, if any role, tachykinin signaling may play in AN etiology. The diacylglycerol kinase gamma (DGKG) gene was additionally found upregulated in AN neurons, which has previously been associated with obesity (Cheung et al. 2010) and chronic stress (Lisowski et al. 2013), suggesting it may potentially play a role in the development of AN. Several other genes also found differentially expressed were connective tissue growth factor (CTCF) and tudor domain containing 10 (TDRD10), which are involved in normal ovarian follicle development/ovulation (Nagashima et al. 2011) and gametogenesis (Hosokawa et al. 2007) respectively. However, as amenorrhea and infertility commonly result from AN (Bulik et al. 2005; Katz and Vollenhoven 2000), these changes in gene expression are more likely to be consequential than causative.
Major depressive disorder
Major depressive disorder (MDD) is the most common psychiatric disorder with a life time prevalence of 10% worldwide (Wittchen et al. 2001). Core symptoms are depressed mood, decreased energy and lack of interest. Suicide is a major complication of MDD, and women suffer about twice as often than men (Wittchen et al. 2011). MDD is a highly heterogeneous disorder, and genetic factors are thought to contribute ~ 50% to disease etiology (Ormel et al. 2019). Recent genome-wide association studies reported only a few replicated common risk gene variants associated with major depression (Ormel et al. 2019). With regards to unipolar or MDD, the focus of iPSC-based studies has been on cells generated from patients with therapy-resistant depression, with our literature search revealing two currently published studies by the same research group. Vadodaria and colleagues generated iPSC-derived forebrain neurons from three excellent selective-serotonin-reuptake-inhibitor (SSRI) responder patients, three non-responder patients and three healthy controls without a history of depression. Calcium signaling was measured as a marker for neuronal activity using the calcium indicator dye Fluo-4. There was no significant difference between the three groups at baseline, however after 5-Hydroxytryptamine (= serotonin, 5-HT) in vitro treatment non-responder forebrain neurons displayed significantly higher activity compared to responder and healthy controls. After further analysis, it was found that SSRI-non-responder neurons displayed 5-HT-induced hyperactivity via upregulated 5-HT2A and 5-HT7 receptors (Vadodaria et al. 2019a). In contrast, serotonergic neurons from three SSRI-responders and three non-responders did not demonstrate significant differences in 5-HT release/reuptake, or in genes related to 5-HT signaling. However, non-responder serotonergic neurons did appear to have altered neurite growth and morphology compared to healthy controls and SSRI-responders. Transcriptome analysis of these neurons showed dysregulated protocadherin alpha gene expression (PCDHA6 and PCDHA8) in non-responders compared to responders and healthy controls. Additionally, knockdown of PCDHA6 and PCDHA8 in HEK cells and serotonergic neurons was found to alter neurite length, suggesting that differences in non-responder serotonergic neuron length may be mediated by decreased PCDHA6 and PCDHA8 gene expression levels (Vadodaria et al. 2019b).
Obsessive–compulsive disorder
The concept of obsessive–compulsive disorder (OCD) has changed during recent years, most notably with the switch of diagnostic categorization from an anxiety disorder in DSM-IV (American Psychiatric Association 2000) to its own diagnostic section of “obsessive–compulsive and related disorders” in DSM-5, which also comprises skin picking disorder, hair pulling disorder, hoarding, and body dysmorphic disorder (American Psychiatric Association 2013). Lifetime prevalence in the United States is estimated to be 2.3% (Ruscio et al. 2010). (Epi)genetic data, functional imaging studies, and neuropsychological data point to a complex interplay between environmental and biological factors in OCD etiology (Pauls et al. 2014), with an estimated heritability of ~ 40% in adult- and up to 65% in childhood-onset OCD (Abramowitz et al. 2009; Taylor 2011). Childhood-onset OCD exhibits a higher genetic load with increased familial risk in comparison to adult-onset OCD (Nestadt et al. 2000; Pauls et al. 1995). A neural network between cortical regions and the basal ganglia, called the cortico-striato-thalamo-cortical (CSTC) circuit, has also been repeatedly implicated in OCD (Graybiel and Rauch 2000; Posner et al. 2014). A meta-analysis of the two previous OCD GWAS showed no genome-wide significant findings, but amongst the top 3 haploblocks was a region comprising the GRID2 gene (IOCDF-GC and OCGAS 2018). Further evidence for an involvement of glutamatergic neurotransmission in OCD comes from magnetic resonance spectroscopy studies, which suggested increased glutamatergic activity in the striatum (Naaijen et al. 2015). The pharmacological effectiveness of SSRIs implies that serotonergic neurotransmission also plays a role in OCD etiology, and a meta-analytic review of the literature revealed disease-associated variants in both the serotonin transporter (5-HTTLPR) and the serotonin receptor gene HTR2A (Taylor 2013). To date, no functional studies using iPSC-derived cell lines from patients with OCD have been published. One group reported the generation of an iPSC line from a 32-year-old male OCD patient from blood cells, and showed that three SNP variants in OCD candidate genes remained unchanged during reprogramming (Wang et al. 2018b). Another group generated an iPSC line from urothelial cells derived from a 29-year old male patient with early-onset OCD and comorbid skin picking and body dysmorphic disorder (Sochacki et al. 2016a). Urothelial cells are a promising source for the generation of iPSCs, as these cells exhibit comparably high reprogramming efficiencies (Drozd et al. 2015; Zhou et al. 2011) and as a non-invasive method might be particularly suitable for psychiatric patients with contamination or needle fears. The development of cerebral organoids needs further progress before complex circuitry of interest, such as the CSTC loop, can be effectively mimicked in vitro. Until then, the focus should be on straightforward strategies for the investigation of alterations in potentially relevant neurotransmission systems to gain new insights into OCD-related cellular alterations. To this end, protocols for the differentiation of both serotonergic (Lu et al. 2016) and glutamatergic neurons (Cao et al. 2017; Gunhanlar et al. 2018) from iPSCs can be used. Additionally, as the overall heritability of OCD is only moderate selection of study subjects will be critical to obtain meaningful insights into cellular mechanisms.
Personality disorders
Personality disorders (PDs) are classified as ‘a pervasive pattern of thought, feeling and behavior that characterize an individual’s unique lifestyle and mode of adaptation, which deviates markedly from the expectations of the individual’s culture’ (American Psychiatric Association 2000). Examples include borderline, paranoid, narcissistic and obsessive–compulsive PDs (Angstman and Rasmussen 2011). Onset is usually during adolescence or early adulthood, symptoms remain stable over time, and lead to severe impairment and distress (Angstman and Rasmussen 2011). Although the etiology of PDs is still relatively unknown, studies have revealed a moderate to strong genetic contribution, with heritability estimated between 30 and 80% (Fontaine and Viding 2008). Genetic studies have so far been sparse, although candidate genes coding for serotonergic and dopaminergic neurotransmitters have been proposed (Ma et al. 2016; Reichborn-Kjennerud 2010). There is currently only one published study using iPSC-based models to investigate PD pathomechanisms. Tiihonen et al. (2019) focused on antisocial personality disorder (ASPD), of which no underlying molecular pathways are known. It is characterised by aggression, hostility, callousness, manipulativeness, deceitfulness, impulsivity, and its most severe symptom; psychopathy (Tiihonen et al. 2019). ASPD has a prevalence rate of 1–3% in the general population, which increases to 40–70% in prison populations. This study generated iPSC lines and cortical neurons from six ASPD violent offenders, three nonviolent substance abusers (to control for the potential confounding variable of substance abuse), and six control subjects. Several genes were identified to have significantly altered expression in ASPD violent offenders, including the ribosomal (RPL10P9) pseudogene, zinc finger protein 132 (ZNF132), cadherin 5 (CDH5) and opioid receptor delta 1 (OPRD1) genes. Moreover, expression of these genes significantly correlated with psychopathy score. Interestingly, all 4 genes have previously been associated with autism (Chiocchetti et al. 2011; Klauck et al. 2006; O'Roak et al. 2012; Redies et al. 2012; Wang et al. 2017), which the authors propose might contribute to the emotional callousness and lack of empathy observed in psychopathic violent offenders. Proteomic analysis showed that the largest effect sizes were observed for opioid-binding protein/cell-adhesion molecule (OPCML), with the highest expression found in ASPD violent offenders, supporting the OPRD1 transcription data. These results support previous research which suggested that a deficient endogenous opioid system contributes to ASPD (Bandelow and Wedekind 2015).
Substance use disorders
Substance use disorders (SUDs) rank among the most common psychiatric syndromes and are often characterised by psychosocial dysfunction, unemployment, social decline and somatic or psychiatric comorbidities resulting in a significantly increased mortality (Rehm and Shield 2019). A variety of studies have been performed on the genetic component to SUDs, mainly alcohol use disorder (AUD), and found the heritability of AUD to be ~ 49% (Verhulst et al. 2015). In linkage and genome-wide association studies (GWAS), genes involved in alcohol metabolism such as alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH) have been identified as major risk variants in the development of AUD (Frank et al. 2012; Takeuchi et al. 2011; Tawa et al. 2016). The limited number of studies using iPSCs to analyze the biological effects of alcohol on neural cells includes an investigation by Lieberman et al., who found that chronic alcohol exposure lead to a compensatory upregulation of NMDA receptors in iPSC-derived neural cells from genetically heterogeneous AUD patients, but not in control cell lines (Lieberman et al. 2012). This effect suggests that heritable traits might contribute to the development of tolerance to the inhibitory effects of alcohol on NMDA receptor activity in AUD patients (Lieberman et al. 2012). In a subsequent study, the AUD-associated risk allele rs279858*C of GABRA2, which encodes for the alpha subunit of the GABAA receptor, was identified to lead to significantly lower receptor expression levels in iPSC-derived neural cell cultures (Lieberman et al. 2015). As the authors could not replicate this result for protein levels in postmortem cortices, they hypothesized that the increased risk for AUD in individuals carrying the polymorphism might result from developmental processes (Lieberman et al. 2015). Additionally, after 21 days alcohol exposure, gene expression of GABRD (another GABAA subunit) was upregulated in iPSC-derived neural cells from genetically heterogeneous AUD patients compared to healthy controls (Lieberman et al. 2018). In a different investigation, De Filippis et al. hypothesized that neuronal inflammation might be involved in the pathophysiology of AUD. They demonstrated how alcohol exposure altered mitochondrial and lysosomal functions, and increased sensitivity to oxidative stress, which ultimately lead to activation of the inflammasome pathway (De Filippis et al. 2016). However, this was solely shown in iPSCs derived from healthy individuals, which limits conclusions on the impact of neuronal inflammation in genetically predisposed AUD patients.
