
Abstract
Common bunt (CB), caused by Tilletia caries and T. foetida, and dwarf bunt (DB), caused by T. controversa, are particularly destructive diseases of wheat grown under organic (low-input) production conditions and negatively affect both grain yield and quality. A total of 16 race specific bunt resistance genes have been proposed to date. Thereof, only Bt9 and Bt10 have been mapped so far. A mapping and two validation populations comprising 176 recombinant inbred lines were evaluated for CB and DB in artificially inoculated field trials. The mapping population was derived from the cross of the Bt12 carrier PI119333 and the susceptible cultivar ‘Rainer’. The population was genotyped with the Illumina 15 K SNP chip and the major QTL QBt.ifa-7DS representing Bt12 was identified on chromosome 7DS, explaining 39% and 14% of the phenotypic variation for CB and DB resistance, respectively. Selected SNP markers were turned into Kompetitive Allele-Specific (KASP) markers and used to validate Bt12 in two independent validation populations. These markers can be used for introgressing Bt12 into regionally adapted elite breeding material.
Similar content being viewed by others


Introduction
Common bunt (CB) and dwarf bunt (DB) are two destructive fungal diseases of wheat (Triticum aestivum L.) occurring worldwide (Goates 1996). While CB is caused by Tilletia caries (syn. T. tritici) and Tilletia foetida (syn. T. laevis), DB is caused by Tilletia controversa. Infection with CB and/or DB leads to the formation of dark bunt sori that replace kernels and are composed almost entirely of Tilletia teliospores. No specific mycotoxins have been identified until now but the high levels of trimethylamine contained in bunt sori cause the typical smell of rotten fish in diseased crops (Chen et al. 2016; Matanguihan et al. 2011). Both diseases result in considerable yield as well as quality losses in grain contaminated with bunt sori at levels as low as 0.01% (Laroche et al. 2000; Menzies et al. 2006). With the introduction of effective fungicides around 50 years ago, most wheat breeding programs shifted their priorities away from the selection for bunt resistance. An increase in organic farming over the last two decades gave rise to a renewed interest in bunt diseases of wheat (Matanguihan et al. 2011). Growing resistant cultivars is the only effective measure available to control bunt diseases of wheat under organic production conditions, and helps to minimize negative environmental impact while maximizing economic efficiency for conventional agriculture (Matanguihan et al. 2011).
We currently face a severe lack of knowledge regarding the genetic basis of bunt resistance in wheat. The fungi causing CB and DB are closely related and resistance to both diseases is controlled by shared genes in wheat (Goates 1996, 2012). For this pathosystem a series of major resistance genes that follow the classic gene-for-gene concept of pathogen–host interaction has been described (Goates 1996, 2012; Metzger and Hoffmann 1978). A total of 16 race specific bunt resistance genes, Bt1 to Bt15 and BtP have been proposed to date; the current set of differential lines used for Tilletia race tests makes use of Bt1 to Bt13 and BtP (Goates 2012). So far, only two Bt genes have been mapped to specific chromosomal regions using genetic mapping in bi-parental populations. Bt10 was mapped to the short arm of chromosome 6D (Menzies et al. 2006) and Bt9 was mapped by Steffan et al. (2017) to the long arm of chromosome 6D. Both genes were validated in independent populations (Singh et al. 2016; Wang et al. 2019), and DNA markers linked to the genes have been published for the purpose of marker-assisted selection (MAS) of Bt9 (Steffan et al. 2017; Wang et al. 2019) and Bt10 (Laroche et al. 2000; Menzies et al. 2006). Bunt resistance is mediated partly by non-race specific quantitative trait loci (QTL), and 24 QTL for bunt resistance have been identified to date by QTL mapping in wheat. More specifically, multiple major CB QTL were mapped to chromosome 1B in several independent populations (Dumalasova et al. 2012; Fofana et al. 2008; Singh et al. 2016; Wang et al. 2009; Zou et al. 2017). Additional minor QTL for CB resistance were identified on 1D, 2A, 3D (Bokore et al. 2017), 3A (Zou et al. 2017), 4B (Singh et al. 2016), 4D (Singh et al. 2016), 5A (Bokore et al. 2017), 5B (Dumalasova et al. 2012; Singh et al. 2016), 7A (Bokore et al. 2017; Dumalasova et al. 2012; Fofana et al. 2008), 7B (Dumalasova et al. 2012; Knox et al. 2013) and 7DL (Singh et al. 2016). A major QTL for DB resistance was identified by Chen et al. (2016) on the short arm of chromosome 7D. Minor DB QTL were identified on 1A (Chen et al. 2016), 2B (Chen et al. 2016), and 7A (Wang et al. 2019). Association mapping studies identified several significant marker traits association for CB. While Bhatta et al. (2018) detected 15 SNPs associated with CB resistance on chromosome 1B, 2A, 2B, 3D, 4A, 7A, and 7B in a set of 125 synthetic hexaploid wheats, Mourad et al. (2018) reported 123 SNPs significantly associated with CB resistance on 14 chromosomes in a diversity panel of 330 Nebraska winter wheat genotypes.