Besides AUD, there have been first promising reports on establishing iPSC research for the investigation of nicotine and opioid use disorders. Oni et al. discovered that the risk allele rs16969968 of CHRNA5 (encoding for the nicotinic receptor alpha 5), which is associated with nicotine addiction, lead to increased response to nicotine and rapid receptor desensitization (Oni et al. 2016). Similar findings have been obtained in iPSC-derived dopaminergic neurons from patients suffering from opioid use disorder, where a variable number tandem repeat (VNTR) polymorphism in the human dopamine transporter hDAT resulted in increased protein expression (Sheng et al. 2016).
These few studies must be considered preliminary groundwork for the various possibilities that the use of iPSCs offers in trying to elucidate the neurobiological background of SUDs. Their results however have to be evaluated carefully, as some included iPSCs obtained from mixed populations with an unknown, genetically heterogeneous background. Others recruited only a very limited number of individuals as cell donors, such as n = 3 (De Filippis et al. 2016; Oni et al. 2016).
Discussion and conclusions
There can be no doubt that the advent of iPSC technology has facilitated significant progress in our understanding of neuropsychiatric disease. However, there remains a number of limitations which must be considered, as well as points of discussion on the best procedures for iPSC use.
The process of reprogramming has been shown to erase the epigenetic memory of cells, which presents a problem for both psychiatric disorders known to have an age of onset later than childhood (e.g. SCZ) (Häfner et al. 1994) and those which are thought to be significantly influenced by environmental risk factors, which facilitate their effects via epigenetic marks (Soliman et al. 2017). Transgenerational epigenetic inheritance due to external factors such as malnutrition and stress is gaining traction as an important risk factor in the development of psychiatric disease, and therefore constitutes an important area of study (Yehuda et al. 2016). One possible solution to overcome the erasure of epigenetic marks is via transdifferentiation, which is the process of directly inducing neuronal cells from fibroblasts, without the intermediate generation of iPSCs (Pfisterer et al. 2011). Bypassing reprogramming in this manner has been shown to reduce disruption to the epigenomic landscape, thereby allowing the production of neuronal cells at a ‘pathogenic age’ (Mertens et al. 2018). Alternatively, if a disease-associated epigenetic mark is already known, there is now the possibility to modify that mark directly in iPSCs using CRISPR-Cas9 technology (Xie et al. 2018).
iPSCs and many iPSC-derived cells are grown as a homogenous 2D monolayer, which although provides numerous advantages (such as ease of downstream analysis, simple protocols, highly producible) (Duval et al. 2017; Ho et al. 2018), also has several disadvantages. Neuronal cell-only cultures are inhibited by the absence of other cell types, limiting their ability to recapitulate in vivo aspects such as network functionality, neuronal development and synaptic pruning (Silva and Haggarty 2019). Other physiological brain characteristics, including spatial organisation and extracellular matrix interactions, are also absent. iPSC-derived neuronal cultures therefore cannot be used to study overall brain function, particularly not at the level of neural circuits (Nakazawa et al. 2019). Culturing in a 2D monolayer additionally results in an altered, flat cell morphology, and cell–cell interactions are limited to adjacent cells only (Logan et al. 2019). Recent studies have also reported that the overall quality of the 2D culture (e.g. cell health and differentiation capacity) can be easily effected by various factors such as individual handling of cells, disease status of the donor, differentiation protocols and cell seeding density (Duval et al. 2017). This has resulted in many researchers questioning the validity of studying a single 2D monolayer cell type in isolation, and some believe that the molecular mechanisms underlying drugs responses and physiological pathways simply cannot be accurately modeled in this manner (Ishii et al. 2019). Taken together, the limitations of 2D culture result in low predictive power, warranting caution when extrapolating in vitro results to an in vivo scenario. Co-culture, 3D- and organoid models represent an opportunity to address some of the limitations associated with 2D culture; however, these systems are also not without their disadvantages. For an in-depth review of these newer models see Logan et al. (2019) (Logan et al. 2019). An additional issue is the fact that as well differentiated 2D neuronal cells as brain organoids cannot be differentiated into very mature stages which further limit their usefulness as a model for an adult brain. However, regarding neurodevelopmental disorders like ASD, ADHD and also the prodromal stages of bipolar disorder and schizophrenia that start in childhood as well, we are still convinced that insights to molecular pathomechanisms could be drawn from those immature models.
One of the endemic problems for the use of iPSC models is that the generation of multiple cell lines can be time-consuming, costly and labor-intensive (Quadrato et al. 2016; Soliman et al. 2017). This has resulted in low biological replicate numbers being analyzed, with some studies publishing findings on as little as one biological replicate (Ishii et al. 2019). This presents a problem as it questions the replicability and therefore the validity of findings. Increased biological replicates are needed to minimize sample variability, which has been suggested to vary greatly in iPSC lines (Studer et al. 2015). However, exactly how large the cohort size should be remains uncertain. A recent study by Schwartzentruber et al. (2018) shed some light on this matter, using a large cohort size of 123 biological replicates to analyze molecular and functional variation between iPSC-derived sensory neurons (Schwartzentruber et al. 2018). Their results suggested that between 20 and 80 individual iPSC lines are required to detect the effects of regulatory variants with a moderate-to-large effect size. Although useful knowledge, this now presents researchers with the issue of how to generate such large cohort sizes in a timely, reproducible, yet cost-effective manner. Additional suggestions for decreasing the number of required biological replicates have been proposed by Hoekstra et al. They suggest that patient donors should be selected based on either being carriers of more rare risk gene variants, or possessing a high load of a specific polygenic risk score. They also recommend the use of isogenic controls using CRISPR/cas9 technology, studying affected and non-affected family members, and investigating phenotypically more homogenous patients and controls (Hoekstra et al. 2017).
Replicability of results is further complicated by the variability seen between clones originating from the same iPSC line. When generating iPSCs most protocols recommend that individual colonies demonstrating ESC-like morphology are manually picked and cultured as clones, as they represent successfully reprogrammed cells and therefore increase iPSC homogeneity (Meissner et al. 2007; Pfannkuche et al. 2010). It is therefore current practice to pick and culture multiple clones from one iPSC donor, and many published studies using iPSC-based models present results originating from two or more clones. Although well-intended, the analysis of more than one clone per iPSC line may actually be creating more problems than it solves, due to the variability arising from technical sources such as researcher-subjective colony selection (Chan et al. 2009). A recent study by Germain and Testa (2017) investigated the potential effects of clone variability on iPSC-based disease modeling (Germain and Testa 2017). Using large transcriptomic datasets, they revealed that the use of more than clone per donor increased the detection of spurious differentially expressed genes and the false discovery rate, thereby decreasing the robustness of data. Moreover, this result did not change even when a higher number of biological replicates was used. These results therefore question the current widespread habit of using multiple clones per iPSC donor, a practice that is additionally entrenched into the guidelines of several journals (e.g. Stem Cell Reports). Possible solutions could be (1) to continue to use multiple clones per line, but account for their interdependence in the statistical analysis, (2) use isogenic controls from the same donor, or (3) use only one clone per donor but increase the overall number of biological replicates (Germain and Testa 2017).
A further option could be to use cultures derived from ‘mixed’ clones, which are derived from several initial colonies combined. Bona fide iPSCs have escaped replicative senescence, demonstrating high proliferation rates, and therefore are likely to outgrow non- or only partially reprogrammed cells during culture expansion (Koch et al. 2013; Lapasset et al. 2011). One study investigating this possibility reported that after several passages no significant differences could be observed in morphology, pluripotency, or gene expression profiles between mixed cultures and clonally-derived cultures (Willmann et al. 2013). The concept of using mixed cultures is attractive, as it would help facilitate the standardization and automaton of iPSC generation, allowing for high throughput operation. Further studies are needed to determine whether manual clonal selection is truly necessary for homogenous iPSCs, or whether mixed cultures will suffice. Moreover, there are now technological options for obtaining highly homogenous iPSC cultures without the need for clonal selection, such as the use of magnetic beads for cell selection/sorting (Gao et al. 2018; Yang et al. 2015). It is therefore possible that the act of manually picking individual colonies and publishing results from multiple iPSC clones may become redundant.
One last solution for decreasing data variability, increasing cohort size and consequently increasing data validity, is the use of newly developed iPSC banks. Such initiatives are aimed at generating and collecting a large number of iPSC lines, purely for the purpose of scientific research. Most are non-profit and government-funded, due to the scale and influence required (Huang et al. 2019). These banks allow researchers to increase biological replicate numbers in a time- and cost-effective manner. Moreover, iPSC lines submitted to the banks are stringently checked for quality and iPSC status, in line with guidelines set out by the Global Alliance for iPSC Therapies (Sullivan et al. 2018). For example, the European Bank for induced pluripotent Stem Cells (EBiSC) has established standard operation procedures (SOPs) to provide quality assurance and quality control, tightly regulating processes such as iPSC generation and maintenance. Consequently, data obtained from using iPSC bank cultures may be highly comparable, with decreased variability. For more information, a comprehensive review of current iPSC banks has recently been published by Huang et al. (2019).
An additional advantage of using cell lines from iPSC banks is that the samples provided are usually connected to a wealth of clinical information regarding the donor. Such data can include psychometric assessments, fluid biomarker sampling, and structural, functional and molecular neuroimaging (Chhatwal et al. 2016; Ghetti et al. 2015; Jack and Holtzman 2013; Jack et al. 2013; Marquie et al. 2015). Access to this type of information will allow for the implementation of integrative approaches, providing a rich dataset from which to interpret in vitro iPSC-based results. Studies utilizing clinically informed iPSC models have already started to reveal that dysfunctional mechanisms observed in the patient in vivo can also be modeled in vitro, demonstrating the validity of using this approach. Moreover, these results suggested that disruption to molecular pathways may occur much earlier in disease development than previously thought (Silva and Haggarty 2019). However, despite efforts to promote data sharing in the academic, industry and funding sections, there is still a reluctance to share important results. This is particularly seen in clinical trials, where up to a third of trials remain unpublished up to 5 years after completion (Stefaniak et al. 2017). Such lack of dissemination inhibits the information available to the scientific community, wasting precious resources and hindering progress, and therefore open science in the field of iPSC research should be encouraged in the future.
As mentioned in the introduction, psychiatric diseases are usually highly polygenic disorders, where multiple SNPs convey a small effect size and act in concert to contribute to disease risk (International Schizophrenia et al. 2009). Identifying significantly disease-associated SNPs therefore requires large sample numbers, which as discussed, can be difficult to obtain for iPSC models. To address this problem, a slightly altered approach to study design could be considered. Instead of the classical ‘healthy control versus disease’ design, researchers could instead aim to use the concept of endophenotypes or polygenic risk scores (PRS); or even a combination of the two. Endophenotypes are measurable traits that exist somewhere along the spectrum between disease and distal genotype, falling under categories such as cognitive, biochemical and neuroanatomical, and are proposed to consist of a simpler genetic component than the complex psychiatric disease itself (Gottesman and Gould 2003). PRS measures the risk of an individual developing a certain disease, based on their genetic makeup (Wray et al. 2007). Using GWAS data, each individual disease-associated SNP is assigned a weight based on their effect size, and the total number of SNPs combined with their weights is summed to calculate an individual’s PRS. This approach has already revealed that PRS can significantly predict endophenotypes in disease and control settings, such as cognition (Richards et al. 2019) and impulsivity (Hari Dass et al. 2019). Moreover, PRS can also identify specific pathways which are involved both individually and cross-disorder. For example, certain SNPs existing in calcium signaling genes have now been associated with several different psychiatric diseases, suggesting these genes may have a pleiotropic effect on psychopathology (Cross-Disorder Group of the Psychiatric Genomics Consortium 2013). PRS can therefore be modeled in vitro using iPSC-based models derived from donors for whom sequencing data is available, and consequent functional outcomes analyzed (e.g. synaptic signaling). Moreover, gene editing can be used to manipulate several SNPs simultaneously (Cong et al. 2013; Minkenberg et al. 2017), allowing the creation of isogenic controls based on PRS. Using these approaches will augment the ability of researchers to capture significant molecular changes pertaining to pathomechanisms, providing valuable into disease etiology and potentially revealing novel drug targets.
To summarize, the use of iPSC-based models has greatly contributed to our understanding of the complex etiology of psychiatric diseases, providing a model that is more relevant to the in vivo setting and increasing validity of results. Of course, no method is without its limitations, and the sole use of iPSC cultures to understand mental illness has several. However, these limitations can be seen as areas to improve upon, rather than true limitations (Logan et al. 2019). In this systematic review, we have not only provided the reader with an insight into the investigation of less well-studied psychiatric disorders using iPSCs, but have also provided several possible avenues to explore to increase the potential of current iPSC methods. We hope that implementation of such ideas will aid the discovery of robust novel findings, facilitating valuable insight into disease etiology and informing drug development. The future of iPSC technology appears bright, and by association, so does the future of psychiatric research.
References
Abramowitz JS, Taylor S, McKay D (2009) Obsessive-compulsive disorder. Lancet 374:491–499. https://doi.org/10.1016/S0140-6736(09)60240-3
Ahmad R, Sportelli V, Ziller M, Spengler D, Hoffmann A (2018) Tracing early neurodevelopment in schizophrenia with induced pluripotent stem cells. Cells. https://doi.org/10.3390/cells7090140
Aigner S, Heckel T, Zhang JD, Andreae LC, Jagasia R (2014) Human pluripotent stem cell models of autism spectrum disorder: emerging frontiers, opportunities, and challenges towards neuronal networks in a dish. Psychopharmacology 231:1089–1104. https://doi.org/10.1007/s00213-013-3332-1
Amatya DN et al (2019) Dynamical Electrical Complexity Is Reduced during neuronal differentiation in autism spectrum disorder. Stem Cell Rep 13:474–484. https://doi.org/10.1016/j.stemcr.2019.08.001
American Psychiatric Association (2000) Diagnostic and statistical manual of mental disorders. 4th edn, text rev. https://doi.org/10.1176/appi.books.9780890423349
American Psychiatric Association (2013) Diagnostic and statistical manual of mental disorders. 5th edn. https://doi.org/10.1176/appi.books.9780890425596
Angstman K, Rasmussen NH (2011) Personality disorders: review and clinical application in daily practice. AFP 84:1253–1260
Arcelus J, Mitchell AJ, Wales J, Nielsen S (2011) Mortality rates in patients with anorexia nervosa and other eating disorders. A meta-analysis of 36 studies. Arch Gen Psychiatry 68:724–731. https://doi.org/10.1001/archgenpsychiatry.2011.74
Arcos-Burgos M et al (2010) A common variant of the latrophilin 3 gene, LPHN3, confers susceptibility to ADHD and predicts effectiveness of stimulant medication. Mol Psychiatry 15:1053–1066. https://doi.org/10.1038/mp.2010.6
Arnerić SP, Kern VD, Stephenson DT (2018) Regulatory-accepted drug development tools are needed to accelerate innovative CNS disease treatments. Biochem Pharmacol 151:291–306. https://doi.org/10.1016/j.bcp.2018.01.043
Balan S, Toyoshima M, Yoshikawa T (2019) Contribution of induced pluripotent stem cell technologies to the understanding of cellular phenotypes in schizophrenia. Neurobiol Dis 131:104162. https://doi.org/10.1016/j.nbd.2018.04.021
Bandelow B, Wedekind D (2015) Possible role of a dysregulation of the endogenous opioid system in antisocial personality disorder. Hum Psychopharmacol 30:393–415. https://doi.org/10.1002/hup.2497
Bartl J, Mori T, Riederer P, Ozawa H, Grunblatt E (2013) Methylphenidate enhances neural stem cell differentiation. J Mol Psychiatry 1:5. https://doi.org/10.1186/2049-9256-1-5
Bavamian S et al (2015) Dysregulation of miR-34a links neuronal development to genetic risk factors for bipolar disorder. Mol Psychiatry 20:573–584. https://doi.org/10.1038/mp.2014.176
Benraiss A, Goldman SA (2011) Cellular therapy and induced neuronal replacement for Huntington's disease. Neurotherapeutics 8:577–590. https://doi.org/10.1007/s13311-011-0075-8
Brandys MK, de Kovel CGF, Kas MJ, van Elburg AA, Adan RAH (2015) Overview of genetic research in anorexia nervosa: The past, the present and the future. Int J Eat Disord 48:814–825. https://doi.org/10.1002/eat.22400
Breiderhoff T et al (2013) Sortilin-related receptor SORCS3 is a postsynaptic modulator of synaptic depression and fear extinction. PLoS ONE 8:e75006. https://doi.org/10.1371/journal.pone.0075006
Brennand KJ, Gage FH (2011) Concise review: the promise of human induced pluripotent stem cell-based studies of schizophrenia. Stem Cells 29:1915–1922. https://doi.org/10.1002/stem.762
Brennand KJ, Gage FH (2012) Modeling psychiatric disorders through reprogramming. Dis Model Mech 5:26–32. https://doi.org/10.1242/dmm.008268
Brennand KJ et al (2011) Modelling schizophrenia using human induced pluripotent stem cells. Nature 473:221–225. https://doi.org/10.1038/nature09915
Brennand KJ, Landek-Salgado MA, Sawa A (2014) Modeling heterogeneous patients with a clinical diagnosis of schizophrenia with induced pluripotent stem cells. Biol Psychiatry 75:936–944. https://doi.org/10.1016/j.biopsych.2013.10.025