Although some variation for bunt resistance is present in elite material and modern cultivars, gene bank accessions are an important source for broadening genetic resistance and to control bunt diseases of wheat in the long-term (Goates and Bockelman 2012).
The genebank accession PI119333 has been described as a carrier of the bunt resistance gene Bt12 and is accordingly the differential cultivar that is used in race identification studies (Goates and Bockelman 2012). Bt12 is highly effective against most known races of bunt and an important source for breeding on its own as well as in combination with other bunt resistance factors.
Using a RIL mapping population derived from a cross of the Bt12 differential line PI119333 to the bunt susceptible cultivar Rainer, and two independent validation populations descending from crosses of PI119333 with the susceptible cultivars Midas and Pannonikus, the main objectives of this study were to 1) map the chromosomal position of Bt12, 2) validate the mapping results in an independent set of validation lines and 3) identify and validate KASP markers suitable for conducting MAS in breeding material segregating for Bt12.
Material and methods
Plant material
Mapping population
A mapping population comprising 80 F5:7 recombinant inbred lines (RILs) was developed by crossing PI119333 to Rainer, which is hereafter referred to as mapping population MP-PR. PI119333 is an awned Turkish winter wheat landrace collected in 1937. PI119333 carries the bunt resistance gene Bt12, which is highly effective against most CB and DB races (Goates 2012). Rainer is an awnless winter wheat variety released by Saatzucht Donau GesmbH & CoKG (Austria) in 2006 and possesses well adapted agronomic traits for cultivation in Austria but is highly susceptibility to both CB and DB.
Validation populations
Aside from the mapping population, two independent populations were used for validating the CB and DB resistance QTL that were identified in this study. These two validation populations consisted of 56 and 40 F5:7 RILs derived from crosses of PI119333 to Midas and PI119333 to Pannonikus, respectively, and are hereafter referred to as VP-PM and VP-PP. Midas and Pannonikus are both awned and locally adapted Austrian winter wheat varieties released by Saatzucht Donau GmbH & CoKG (Austria) in 2008 and highly susceptible to CB and DB.
Bunt differential lines
The full bunt differential set was used to monitor the virulence spectrum of the T. caries and T. controversa races applied for artificial inoculation of the field trials. The bunt differential set was kindly provided by Blair Goates and comprises 14 winter wheat lines, each carrying one of the 14 bunt resistance genes Bt1 to Bt13 and BtP (Goates 2012) with PI119333 being part of this set as a carrier of Bt12. In accordance to Goates (2012) the reaction of the spore mixtures were considered avirulent to a specific bunt differential line when 10% or less of the spikes were diseased, and virulent if the disease exceeded 10%.