Brennand K et al (2015) Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry 20:361–368. https://doi.org/10.1038/mp.2014.22
Brito A, Russo FB, Muotri AR, Beltrao-Braga PCB (2018) Autism spectrum disorders and disease modeling using stem cells. Cell Tissue Res 371:153–160. https://doi.org/10.1007/s00441-017-2685-x
Brown AS, Derkits EJ (2010) Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry 167:261–280. https://doi.org/10.1176/appi.ajp.2009.09030361
Bulik CM, Reba L, Siega-Riz A-M, Reichborn-Kjennerud T (2005) Anorexia nervosa: definition, epidemiology, and cycle of risk. Int J Eat Disord. https://doi.org/10.1002/eat.20107
Bulik CM, Berkman ND, Brownley KA, Sedway JA, Lohr KN (2007) Anorexia nervosa treatment: a systematic review of randomized controlled trials. Int J Eat Disord 40:310–320. https://doi.org/10.1002/eat.20367
Bundo M et al (2014) Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron 81:306–313. https://doi.org/10.1016/j.neuron.2013.10.053
Cao SY et al (2017) Enhanced derivation of human pluripotent stem cell-derived cortical glutamatergic neurons by a small molecule. Sci Rep 7:3282. https://doi.org/10.1038/s41598-017-03519-w
Castellanos FX, Tannock R (2002) Neuroscience of attention-deficit/hyperactivity disorder: the search for endophenotypes. Nat Rev Neurosci 3:617–628. https://doi.org/10.1038/nrn896
Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27:275–280. https://doi.org/10.1038/nbt.1529
Chan EM et al (2009) Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat Biotechnol 27:1033–1037. https://doi.org/10.1038/nbt.1580
Chen HM, DeLong CJ, Bame M, Rajapakse I, Herron TJ, McInnis MG, O'Shea KS (2014) Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients. Transl Psychiatry 4:e375. https://doi.org/10.1038/tp.2014.12
Chen YC et al (2016) Foxp2 controls synaptic wiring of corticostriatal circuits and vocal communication by opposing Mef2c. Nat Neurosci 19:1513–1522. https://doi.org/10.1038/nn.4380
Cheng C, Fass DM, Folz-Donahue K, MacDonald ME, Haggarty SJ (2017) Highly expandable human ips cell–derived neural progenitor cells (npc) and neurons for central nervous system disease modeling and high-throughput screening. Curr Protoc Human Genet. https://doi.org/10.1002/cphg.33
Cheung CYY et al (2010) Obesity susceptibility genetic variants identified from recent genome-wide association studies: implications in a chinese population. J Clin Endocrinol Metab 95:1395–1403. https://doi.org/10.1210/jc.2009-1465
Chhatwal JP et al (2016) Temporal T807 binding correlates with CSF tau and phospho-tau in normal elderly. Neurology 87:920–926. https://doi.org/10.1212/WNL.0000000000003050
Chiocchetti A, Pakalapati G, Duketis E, Wiemann S, Poustka A, Poustka F, Klauck SM (2011) Mutation and expression analyses of the ribosomal protein gene RPL10 in an extended German sample of patients with autism spectrum disorder. Am J Med Genet A 155A:1472–1475. https://doi.org/10.1002/ajmg.a.33977
Cong L et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. https://doi.org/10.1126/science.1231143
Cook EH Jr, Stein MA, Krasowski MD, Cox NJ, Olkon DM, Kieffer JE, Leventhal BL (1995) Association of attention-deficit disorder and the dopamine transporter gene. Am J Hum Genet 56:993–998
Coskun V, Lombardo DM (2016) Studying the pathophysiologic connection between cardiovascular and nervous systems using stem cells. J Neurosci Res 94:1499–1510. https://doi.org/10.1002/jnr.23924
Costain G et al (2013) Pathogenic rare copy number variants in community-based schizophrenia suggest a potential role for clinical microarrays. Hum Mol Genet 22:4485–4501. https://doi.org/10.1093/hmg/ddt297
Craddock N, Jones I (1999) Genetics of bipolar disorder. J Med Genet 36:585–594
Cross-Disorder Group of the Psychiatric Genomics Consortium (2013) Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381:1371–1379. https://doi.org/10.1016/S0140-6736(12)62129-1
International Schizophrenia C et al (2009) Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460:748–752. https://doi.org/10.1038/nature08185
Cummings J (2018) Lessons learned from Alzheimer disease: clinical trials with negative outcomes. Clin Transl Sci 11:147–152. https://doi.org/10.1111/cts.12491
DAiuto L et al (2015) Persistent infection by HSV is associated with changes in functional architecture of iPSC-derived neurons and brain activation patterns underlying working memory performance Schizophrenia. Bulletin. https://doi.org/10.1093/schbul/sbu032
Das DK et al (2015) Genetic and morphological features of human iPSC-derived neurons with chromosome 15q11.2 (BP1-BP2) deletions. Mol Neuropsychiatry 1:116–123. https://doi.org/10.1159/000430916
Demontis D et al (2019) Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet 51(1):63–75. https://doi.org/10.1038/s41588-018-0269-7
Dolmetsch R, Geschwind DH (2011) The human brain in a dish: the promise of iPSC-derived neurons. Cell 145:831–834. https://doi.org/10.1016/j.cell.2011.05.034
Drew LJ et al (2011) The 22q11.2 microdeletion: fifteen years of insights into the genetic and neural complexity of psychiatric disorders. Int J Dev Neurosci 29:259–281. https://doi.org/10.1016/j.ijdevneu.2010.09.007
Drozd AM, Walczak MP, Piaskowski S, Stoczynska-Fidelus E, Rieske P, Grzela DP (2015) Generation of human iPSCs from cells of fibroblastic and epithelial origin by means of the oriP/EBNA-1 episomal reprogramming system. Stem Cell Res Ther 6:122. https://doi.org/10.1186/s13287-015-0112-3
Duval K, Grover H, Han LH, Mou Y, Pegoraro AF, Fredberg J, Chen Z (2017) Modeling physiological events in 2D vs. 3D. Cell Culture Physiol 32:266–277. https://doi.org/10.1152/physiol.00036.2016
Elia J et al (2012) Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat Genet 44:78–84. https://doi.org/10.1038/ng.1013
Essayan-Perez S, Zhou B, Nabet AM, Wernig M, Huang YA (2019) Modeling Alzheimer's disease with human iPS cells: advancements, lessons, and applications. Neurobiol Dis 130:104503. https://doi.org/10.1016/j.nbd.2019.104503
Estes ML, McAllister AK (2016) Maternal immune activation: Implications for neuropsychiatric disorders. Science 353:772–777. https://doi.org/10.1126/science.aag3194
Faraone SV, Larsson H (2019) Genetics of attention deficit hyperactivity disorder. Mol Psychiatry 24:562–575. https://doi.org/10.1038/s41380-018-0070-0
Faraone SV, Biederman J, Mick E (2006) The age-dependent decline of attention deficit hyperactivity disorder: a meta-analysis of follow-up studies. Psychol Med 36:159–165. https://doi.org/10.1017/S003329170500471X
Faundez V et al (2018) Translating molecular advances in Down syndrome and Fragile X syndrome into therapies. Eur Neuropsychopharmacol 28:675–690. https://doi.org/10.1016/j.euroneuro.2018.03.006
De Filippis L, Halikere A, McGowan H, Moore JC, Tischfield JA, Hart RP, Pang ZP (2016) Ethanol-mediated activation of the NLRP3 inflammasome in iPS cells and iPS cells-derived neural progenitor cells. Mol Brain 9:51. https://doi.org/10.1186/s13041-016-0221-7
Fontaine N, Viding E (2008) Genetics of personality disorders. Psychiatry 7:137–141. https://doi.org/10.1016/j.mppsy.2008.01.002
Forrest MP et al (2017) Open Chromatin Profiling in hiPSC-derived neurons prioritizes functional noncoding psychiatric risk variants and highlights neurodevelopmental loci. Cell Stem Cell 21:305–318.e308. https://doi.org/10.1016/j.stem.2017.07.008
Frank J et al (2012) Genome-wide significant association between alcohol dependence and a variant in the ADH gene cluster. Addict Biol 17:171–180. https://doi.org/10.1111/j.1369-1600.2011.00395.x
Freitas BC, Trujillo CA, Carromeu C, Yusupova M, Herai RH, Muotri AR (2014) Stem cells and modeling of autism spectrum disorders. Exp Neurol 260:33–43. https://doi.org/10.1016/j.expneurol.2012.09.017
Gao X, Sprando RL, Yourick JJ (2018) A rapid and highly efficient method for the isolation purification, and passaging of human-induced pluripotent. Stem Cells Cell Reprogr 20:282–288. https://doi.org/10.1089/cell.2018.0022
Georgieva L et al (2014) De novo CNVs in bipolar affective disorder and schizophrenia. Hum Mol Genet 23:6677–6683. https://doi.org/10.1093/hmg/ddu379
Germain P-L, Testa G (2017) Taming human genetic variability: transcriptomic meta-analysis guides the experimental design and interpretation of ipsc-based disease modeling. Stem Cell Rep 8:1784–1796. https://doi.org/10.1016/j.stemcr.2017.05.012
Gervasini G, Gordillo I, García-Herráiz A, Flores I, Jiménez M, Monge M, Carrillo JA (2013) Influence of dopamine polymorphisms on the risk for anorexia nervosa and associated psychopathological features. J Clin Psychopharmacol 33:551–555. https://doi.org/10.1097/JCP.0b013e3182970469
Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M (2015) Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol 41:24–46. https://doi.org/10.1111/nan.12213
Giegling I et al (2017) Genetics of schizophrenia: a consensus paper of the WFSBP Task Force on Genetics. World J Biol Psychiatry 18:492–505. https://doi.org/10.1080/15622975.2016.1268715
Gjervan B, Torgersen T, Nordahl HM, Rasmussen K (2012) Functional impairment and occupational outcome in adults with ADHD. J Atten Disord 16:544–552. https://doi.org/10.1177/1087054711413074
Golas MM, Sander B (2016) Use of human stem cells in Huntington disease modeling and translational research. Exp Neurol 278:76–90. https://doi.org/10.1016/j.expneurol.2016.01.021
Goldstein LS, Reyna S, Woodruff G (2015) Probing the secrets of Alzheimer's disease using human-induced pluripotent stem cell technology. Neurotherapeutics 12:121–125. https://doi.org/10.1007/s13311-014-0326-6
Gottesman II, Gould TD (2003) The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry 160:636–645. https://doi.org/10.1176/appi.ajp.160.4.636
Gratten J, Wray NR, Keller MC, Visscher PM (2014) Large-scale genomics unveils the genetic architecture of psychiatric disorders. Nat Neurosci 17:782–790. https://doi.org/10.1038/nn.3708
Graybiel AM, Rauch SL (2000) Toward a neurobiology of obsessive-compulsive disorder. Neuron 28:343–347. https://doi.org/10.1016/s0896-6273(00)00113-6
Griesi-Oliveira K et al (2015) Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry 20:1350–1365. https://doi.org/10.1038/mp.2014.141
Grünblatt E, Bartl J, Marinova Z, Walitza S (2013) vitro study methodologies to investigate genetic aspects and effects of drugs used in attention-deficit hyperactivity disorder. J Neural Trans 120:131–139. https://doi.org/10.1007/s00702-012-0869-9
Grunwald L-M et al (2019) Comparative characterization of human induced pluripotent stem cells (hiPSC) derived from patients with schizophrenia and autism. Transl Psychiatry 9:1–11. https://doi.org/10.1038/s41398-019-0517-3
Gunhanlar N et al (2018) A simplified protocol for differentiation of electrophysiologically mature neuronal networks from human induced pluripotent stem cells. Mol Psychiatry 23:1336–1344. https://doi.org/10.1038/mp.2017.56
Häfner H et al (1994) The epidemiology of early schizophrenia. Influence of age and gender on onset and early course. Br J Psychiatry 164:29–38
Hari Dass SA et al (2019) A biologically-informed polygenic score identifies endophenotypes and clinical conditions associated with the insulin receptor function on specific brain regions. EBioMedicine 42:188–202. https://doi.org/10.1016/j.ebiom.2019.03.051
Hashimoto-Torii K et al (2014) Roles of heat shock factor 1 in neuronal response to fetal environmental risks and its relevance to brain disorders. Neuron 82:560–572. https://doi.org/10.1016/j.neuron.2014.03.002
Hilker R et al (2018) Heritability of schizophrenia and schizophrenia spectrum based on the nationwide Danish Twin register. Biol Psychiatry 83:492–498. https://doi.org/10.1016/j.biopsych.2017.08.017
Hirschfeld RM et al (2003) Screening for bipolar disorder in the community. J Clin Psychiatry 64:53–59. https://doi.org/10.4088/jcp.v64n0111
Ho BX, Pek NMQ, Soh BS (2018) Disease modeling using 3d organoids derived from human induced pluripotent stem cells. Int J Mol Sci. https://doi.org/10.3390/ijms19040936
Hoekstra SD, Stringer S, Heine VM, Posthuma D (2017) Genetically-informed patient selection for iPSC studies of complex diseases may aid in reducing cellular heterogeneity. Front Cell Neurosci 11:164. https://doi.org/10.3389/fncel.2017.00164
Hoffman GE, Schrode N, Flaherty E, Brennand KJ (2019) New considerations for hiPSC-based models of neuropsychiatric disorders. Mol Psychiatry 24:49–66. https://doi.org/10.1038/s41380-018-0029-1
Hoffmann A, Sportelli V, Ziller M, Spengler D (2018) From the psychiatrist's couch to induced pluripotent stem cells: bipolar disease in a dish. Int J Mol Sci 19: https://doi.org/10.3390/ijms19030770
Hosokawa M, Shoji M, Kitamura K, Tanaka T, Noce T, Chuma S, Nakatsuji N (2007) Tudor-related proteins TDRD1/MTR-1, TDRD6 and TDRD7/TRAP: domain composition, intracellular localization, and function in male germ cells in mice. Dev Biol 301:38–52. https://doi.org/10.1016/j.ydbio.2006.10.046
Hu H et al (2011) ST3GAL3 mutations impair the development of higher cognitive functions. Am J Hum Genet 89:407–414. https://doi.org/10.1016/j.ajhg.2011.08.008
Huang CY, Liu CL, Ting CY, Chiu YT, Cheng YC, Nicholson MW, Hsieh PCH (2019) Human iPSC banking: barriers and opportunities. J Biomed Sci 26:87. https://doi.org/10.1186/s12929-019-0578-x
IOCDF-GC and OCGAS (2018) Revealing the complex genetic architecture of obsessive-compulsive disorder using meta-analysis. Molecular psychiatry 23:1181–1188. https://doi.org/10.1038/mp.2017.154
Ishii K, Kubo KI, Nakajima K (2016) Reelin and neuropsychiatric disorders. Front Cell Neurosci 10:229. https://doi.org/10.3389/fncel.2016.00229
Ishii T et al (2019a) In vitro modeling of the bipolar disorder and schizophrenia using patient-derived induced pluripotent stem cells with copy number variations of PCDH15 and RELN. eNeuro. https://doi.org/10.1523/ENEURO.0403-18.2019
Ivanov HY, Stoyanova VK, Popov NT, Vachev TI (2015) Autism spectrum disorder - a complex genetic disorder. Folia Med (Plovdiv) 57:19–28. https://doi.org/10.1515/folmed-2015-0015
Jack CR Jr, Holtzman DM (2013) Biomarker modeling of Alzheimer's disease. Neuron 80:1347–1358. https://doi.org/10.1016/j.neuron.2013.12.003
Jack CR Jr et al (2013) Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12:207–216. https://doi.org/10.1016/S1474-4422(12)70291-0
Jansch C et al (2018) Generation of a human induced pluripotent stem cell (iPSC) line from a 51-year-old female with attention-deficit/hyperactivity disorder (ADHD) carrying a duplication of SLC2A3. Stem Cell Res 28:136–140. https://doi.org/10.1016/j.scr.2018.02.005
Katz MG, Vollenhoven B (2000) The reproductive endocrine consequences of anorexia nervosa. BJOG 107:707–713. https://doi.org/10.1111/j.1471-0528.2000.tb13329.x
Kaye WH, Gwirtsman HE, George DT, Jimerson DC, Ebert MH (1988) CSF 5-HIAA concentrations in anorexia nervosa: reduced values in underweight subjects normalize after weight gain. Biol Psychiatry 23:102–105. https://doi.org/10.1016/0006-3223(88)90113-8
Kaye WH, Frank GK, Bailer UF, Henry SE (2005) Neurobiology of anorexia nervosa: clinical implications of alterations of the function of serotonin and other neuronal systems. Int J Eat Disord. https://doi.org/10.1002/eat.20109
Kim KH et al (2015) Transcriptomic analysis of induced pluripotent stem cells derived from patients with bipolar disorder from an old order Amish Pedigree. PLoS ONE 10:e0142693. https://doi.org/10.1371/journal.pone.0142693
Kinch MS (2015) An analysis of FDA-approved drugs for neurological disorders. Drug Discov Today 20:1040–1043. https://doi.org/10.1016/j.drudis.2015.02.003
Kittel-Schneider S et al (2014) Influence of DGKH variants on amygdala volume in patients with bipolar affective disorder and schizophrenia. Eur Arch Psychiatry Clin Neurosci. https://doi.org/10.1007/s00406-014-0513-9
Kittel-Schneider S, Lorenz C, Auer J, Weissflog L, Reif A (2016) DGKH genetic risk variant influences gene expression in bipolar affective disorder. J Affect Disord 198:148–157. https://doi.org/10.1016/j.jad.2016.03.041
Klauck SM et al (2006) Mutations in the ribosomal protein gene RPL10 suggest a novel modulating disease mechanism for autism. Mol Psychiatry 11:1073–1084. https://doi.org/10.1038/sj.mp.4001883
Koch P, Opitz T, Steinbeck JA, Ladewig J, Brüstle O (2009) A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc Natl Acad Sci USA 106:3225–3230. https://doi.org/10.1073/pnas.0808387106
Koch CM et al (2013) Pluripotent stem cells escape from senescence-associated DNA methylation changes. Genome Res 23:248–259. https://doi.org/10.1101/gr.141945.112
Korhonen P, Malm T, White AR (2018) 3D human brain cell models: new frontiers in disease understanding and drug discovery for neurodegenerative diseases. Neurochem Int 120:191–199. https://doi.org/10.1016/j.neuint.2018.08.012
Kustanovich V et al (2004) Transmission disequilibrium testing of dopamine-related candidate gene polymorphisms in ADHD: confirmation of association of ADHD with DRD4 and DRD5. Mol Psychiatry 9:711–717. https://doi.org/10.1038/sj.mp.4001466
Lapasset L et al (2011) Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev 25:2248–2253. https://doi.org/10.1101/gad.173922.111
Lee IS et al (2015) Characterization of molecular and cellular phenotypes associated with a heterozygous CNTNAP2 deletion using patient-derived hiPSC neural cells. NPJ Schizophr https://doi.org/10.1038/npjschz.2015.19
Lee CT, Bendriem RM, Wu WW, Shen RF (2017) 3D brain Organoids derived from pluripotent stem cells: promising experimental models for brain development and neurodegenerative disorders. J Biomed Sci 24:59. https://doi.org/10.1186/s12929-017-0362-8
Lesch KP, Merker S, Reif A, Novak M (2012) Dances with black widow spiders: dysregulation of glutamate signalling enters centre stage in ADHD. Eur Neuropsychopharmacol. https://doi.org/10.1016/j.euroneuro.2012.07.013
Levine J, Chengappa KN, Brar JS, Gershon S, Yablonsky E, Stapf D, Kupfer DJ (2000) Psychotropic drug prescription patterns among patients with bipolar I disorder. Bipolar Disord 2:120–130. https://doi.org/10.1034/j.1399-5618.2000.020205.x