Field experiments and disease evaluations
The mapping population MP-PR was tested for CB resistance in four artificially inoculated field trials in 2014, 2015, 2016 and 2018 (CB14, CB15, CB16, and CB18) and for DB resistance in one artificially inoculated field trial in 2018 (DB18). The validation populations VP-PM and VP-PP were evaluated for CB and DB resistance in field trials CB18 and DB18. In all experiments, RILs of each population were grown alongside the parental and full set of bunt differential lines.
Phenotypic evaluations for CB resistance were conducted at the experimental station of the Department of Agrobiotechnology in Tulln, Austria (48°19′08′’N 16°04′16′’E, elevation: 177 m). All tested genotypes were seed inoculated with a race mix of T. caries teliospores that represented the CB races which is currently prevalent in Eastern and Western Austria. The isolates were collected at three different locations in Austria from naturally infected wheat spikes. CB spores for seed inoculation were harvested from previous season`s infected heads of a diverse set of susceptible genotypes and stored under dry conditions at room temperature. Seeds were inoculated according to the protocol outlined by Goates (1996) with a concentration of 0.75 g teliospores per 100 g seeds. All field trials were laid out as randomized complete block designs with two replicates. Plot size was 0.75 m2 consisting of two 1.5 m rows spaced 25 cm apart, with approximately 60 plants in each row. CB nurseries were established in early November by sowing 6 g of inoculated spore coated seeds per plot.
DB resistance was evaluated at the Utah State University Research Farm in Logan, Utah, USA (41°45′46.46"N 111°48′54.98"W, elevation: 1400 m), where sowing took place in the beginning of October. Each genotype was tested in a 1 m single row at a seeding rate of 2 g per row. According to the protocol developed by Goates (1996), the disease nursery was inoculated after seedling emergence prior to snow cover in early November by spraying approximately 100 ml per 1 m plot of an aqueous suspension containing 1.3 g of a T. controversa teliospore race mix that represented the virulence spectrum of races found in the United States (Chen et al. 2016). The field trial was conducted as randomized complete blocks with two replicates.
CB and DB incidence were determined as the percentage of infected spikes within a given plot at plant maturity. A spike was considered infected when it contained at least one bunted spikelet. In addition to CB and DB incidence, plant height was recorded in CB14, CB15 and CB16 at plant maturity in centimetres excluding awns. Lodging was scored visually on a scale from 1 (no lodging) to 9 (complete lodging) in CB14, CB15 and CB16. Date of heading in days from January 1st was recorded for CB15 and CB16. Powdery mildew (Blumeria graminis f. sp. tritici DC.) severity was evaluated in two years of natural occurrence in CB15 and CB16 on a scale from 1 (resistant) to 9 (highly susceptible), with the average of two time points being taken for further analyses. Finally, stripe rust (Puccinia striiformis f. sp. tritici) severity was determined visually in two years of natural occurrence in CB14 and CB16, and estimated as the average percentage of diseased leaf area across three time points.
Phenotypic analysis
For each genotype tested and trait investigated, best linear unbiased estimates (BLUEs) of each individual environment were first calculated with a linear mixed model of the form:
where Pik denotes the observed phenotypic value, μ the population mean, Gi the effect of the ith genotype, Rk the effect of the kth replicate and eik the residual effect. The model was subsequently extended for the across environment analysis to:
where Pijk designates the observed phenotypic value, μ the population mean, Gi the effect of the ith genotype, Ej the effect of the jth environment, Ej(Rk) the effect of the kth replicate within the jth environment, Gi × Ej the ijth effect of the genotype-by-environment interaction and eijk the residual effect. The genotype effect was treated as fixed to derive BLUEs and random to estimate the genetic variance, while all other effects were modelled as random in both models. Fixed and random effects of the models were tested one by one using the Wald F-test. Broad-sense heritability (H2) was computed as suggested by Piepho and Möhring (2007):
where σ2G designates the genetic variance and MVD the mean variance of a difference of the BLUEs. All phenotypic analyses were conducted using the statistical package ASReml (Gilmour et al. 2015) for the R programming environment (R development core team 2016).