Lieberman R, Levine ES, Kranzler HR, Abreu C, Covault J (2012) Pilot study of iPS-derived neural cells to examine biologic effects of alcohol on human neurons in vitro. Alcohol Clin Exp Res 36:1678–1687. https://doi.org/10.1111/j.1530-0277.2012.01792.x
Lieberman R, Kranzler HR, Joshi P, Shin DG, Covault J (2015) GABRA2 alcohol dependence risk allele is associated with reduced expression of chromosome 4p12 GABAA subunit genes in human neural cultures. Alcohol Clin Exp Res 39:1654–1664. https://doi.org/10.1111/acer.12807
Lieberman R, Kranzler HR, Levine ES, Covault J (2018) Examining the effects of alcohol on GABAA receptor mRNA expression and function in neural cultures generated from control and alcohol dependent donor induced pluripotent stem cells. Alcohol 66:45–53. https://doi.org/10.1016/j.alcohol.2017.08.005
Lin M et al (2016) Integrative transcriptome network analysis of iPSC-derived neurons from schizophrenia and schizoaffective disorder patients with 22q11.2 deletion. BMC Syst Biol 10:105. https://doi.org/10.1186/s12918-016-0366-0
Lisowski P, Wieczorek M, Goscik J, Juszczak GR, Stankiewicz AM, Zwierzchowski L, Swiergiel AH (2013) Effects of chronic stress on prefrontal cortex transcriptome in mice displaying different genetic backgrounds. J Mol Neurosci 50:33–57. https://doi.org/10.1007/s12031-012-9850-1
Logan S, Arzua T, Canfield SG, Seminary ER, Sison SL, Ebert AD, Bai X (2019) Studying human neurological disorders using induced pluripotent stem cells: from 2d monolayer to 3d organoid and blood brain barrier models. Compr Physiol 9:565–611. https://doi.org/10.1002/cphy.c180025
Lu J et al (2016) Generation of serotonin neurons from human pluripotent stem cells. Nat Biotechnol 34:89–94. https://doi.org/10.1038/nbt.3435
Ma G, Fan H, Shen C, Wang W (2016) Genetic and neuroimaging features of personality disorders: state of the art. Neurosci Bull 32:286–306. https://doi.org/10.1007/s12264-016-0027-8
Madison JM et al (2015) Characterization of bipolar disorder patient-specific induced pluripotent stem cells from a family reveals neurodevelopmental and mRNA expression abnormalities. Mol Psychiatry 20:703–717. https://doi.org/10.1038/mp.2015.7
Marchetto MC et al (2017) Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry 22:820–835. https://doi.org/10.1038/mp.2016.95
Mariani J et al (2015) FOXG1-Dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 162:375–390. https://doi.org/10.1016/j.cell.2015.06.034
Marquie M et al (2015) Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol 78:787–800. https://doi.org/10.1002/ana.24517
Marshall CR et al (2017) Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet 49:27–35. https://doi.org/10.1038/ng.3725
Maussion G, Demirova I, Gorwood P, Ramoz N (2019) Induced pluripotent stem cells; new tools for investigating molecular mechanisms in anorexia nervosa. Front Nutr https://doi.org/10.3389/fnut.2019.00118
McGuffin P, Rijsdijk F, Andrew M, Sham P, Katz R, Cardno A (2003) The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch Gen Psychiatry 60:497–502. https://doi.org/10.1001/archpsyc.60.5.497
Meissner A, Wernig M, Jaenisch R (2007) Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nat Biotechnol 25:1177–1181. https://doi.org/10.1038/nbt1335
Mertens J et al (2015) Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 527:95–99. https://doi.org/10.1038/nature15526
Mertens J et al (2016) Erratum: differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 530:242. https://doi.org/10.1038/nature16182
Mertens J, Reid D, Lau S, Kim Y, Gage FH (2018) Aging in a dish: iPSC-derived and directly induced neurons for studying brain aging and age-related neurodegenerative diseases. Annu Rev Genet 52:271–293. https://doi.org/10.1146/annurev-genet-120417-031534
Millar JK et al (2000) Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet 9:1415–1423. https://doi.org/10.1093/hmg/9.9.1415
Miller ND, Kelsoe JR (2017) Unraveling the biology of bipolar disorder using induced pluripotent stem-derived neurons. Bipolar Disord 19:544–551. https://doi.org/10.1111/bdi.12535
Minkenberg B, Wheatley M, Yang Y (2017) CRISPR/Cas9-Enabled multiplex genome editing and its application. Prog Mol Biol Transl Sci 149:111–132. https://doi.org/10.1016/bs.pmbts.2017.05.003
Moher D, Liberati A, Tetzlaff J, Altman DG, Group P (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ 339:b2535. https://doi.org/10.1136/bmj.b2535
Moore D et al (2019) Downregulation of an evolutionary young mir-1290 in an ipsc-derived neural stem cell model of autism spectrum disorder. Stem Cells Int 2019:8710180. https://doi.org/10.1155/2019/8710180
Muñoz M, Coveñas R (2014) Involvement of substance P and the NK-1 receptor in human pathology. Amino Acids 46:1727–1750. https://doi.org/10.1007/s00726-014-1736-9
Murai K et al (2016) The TLX-miR-219 cascade regulates neural stem cell proliferation in neurodevelopment and schizophrenia iPSC model. Nat Commun 7:10965. https://doi.org/10.1038/ncomms10965
Muratore CR, Srikanth P, Callahan DG, Young-Pearse TL (2014) Comparison and optimization of hiPSC forebrain cortical differentiation protocols. PLoS ONE 9:e105807. https://doi.org/10.1371/journal.pone.0105807
Naaijen J, Lythgoe DJ, Amiri H, Buitelaar JK, Glennon JC (2015) Fronto-striatal glutamatergic compounds in compulsive and impulsive syndromes: a review of magnetic resonance spectroscopy studies. Neurosci Biobehav Rev 52:74–88. https://doi.org/10.1016/j.neubiorev.2015.02.009
Nagashima T, Kim J, Li Q, Lydon JP, DeMayo FJ, Lyons KM, Matzuk MM (2011) Connective tissue growth factor is required for normal follicle development and ovulation. Mol Endocrinol 25:1740–1759. https://doi.org/10.1210/me.2011-1045
Nakazawa T et al (2017) Differential gene expression profiles in neurons generated from lymphoblastoid B-cell line-derived iPS cells from monozygotic twin cases with treatment-resistant schizophrenia and discordant responses to clozapine. Schizophr Res 181:75–82. https://doi.org/10.1016/j.schres.2016.10.012
Nakazawa T, Hashimoto R, Takuma K, Hashimoto H (2019) Modeling of psychiatric disorders using induced pluripotent stem cell-related technologies. J Pharmacol Sci 140:321–324. https://doi.org/10.1016/j.jphs.2019.06.002
Negraes PD et al (2017) Modeling anorexia nervosa: transcriptional insights from human iPSC-derived neurons. Transl Psychiatry 7:e1060–e1060. https://doi.org/10.1038/tp.2017.37
Nestadt G et al (2000) A family study of obsessive-compulsive disorder. Arch Gen Psychiatry 57:358–363. https://doi.org/10.1001/archpsyc.57.4.358
Nestler EJ, Hyman SE (2010) Animal models of neuropsychiatric disorders. Nat Neurosci 13:1161–1169. https://doi.org/10.1038/nn.2647
O'Hara SK, Smith KC (2007) Presentation of eating disorders in the news media: What are the implications for patient diagnosis and treatment? Patient Educ Couns 68:43–51. https://doi.org/10.1016/j.pec.2007.04.006
Oni EN et al (2016) Increased nicotine response in iPSC-derived human neurons carrying the CHRNA5 N398 allele. Sci Rep 6:34341. https://doi.org/10.1038/srep34341
Ooi L, Sidhu K, Poljak A, Sutherland G, O'Connor MD, Sachdev P, Munch G (2013) Induced pluripotent stem cells as tools for disease modelling and drug discovery in Alzheimer's disease. J Neural Transm (Vienna) 120:103–111. https://doi.org/10.1007/s00702-012-0839-2
Ormel J, Hartman CA, Snieder H (2019) The genetics of depression: successful genome-wide association studies introduce new challenges. Transl Psychiatry 9:114. https://doi.org/10.1038/s41398-019-0450-5
O'Roak BJ et al (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485:246–250. https://doi.org/10.1038/nature10989
O'Shea KS, McInnis MG (2015) Induced pluripotent stem cell (iPSC) models of bipolar disorder. Neuropsychopharmacology 40:248–249. https://doi.org/10.1038/npp.2014.221
Owen MJ, Sawa A, Mortensen PB (2016) Schizophrenia. Lancet 388:86–97. https://doi.org/10.1016/S0140-6736(15)01121-6
Palladino VS, Subrata NOC, Geburtig-Chiocchetti A, McNeill R, Hoffmann P, Reif A, Kittel-Schneider S (2018) Generation of human induced pluripotent stem cell lines (hiPSC) from one bipolar disorder patient carrier of a DGKH risk haplotype and one non-risk-variant-carrier bipolar disorder patient. Stem Cell Res 32:104–109. https://doi.org/10.1016/j.scr.2018.09.008
Parmar M, Grealish S, Henchcliffe C (2020) The future of stem cell therapies for Parkinson disease. Nat Rev Neurosci 21:103–115. https://doi.org/10.1038/s41583-019-0257-7
Pauls DL, Alsobrook JP 2nd, Goodman W, Rasmussen S, Leckman JF (1995) A family study of obsessive-compulsive disorder. Am J Psychiatry 152:76–84. https://doi.org/10.1176/ajp.152.1.76
Pauls DL, Abramovitch A, Rauch SL, Geller DA (2014) Obsessive-compulsive disorder: an integrative genetic and neurobiological perspective. Nat Rev Neurosci 15:410–424. https://doi.org/10.1038/nrn3746
Paulsen BdS et al (2012) Altered oxygen metabolism associated to neurogenesis of induced pluripotent stem cells derived from a schizophrenic patient. Cell Transpl 21:1547–1559. https://doi.org/10.3727/096368911X600957
Pedrosa E et al (2011) Development of patient-specific neurons in schizophrenia using induced pluripotent stem cells. J Neurogenet 25:88–103. https://doi.org/10.3109/01677063.2011.597908