Molecular marker data
Genomic DNA was extracted from fresh leaves of 10 pooled plants of each line using a modified CTAB method (Saghai-Maroof et al. 1984). High density genotyping of all individuals was performed using the Illumina Infinium 15 K wheat SNP array by Trait Genetics GmbH (Gatersleben, Germany, https://www.traitgenetics.de) comprising 12,907 gene-associated SNPs. Marker data were quality checked prior to linkage map construction and QTL mapping by discarding markers that showed significant segregation distortion (p < 0.001) and more than 20% missing data points. Additionally, RILs with more than 20% missing marker data points were removed, while RILs that had more than 95% of marker allele calls in common were combined.
Linkage map construction
Linkage groups were constructed using the statistical package ASMap v0.4 (Taylor and Butler 2017) for the R programming environment (R development core team 2016). The objective function was set to minimize the sum of recombination events between markers for map construction. Firstly, robust linkage groups where constructed using a stringent threshold (p < 1 × 10−8) and assigned to particular wheat chromosomes based on the hexaploid wheat consensus map by Wang et al. (2014). Within linkage groups, markers were reordered at a less stringent threshold (p < 1 × 10−6), and map distances were calculated with the Kosambi mapping function. Each linkage group was oriented with respect to the short (S) and long (L) chromosome arms based on the information of the hexaploid wheat consensus map (Wang et al. 2014), and linkage groups were named according to the wheat genome nomenclature followed by the chromosome number. Linkage maps, including graphical representation of linkage groups and QTL positions, were drawn with MapChart 2.2. (Voorrips 2002).
QTL analysis
QTL mapping for bunt resistance in the population MP-PR was performed with the R package R/qtl (Broman et al. 2003) using BLUEs calculated for each individual CB and DB environment as well as BLUEs from the across environment analysis. Missing genotypic information was imputed using the multiple imputation method of Sen and Churchill (2001). In a first step, the main effect QTL were detected by composite interval mapping (CIM) using the Haley–Knott regression method employing a window size of 10 cM and choosing the number of marker covariates by forward selection. LOD thresholds at α = 0.05 and α = 0.10 significance levels were determined for each trait and experiment based on a permutation test with 1000 replicates (Churchill and Doerge 1994), which were used to declare significance of the marker-trait associations. In a second step, the significant QTL were fit in a multiple QTL model using the addqtl and addint functions of R/qtl to test for the presence of further QTL and QTL-by-QTL interactions. LOD scores, proportion of phenotypic variance explained and additive effects of each QTL were accordingly derived from the resulting final multiple QTL model. Confidence intervals were determined for each QTL as the 1.5-LOD drop off support interval following van Ooijen (1992). Furthermore, QTL analyses were performed using the BLUEs calculated for individual environments and across environments for all additional traits according to the above described algorithm. CB and DB incidence of RILs grouped by their QTL combinations were compared using the Tukey HSD test at p < 0.05.
KASP marker development
Sequence information of Kompetitive Allele-Specific PCR (KASP) assays for breeder-friendly and efficient selection of favourable allelic variants of the mapped resistance loci were derived from the publicly available data set hosted on the Cereals DB website (www.cerealsdb.uk.net; Wilkinson et al. 2016). KASP assays for SNP markers were selected based on their genetic locations on the linkage map obtained from marker data of the Illumina Infinium 15 K wheat SNP array and on the physical Mbp positions on the Chinese Spring reference sequence (IWGSC RefSeq v1.0; (Appels et al. 2018). KASP assays were screened for discrimination between the alleles of the resistance donor PI119333 and the recipient parents Rainer, Midas and Pannonikus and assessed for co-segregation with the corresponding SNP genotype calls from the wheat SNP array in mapping population MP-PR. Moreover, the full set of winter wheat bunt differential lines, and a diverse set of 52 genotypes (European and international wheat cultivars, gene bank accessions and experimental lines) were screened with these KASP assays to verify their applicability for MAS and gene pyramiding (Table S1).
QTL validation
KASP markers that mapped within the QTL support intervals were selected to characterize the RILs of the validation populations VP-PM and VP-PP. Single marker regression analysis was applied to test for association between KASP markers and CB and DB incidence means from season 2018 as described by Broman and Sen (2009). Heterogeneous genotypes and genotypes with ambiguous marker scores were excluded from the analysis. Regression analyses were performed for each validation population separately as well as across both validation populations. CB and DB incidence of RILs grouped by their QTL combinations were compared using the Tukey HSD test at p < 0.05.
Results
Race spectrum
Based on the reaction of the bunt differential lines to the employed T. caries inoculum, the same virulence pattern was evident in all four CB environments. The inoculum was virulent towards the Bt2 and Bt7 differential lines with overall means of 57% and 63% bunt incidence, respectively. The T. controversa race mix used for inoculation of the DB field trial was virulent to Bt3, Bt5 and Bt9 (20–35% bunt incidence) and highly virulent to Bt1, Bt2, Bt4, Bt6 and Bt7 (> 80% bunt incidence). Notably, virulence to the gene of interest, Bt12, was neither observed in the CB nor the DB inoculated environments (Table S2).
Trait variation and correlation
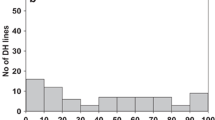
Bunt symptoms were observed in all investigated populations and environments. PI119333 was completely resistant against CB and DB, whereas Rainer showed high susceptibility with a mean score of 74% for CB and 90% for DB incidence (Table 1). Frequency distributions of the 80 RILs of MP-PR for CB and DB incidence were similarly shaped, i.e. positively skewed, in all test environments (Fig. 1). Disease ratings ranged from 0–84% and 0–90% in the CB and DB environments and population means for bunt incidence were always significantly lower than the mid-parental values. Variance component analysis revealed genotype as the main source of variation for CB and DB traits with σG2 explaining 79% and 91% of total phenotypic variation (PV), respectively (Table S3). High values of σG2 as compared to σGE2 resulted in a high broad-sense heritability coefficient of H2 = 0.97 for the CB across environment analysis, indicating a strong consistency of disease resistance across environments. Accordingly, phenotypic correlations between individual CB environments were highly significant (r = 0.71–0.95). CB incidences were highly correlated with DB incidence (r = 0.77–0.83) (Table S4). Averaged across environments, PI119333 had a slightly later heading date, was more than 30 cm taller and prone to lodging as opposed to Rainer. PI119333 was furthermore highly susceptible to stripe rust, while Rainer was barely affected (Table 1). Significant genotypic variation was observed for all the additional analysed traits, i.e. plant height, lodging, heading date, powdery mildew and stripe rust severity (Table S3). No significant associations were detected between bunt incidence and any of these traits (Table S5).

Scatterplot of overall means for common bunt incidence against dwarf bunt incidence with marginal histograms of their frequency distributions in mapping population MP-PR
QTL analysis
Linkage map
After quality check, 79 of originally 80 RILs of mapping population MP-PR and 5005 of 12907 SNP markers from the wheat 15 K SNP chip were available for linkage map construction. The resulting genetic linkage map comprised 42 linkage groups (LGs) representing all 21 wheat chromosomes (Table S6). The whole linkage map covered 2713 cM, with an average chromosome length of 65 cM, an average marker distance of 2 cM and a maximum spacing of 23 cM between markers. 13 SNP markers could not be attributed to specific chromosomes.