Pfannkuche K, Fatima A, Gupta MK, Dieterich R, Hescheler J (2010) Initial colony morphology-based selection for iPS cells derived from adult fibroblasts is substantially improved by temporary UTF1-based selection. PLoS ONE 5:e9580. https://doi.org/10.1371/journal.pone.0009580
Pfisterer U et al (2011) Direct conversion of human fibroblasts to dopaminergic neurons. Proc Natl Acad Sci USA 108:10343–10348. https://doi.org/10.1073/pnas.1105135108
Phillipou A, Rossell SL, Castle DJ (2014) The neurobiology of anorexia nervosa: a systematic review The. Aust N Z J Psychiatry 48:128–152. https://doi.org/10.1177/0004867413509693
Pillidge K, Heal DJ, Stanford SC (2016) The NK1R-/- mouse phenotype suggests that small body size, with a sex- and diet-dependent excess in body mass and fat, are physical biomarkers for a human endophenotype with vulnerability to attention deficit hyperactivity disorder. J Psychopharmacol 30:848–855. https://doi.org/10.1177/0269881116658992
Posner J, Marsh R, Maia TV, Peterson BS, Gruber A, Simpson HB (2014) Reduced functional connectivity within the limbic cortico-striato-thalamo-cortical loop in unmedicated adults with obsessive-compulsive disorder. Hum Brain Mapp 35:2852–2860. https://doi.org/10.1002/hbm.22371
Prytkova I, Goate A, Hart RP, Slesinger PA (2018) Genetics of alcohol use disorder: a role for induced pluripotent stem cells? Alcohol Clin Exp Res 42:1572–1590. https://doi.org/10.1111/acer.13811
Purcell SM et al (2014) A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506:185–190. https://doi.org/10.1038/nature12975
Quadrato G, Brown J, Arlotta P (2016) The promises and challenges of human brain organoids as models of neuropsychiatric disease. Nat Med 22:1220–1228. https://doi.org/10.1038/nm.4214
Rask-Andersen M, Olszewski PK, Levine AS, Schiöth HB (2010) Molecular mechanisms underlying anorexia nervosa: focus on human gene association studies and systems controlling food intake. Brain Res Rev 62:147–164. https://doi.org/10.1016/j.brainresrev.2009.10.007
Re S, Dogan AA, Ben-Shachar D, Berger G, Werling AM, Walitza S, Grunblatt E (2018) Improved generation of induced pluripotent stem cells from hair derived keratinocytes a tool to study neurodevelopmental disorders as ADHD. Front Cell Neurosci 12:321. https://doi.org/10.3389/fncel.2018.00321
Reas DL, Rø Ø (2018) Less symptomatic, but equally impaired: Clinical impairment in restricting versus binge-eating/purging subtype of anorexia nervosa. Eat Behav 28:32–37. https://doi.org/10.1016/j.eatbeh.2017.12.004
Redies C, Hertel N, Hübner CA (2012) Cadherins and neuropsychiatric disorders. Brain Res 1470:130–144. https://doi.org/10.1016/j.brainres.2012.06.020
Rehbach K, Fernando MB, Brennand KJ (2020) Integrating CRISPR Engineering and hiPSC-Derived 2D Disease Modeling Systems. J Neurosci 40:1176–1185. https://doi.org/10.1523/JNEUROSCI.0518-19.2019
Rehm J, Shield KD (2019) Global burden of disease and the impact of mental and addictive disorders. Curr Psychiatry Rep 21:10. https://doi.org/10.1007/s11920-019-0997-0
Reichborn-Kjennerud T (2010) The genetic epidemiology of personality disorders. Dialog Clin Neurosci 12:103–114
Richards AL et al (2019) The relationship between polygenic risk scores and cognition in schizophrenia. Schizophr Bull. https://doi.org/10.1093/schbul/sbz061
Robicsek O et al (2013) Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Mol Psychiatry 18:1067–1076. https://doi.org/10.1038/mp.2013.67
Robicsek O et al (2018) Isolated mitochondria transfer improves neuronal differentiation of schizophrenia-derived induced pluripotent stem cells and rescues deficits in a rat model of the disorder. Schizophr Bull 44:432–442. https://doi.org/10.1093/schbul/sbx077
Ross PJ et al (2019) Synaptic dysfunction in human neurons with autism-associated deletions in PTCHD1-AS. Biol Psychiatry. https://doi.org/10.1016/j.biopsych.2019.07.014
Ruscio AM, Stein DJ, Chiu WT, Kessler RC (2010) The epidemiology of obsessive-compulsive disorder in the National Comorbidity Survey Replication. Mol Psychiatry 15:53–63. https://doi.org/10.1038/mp.2008.94
Russo FB, Brito A, de Freitas AM, Castanha A, de Freitas BC, Beltrao-Braga PCB (2019) The use of iPSC technology for modeling Autism Spectrum Disorders. Neurobiol Dis 130:104483. https://doi.org/10.1016/j.nbd.2019.104483
Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman C, Larsson H, Reichenberg A (2017) The heritability of autism spectrum disorder. JAMA 318:1182–1184. https://doi.org/10.1001/jama.2017.12141
Schank JR (2014) The neurokinin-1 receptor in addictive processes. J Pharmacol Exp Ther 351:2–8. https://doi.org/10.1124/jpet.113.210799
Schwamborn JC (2018) Is Parkinson's disease a neurodevelopmental disorder and will brain organoids help us to understand it? Stem Cells Dev 27:968–975. https://doi.org/10.1089/scd.2017.0289
Schwartzentruber J et al (2018) Molecular and functional variation in iPSC-derived sensory neurons. Nat Genet 50:54–61. https://doi.org/10.1038/s41588-017-0005-8
Schwarz R et al (2014) A preliminary study on methylphenidate-regulated gene expression in lymphoblastoid cells of ADHD patients. World J Biol Psychiatry. https://doi.org/10.3109/15622975.2014.948064
Seok J et al (2013) Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA 110:3507–3512
Sharan P, Sundar AS (2015) Eating disorders in women. Indian J Psychiatry 57:S286–295. https://doi.org/10.4103/0019-5545.161493
Shastry BS (2005) Bipolar disorder: an update. Neurochem Int 46:273–279
Sheng Y et al (2016) Using iPSC-derived human DA neurons from opioid-dependent subjects to study dopamine dynamics. Brain Behav 6:e00491. https://doi.org/10.1002/brb3.491
Silva MC, Haggarty HJ (2019) Human pluripotent stem cell-derived models and drug screening in CNS precision medicine. Ann N Y Acad Sci. https://doi.org/10.1111/nyas.14012
Silva MC et al (2016) Human iPSC-derived neuronal model of tau-A152T frontotemporal dementia reveals tau-mediated mechanisms of neuronal vulnerability. Stem Cell Rep 7:325–340. https://doi.org/10.1016/j.stemcr.2016.08.001
Smink FRE, van Hoeken D, Hoek HW (2012) Epidemiology of eating disorders: incidence, prevalence and mortality rates. Curr Psychiatry Rep 14:406–414. https://doi.org/10.1007/s11920-012-0282-y
Sochacki J, Devalle S, Reis M, Fontenelle LF, Rehen S (2016a) Generation of urine iPS cell line from a patient with obsessive-compulsive disorder using a non-integrative method. Stem Cell Res 17:107–110
Sochacki J, Devalle S, Reis M, Mattos P, Rehen S (2016b) Generation of urine iPS cell lines from patients with Attention Deficit Hyperactivity Disorder (ADHD) using a non-integrative method. Stem Cell Res 17:102–106
Soliman MA, Aboharb F, Zeltner N, Studer L (2017) Pluripotent stem cells in neuropsychiatric disorders. Mol Psychiatry 22:1241–1249. https://doi.org/10.1038/mp.2017.40
Srikanth P et al (2015) Genomic DISC1 disruption in hiPSCs alters wnt signaling and neural cell fate. Cell Rep 12:1414–1429. https://doi.org/10.1016/j.celrep.2015.07.061
St Clair D et al (2005) Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959–1961. JAMA 294:557–562. https://doi.org/10.1001/jama.294.5.557
Stahl EA et al (2019) Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet 51:793–803. https://doi.org/10.1038/s41588-019-0397-8
Stefaniak JD, Lam TCH, Sim NE, Al-Shahi Salman R, Breen DP (2017) Discontinuation and non-publication of neurodegenerative disease trials: a cross-sectional analysis. Eur J Neurol 24:1071–1076. https://doi.org/10.1111/ene.13336
Stefansson H et al (2008) Large recurrent microdeletions associated with schizophrenia. Nature 455:232–236. https://doi.org/10.1038/nature07229
Stern S et al (2018) Neurons derived from patients with bipolar disorder divide into intrinsically different sub-populations of neurons, predicting the patients' responsiveness to lithium. Mol Psychiatry 23:1453–1465. https://doi.org/10.1038/mp.2016.260
Studer L, Vera E, Cornacchia D (2015) Programming and reprogramming cellular age in the Era of induced pluripotency. Cell Stem Cell 16:591–600. https://doi.org/10.1016/j.stem.2015.05.004
Sullivan PF, Kendler KS, Neale MC (2003) Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry 60:1187–1192. https://doi.org/10.1001/archpsyc.60.12.1187
Sullivan S et al (2018) Quality control guidelines for clinical-grade human induced pluripotent stem cell lines. Regen Med 13:859–866. https://doi.org/10.2217/rme-2018-0095
Susser ES, Lin SP (1992) Schizophrenia after prenatal exposure to the Dutch Hunger Winter of 1944–1945. Arch Gen Psychiatry 49:983–988. https://doi.org/10.1001/archpsyc.1992.01820120071010
Susser E, Neugebauer R, Hoek HW, Brown AS, Lin S, Labovitz D, Gorman JM (1996) Schizophrenia after prenatal famine. Further evidence. Arch Gen Psychiatry 53:25–31. https://doi.org/10.1001/archpsyc.1996.01830010027005
Swanson JM et al (1998) Association of the dopamine receptor D4 (DRD4) gene with a refined phenotype of attention deficit hyperactivity disorder (ADHD): a family-based approach. Mol Psychiatry 3:38–41. https://doi.org/10.1038/sj.mp.4000354