QTL analysis for bunt resistance
QTL analysis identified a major effect QTL on the short arm of chromosome 7D and a minor effect QTL on chromosome 4B that were designated as QBt.ifa-7DS and QBt.ifa-4B (Table 2). For both QTL, the resistance improving allele was derived from PI119333. The large effect QTL QBt.ifa-7DS mapped to LG 7D.1, which comprised 13 completely linked, co-segregating markers and most likely relates to the gene Bt12. These 13 markers correspond to a physical size of ~ 4.3 Mbp ranging from Mbp positions 6.47 to 10.84 (Fig. 2). Aiming to link the LG 7D.1 of the QBt.ifa-7DS|Bt12 cluster with the 55 Mbp more proximal LG 7D.2 we selected 26 KASP assays derived from SNP markers from the Infinium 90 K wheat SNP chip that were located within this interval. The attempt to connect these two LGs for a better resolution of chromosome 7DS failed, since all tested markers were monomorphic between our parental lines (data not shown). QBt.ifa-7DS|Bt12 was consistently detected in all CB environments, where it explained 17–39% of PV for individual environments and 39% of PV for the overall mean across CB environments. Moreover, QBt.ifa-7DS|Bt12 was significantly associated with DB incidence accounting for 14% of PV. The smaller effect QTL, QBt.ifa-4B was only significant in experiments CB15 and CB16 and explained 11% of PV (Table 2).

Comparison of position of QTL and markers on wheat chromosome 7DS. a Marker cluster of linkage groups LG 7D.1 and the closest more proximal markers of linkage group LG 7D.2, gap between linkage groups is symbolized by dashed lines bordering the chromosome bar. b Physical Mbp positions of markers on LG 7D.1, LG 7D.2 and on the 7DS map published by Chen et al. (2016) c 7DS map reported by Chen et al. (2016). Only the segment of the more distal 7DS chromosome arm is shown. Bar segments highlighted in yellow refer to the QTL interval of QBt.ifa-7DS and red bar segments refer to QDB.ui-7DS
Pairwise group comparison of means over all experiments showed that RILs without resistance QTL had the highest disease incidence for CB and for DB and were significantly different from all other groups, while RILs with resistance alleles at QBt.ifa-7D were highly and equally resistant as RILs that carried the favourable allele at both QTL. Tukey’s tests revealed a significant resistance improvement for RILs having the positive alleles only at QBt.ifa-4B, although its effect was not as strong as the effect of QBt.ifa-7D (Fig. 3). This is in contrast to results of the QTL analysis, where the QBt.ifa-4B did not surpass the significance threshold possibly due to the relatively small size of the mapping population.

Box plot distributions of RILs according to their QTL combinations at QBt-ifa-7DS|Bt12 and QBt.ifa-4B for common (white boxes) and dwarf bunt incidence (gray boxes) in (a) mapping population MP-PR and (b) validation population VP-PM&PP. Medians are indicated by solid bold lines, means by crosses, open circles represent outliers. For each group, the number of lines, mean values and standard deviations (SD) of bunt incidence are provided. Groups of different letters are significantly different (p < 0.05) based on Tukey HSD test. Boxplots of MP-PR are based on means over all CB experiments and dwarf bunt experiment DB18, boxplots of VP-PM&VP-PP are derived from experiment CB18 and DB18
QTL analysis for additional traits
One QTL on 7B associated with plant height contributed 22% to PV. Three QTL were associated with lodging on 2D, 4A and 7D (LG 7D.2) and contributed 40%, 12% and 14% to PV, respectively. In all cases, the PI119333 allele increased plant height and lodging.
Minor QTL for heading date mapped to 2A, 3A, 3B, 4B and 7B and contributed 7–16% of PV. In the majority of cases, alleles decreasing heading date were contributed by Rainer. One major and two minor QTL for stripe rust severity were detected on 2A, 1A and 6A, responsible for 61%, 11% and 7% of PV, respectively. The favourable stripe rust resistance increasing allele was contributed by Rainer in all cases (Table S6). No associations between the bunt resistance QTL on QBt.ifa-7DS|Bt12 and plant height, lodging, heading date or stripe rust severity were detected.
KASP markers and QTL validation
Seven and 12 KASP markers assigned to the QBt.ifa-7DS|Bt12 and QBt.ifa-4B QTL regions, respectively, were screened for their ability to discriminate between the alleles of the resistance donor parent PI119333 and recipient parents, the bunt differential lines and a diverse set of 52 genotypes (Table S8).
Four KASP markers at QBt.ifa-7DS|Bt12 were unique for PI119333 (Bt12 carrier) among the bunt differential lines, by contrast selected KASP markers for QBt.ifa-4B were not informative to differentiate between the allele of the resistance donor PI119333 and the alleles for most or all of the bunt differential lines (Table S8). The allele calls for the selected KASP markers were in 72–94% (for QBt.ifa-7DS|Bt12) and in 27–88% (for QBt.ifa-4B) unique for PI11933 within the entire evaluated wheat panel (Table S8). The KASP assays for QBt.ifa-7DS|Bt12 at RAC875_c99892_81 (IWB61302) and Ra_c11887_1826 (IWB50978) were the most discriminating ones, as among the 68 tested genotypes only 4 had the same allele call as the resistant donor PI11933 (Table S8).
Seven and 4 KASP markers unambiguously discriminated between the resistance donor and recipients for QBt.ifa-7DS|Bt12 and QBt.ifa-4B, respectively, and were therefore used for genotyping an independent set of 97 lines of validation populations VP-PM (57 RILs) and VP-PP (40 RILs). VP-PM and VP-PP were evaluated for CB incidence in CB18, and subsets of VP-PM and VP-PP (20 RILs each) were additionally evaluated for DB incidence in DB18. The KASP markers at QBt.ifa-7DS|Bt12 explained 29%, 23%, and 27% of PV for CB in VP-PM, VP-PP and the full set of 97 validation lines (Table 3). For DB QBt.ifa-7DS|Bt12 explained 24%, 46%, and 33% of PV for VP-PM, VP-PP and the full set of validation lines. In accordance with these results, highly significant differences in CB and DB incidence means between groups of lines carrying susceptible and resistant variants of QBt.ifa-7DS|Bt12 were observed (Fig. 3). The effect of the minor QTL QBt.ifa-4B was in contrast neither evident in experiments CB18 nor DB18 (Table 3, Fig. 3).
Discussion
The limited knowledge regarding the genetic basis of bunt resistance impedes the application of MAS for the rapid development and adoption of bunt resistant cultivars, which are needed for organic and low input agricultural production of wheat. In this study, we dissected the genetic architecture of bunt differential line PI119333, carrier and differential line of the bunt resistance gene Bt12. Bt12 confers resistance to most CB and DB races that are currently known (Goates and Bockelman 2012), making it a favourable target for introgression into elite breeding material.
Quantitative variation was evident for CB and DB incidence in all trials, which generally followed a positively skewed continuous distribution, with a large proportion of lines in the low or not infected groups (Fig. 1). The high level of bunt incidence observed for the susceptible cultivars Rainer, Midas and Pannonikus as well as some RILs of the tested populations indicated a generally high disease pressure (Table 1). PI119333 was confirmed as resistant to CB and DB, whereas Rainer, Midas and Pannonikus were among the most susceptible genotypes. The skewed nature of the distribution of the RIL progeny with a high proportion of lines expressing a high though incomplete resistance to CB and DB suggested that besides Bt12 inherited from PI19333 additional minor resistance factors might be segregating in the mapping population. Similar distributions for bunt resistance have been commonly observed in bunt studies, revealing either the presence of a single major gene (Steffan et al. 2017; Wang et al. 2009) or a major bunt resistance QTL acting in combination with other minor resistance factors (Chen et al. 2016; Fofana et al. 2008; Singh et al. 2016; Wang et al. 2019).