Takahashi K, Yamanaka S (2006) Induction of Pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676. https://doi.org/10.1016/j.cell.2006.07.024
Takeuchi F et al (2011) Confirmation of ALDH2 as a Major locus of drinking behavior and of its variants regulating multiple metabolic phenotypes in a Japanese population. Circ J 75:911–918. https://doi.org/10.1253/circj.cj-10-0774
Tamiya H, Ouchi A, Chen R, Miyazawa S, Akimoto Y, Kaneda Y, Sora I (2018) Neurocognitive impairments are more severe in the binge-eating/purging anorexia nervosa subtype than in the restricting subtype. Front Psychiatry. https://doi.org/10.3389/fpsyt.2018.00138
Tawa EA, Hall SD, Lohoff FW (2016) Overview of the genetics of alcohol use disorder. Alcohol Alcohol 51:507–514. https://doi.org/10.1093/alcalc/agw046
Taylor S (2011) Etiology of obsessions and compulsions: a meta-analysis and narrative review of twin studies. Clin Psychol Rev 31:1361–1372. https://doi.org/10.1016/j.cpr.2011.09.008
Taylor S (2013) Molecular genetics of obsessive-compulsive disorder: a comprehensive meta-analysis of genetic association studies. Mol Psychiatry 18:799–805. https://doi.org/10.1038/mp.2012.76
Tian A, Muffat J, Li Y (2020) Studying human neurodevelopment and diseases using 3D brain organoids. J Neurosci 40:1186–1193. https://doi.org/10.1523/JNEUROSCI.0519-19.2019
Tiihonen J et al (2019) Neurobiological roots of psychopathy. Mol Psychiatry. https://doi.org/10.1038/s41380-019-0488-z
Tobe BT, Snyder EY, Nye JS (2011) Modeling complex neuropsychiatric disorders with human induced pluripotent stem cells. Curr Opin Pharmacol 11:521–527. https://doi.org/10.1016/j.coph.2011.05.007
Tobe BTD et al (2017) Probing the lithium-response pathway in hiPSCs implicates the phosphoregulatory set-point for a cytoskeletal modulator in bipolar pathogenesis. Proc Natl Acad Sci USA 114:E4462–E4471. https://doi.org/10.1073/pnas.1700111114
Tong J et al (2019) Generation of four iPSC lines from peripheral blood mononuclear cells (PBMCs) of an attention deficit hyperactivity disorder (ADHD) individual and a healthy sibling in an Australia-Caucasian family. Stem Cell Res 34:101353. https://doi.org/10.1016/j.scr.2018.11.014
Topol A, Zhu S, Tran N, Simone A, Fang G, Brennand KJ (2015) Altered WNT signaling in human induced pluripotent stem cell neural progenitor cells derived from four schizophrenia patients. Biol Psychiatry 78:e29–34. https://doi.org/10.1016/j.biopsych.2014.12.028
Topol A et al (2016) Dysregulation of miRNA-9 in a subset of schizophrenia patient-derived neural progenitor cells. Cell Rep 15:1024–1036. https://doi.org/10.1016/j.celrep.2016.03.090
Toyoshima M et al (2016) Analysis of induced pluripotent stem cells carrying 22q11.2 deletion. Trans Psychiatry 6:e934. https://doi.org/10.1038/tp.2016.206
Vaccarino FM et al (2011) Annual research review: the promise of stem cell research for neuropsychiatric disorders. J Child Psychol Psychiatry 52:504–516. https://doi.org/10.1111/j.1469-7610.2010.02348.x
Vadodaria KC et al (2019a) Serotonin-induced hyperactivity in SSRI-resistant major depressive disorder patient-derived neurons. Mol Psychiatry 24:795–807. https://doi.org/10.1038/s41380-019-0363-y
Vadodaria KC et al (2019b) Altered serotonergic circuitry in SSRI-resistant major depressive disorder patient-derived neurons. Mol Psychiatry 24:808–818. https://doi.org/10.1038/s41380-019-0377-5
Verhulst B, Neale MC, Kendler KS (2015) The heritability of alcohol use disorders: a meta-analysis of twin and adoption studies. Psychol Med 45:1061–1072. https://doi.org/10.1017/S0033291714002165
Viswanath B et al (2015) Cellular models to study bipolar disorder: a systematic review. J Affect Disord 184:36–50. https://doi.org/10.1016/j.jad.2015.05.037
Vizlin-Hodzic D et al (2017) Early onset of inflammation during ontogeny of bipolar disorder: the NLRP2 inflammasome gene distinctly differentiates between patients and healthy controls in the transition between iPS cell and neural stem cell stages. Transl Psychiatry 7:e1010. https://doi.org/10.1038/tp.2016.284
Wang K et al (2011) A genome-wide association study on common SNPs and rare CNVs in anorexia nervosa. Mol Psychiatry 16:949–959. https://doi.org/10.1038/mp.2010.107
Wang P, Mokhtari R, Pedrosa E, Kirschenbaum M, Bayrak C, Zheng D, Lachman HM (2017) CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol Autism 8:11. https://doi.org/10.1186/s13229-017-0124-1
Wang Y, Fen Q, Yu H, Qiu H, Ma X, Lei Y, Zhao J (2018a) Establishment of TUSMi005-A, an induced pluripotent stem cell (iPSC) line from a 32-year old Chinese Han patient with Bipolar Disorder (BD). Stem Cell Res 33:65–68. https://doi.org/10.1016/j.scr.2018.10.014
Wang Y, Kang C, Yu H, Fen J, Ma X, Lei Y, Zhao J (2018b) Establishment of TUSMi004-A, an induced pluripotent stem cell (iPSC) line from a 32-year old Chinese Han patient with Obsessive-Compulsive Disorder (OCD). Stem Cell Res 32:83–86. https://doi.org/10.1016/j.scr.2018.08.020
Watmuff B, Berkovitch SS, Huang JH, Iaconelli J, Toffel S, Karmacharya R (2016) Disease signatures for schizophrenia and bipolar disorder using patient-derived induced pluripotent stem cells. Mol Cell Neurosci 73:96–103. https://doi.org/10.1016/j.mcn.2016.01.003
Watson HJ, Bulik CM (2013) Update on the treatment of anorexia nervosa: review of clinical trials, practice guidelines and emerging interventions. Psychol Med 43:2477–2500. https://doi.org/10.1017/S0033291712002620
Weber H et al (2011) Cross-disorder analysis of bipolar risk genes: further evidence of DGKH as a risk gene for bipolar disorder, but also unipolar depression and adult ADHD. Neuropsychopharmacology 36:2076–2085. https://doi.org/10.1038/npp.2011.98
Weber H et al (2015) On the role of NOS1 ex1f-VNTR in ADHD-allelic, subgroup, and meta-analysis. Am J Med Genet B Neuropsychiatr Genet 168:445–458. https://doi.org/10.1002/ajmg.b.32326
Wen Z et al (2014) Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515:414–418. https://doi.org/10.1038/nature13716
Willmann CA et al (2013) To clone or not to clone? Induced pluripotent stem cells can be generated in bulk culture. PLoS One 8:e65324. https://doi.org/10.1371/journal.pone.0065324
Wittchen HU, Hofler M, Meister W (2001) Prevalence and recognition of depressive syndromes in German primary care settings: poorly recognized and treated? Int Clin Psychopharmacol 16:121–135
Wittchen HU et al (2011) The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur Neuropsychopharmacol 21:655–679. https://doi.org/10.1016/j.euroneuro.2011.07.018
Wray NR, Goddard ME, Visscher PM (2007) Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res 17:1520–1528. https://doi.org/10.1101/gr.6665407
Wu YY, Chiu FL, Yeh CS, Kuo HC (2019) Opportunities and challenges for the use of induced pluripotent stem cells in modelling neurodegenerative disease. Open Biol 9:180177. https://doi.org/10.1098/rsob.180177
Xie N, Zhou Y, Sun Q, Tang B (2018) Novel epigenetic techniques provided by the CRISPR/Cas9 System. Stem Cells Int. https://doi.org/10.1155/2018/7834175
Yang W, Liu Y, Slovik KJ, Wu JC, Duncan SA, Rader DJ, Morrisey EE (2015) Generation of iPSCs as a pooled culture using magnetic activated cell sorting of newly reprogrammed cells. PLoS ONE. https://doi.org/10.1371/journal.pone.0134995
Ye F et al (2017) DISC1 regulates neurogenesis via modulating kinetochore attachment of Ndel1/Nde1 during mitosis. Neuron 96:1041–1054.e1045. https://doi.org/10.1016/j.neuron.2017.10.010
Yehuda R, Daskalakis NP, Bierer LM, Bader HN, Klengel T, Holsboer F, Binder EB (2016) Holocaust exposure induced intergenerational effects on FKBP5 methylation. Biol Psychiatry 80:372–380. https://doi.org/10.1016/j.biopsych.2015.08.005
Yoo SW, Motari MG, Susuki K, Prendergast J, Mountney A, Hurtado A, Schnaar RL (2015) Sialylation regulates brain structure and function. FASEB J 29:3040–3053. https://doi.org/10.1096/fj.15-270983
Yoon K-J et al (2014) Modeling a genetic risk for schizophrenia in iPSCs and mice reveals neural stem cell deficits associated with adherens junctions and polarity. Cell Stem Cell 15:79–91. https://doi.org/10.1016/j.stem.2014.05.003
Zhao D et al (2015) MicroRNA profiling of neurons generated using induced pluripotent stem cells derived from patients with schizophrenia and schizoaffective disorder, and 22q11.2 Del. PLoS ONE. https://doi.org/10.1371/journal.pone.0132387
Zhou T et al (2011) Generation of induced pluripotent stem cells from urine. J Am Soc Nephrol 22:1221–1228. https://doi.org/10.1681/ASN.2011010106
Acknowledgements
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SKS has received author’s and speaker`s honoraria from Takeda/Shire and Medice Arzneimittel Pütter GmbH. All other authors have no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
McNeill, R.V., Ziegler, G.C., Radtke, F. et al. Mental health dished up—the use of iPSC models in neuropsychiatric research. J Neural Transm 127, 1547–1568 (2020). https://doi.org/10.1007/s00702-020-02197-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-020-02197-9