The fungi causing CB and DB are highly related, thus it can be hypothesized that host plant resistance for both diseases is controlled by the same (shared) genes in wheat (Goates 2012). However, so far published studies focused either on response to CB or DB and did not evaluate the populations for both diseases in parallel. We mapped the major bunt resistance QTL QBt.ifa-7DS which refers to Bt12, close to the distal end of chromosome arm 7DS. QBt.ifa-7DS|Bt12 was highly significant in all experiments and was associated with both, CB and DB resistance, supporting the assumption that common and dwarf bunt are largely under the same genetic control. The effect of Bt12 on CB incidence varied to some extent among experiments, being larger in trials CB14, CB15 and CB16 compared to CB18 presumably due to a lower infection level for the latter experiment, resulting in lower total variation. Variations regarding QTL effects of major bunt resistance genes among test environments were also observed in previous QTL studies (Chen et al. 2016; Singh et al. 2016; Steffan et al. 2017; Wang et al. 2019). QTL for bunt resistance on chromosome 7D were already reported by Chen et al. (2016) and Singh et al. (2016). Chen et al. (2016) identified a major QTL conferring DB resistance in the breeding line Idaho444, whereas Singh et al. (2016) identified a minor QTL for CB resistance derived from cultivar Carberry. QBt.ifa-7DS|Bt12 appears different from the QTL reported by Singh et al. (2016) as the Carberry 7D QTL explained a mere 6% of PV while the effect of QBt.ifa-7DS|Bt12 under CB conditions was a major one conferring almost complete resistance. Furthermore, according map comparison with the high-density linkage map of the 7D chromosome (Ishikawa et al. 2018) the Carberry 7D QTL is located on the long arm and thus unlinked to QBt.ifa-7DS|Bt12. QBt.ifa-7DS|Bt12 in our study mapped to the distal end of the 7D short arm between Mbp 6.5 and 10.8 to a cluster of 13 co-segregating SNP markers. The Idaho444 QTL Q.DB.ui-7DS mapped to the marker wPt-2565 (X116197) at Mbp 5.5 Mbp (IWGSC refSeq V1.0), hence, peak markers of QBt.ifa-7DS|Bt12 and Q.DB.ui-7DS are separated by ~ 1 Mbp only. However, due to absence of marker polymorphism over a large physical distance no precise information on the true location of these QTL can be given. Similar to Chen et al. (2016), we observed an extremely low marker coverage for chromosome 7DS (Table S6). The genetic map of the 7D chromosome reported by Chen et al. (2016)—just as the map of our mapping population—had a large gap of more than 50 cM between Q.DB.ui-7DS and the nearest more proximal marker (Fig. 2). Notably, among a series of markers developed by Prof. J. Chen`s research group at the University of Idaho all markers distal to wPt-2565(Q.DB.ui-7DS) shared the same allele for PI119333 and Idaho444 (Jianli Chen, personal communication). Unfortunately, all markers informative for the Q.DB.ui-7DS were monomorphic for PI119333 and the three recipient parents in our study (data not shown). For given reasons, it remained so far impossible to unambiguously elucidate, whether or not QBt.ifa-7DS|Bt12 and Q.DB.ui-7DS refer to the same resistance gene, namely Bt12. The phenotypic data suggests that QBt.ifa-7DS|Bt12 is tightly but not completely linked with the Bt12 resistance gene, as six RILs with the marker haplotype of the resistance donor PI119333 displayed a susceptible phenotype (> 10% incidence scoring) and likewise six lines with low disease severity possessed the marker haplotype from Rainer. This could possible explain the lower effect size of QBt.ifa-7DS|Bt12 under DB conditions in comparison with the Idaho444 QTL Q.DB.ui-7DS (Chen et al. 2016). More precise fine mapping the position of QBt.ifa-7DS|Bt12 would require a new mapping population that displays a higher degree of marker polymorphism on the short arm of chromosome 7D. Such a population may be difficult to find, because low polymorphism in this region seems a general feature, and even the populations used in this study descending from crosses of a landrace (PI119333) and three modern cultivars showed this phenomenon.
Seven KASP markers were developed and proofed suitable for validating QBt.ifa-7DS|Bt12 in two validation populations. These KASP markers can assist in transferring Bt12 into regionally adapted breeding material. Among the tested KASP markers, the markers for IWB61302 and for IWB61302 appeared to particularly informative, as the SNP haplotypes of these markers were unique for PI11933 among all bunt differential lines and almost unique among the tested genotypes (Table S8). Although a range of markers are now available that are useful for tracking Bt12 in diverse wheat germplasm, further work is necessary to fine-map and ultimately clone Bt12 in order to obtain more tightly linked or even diagnostic markers, which would allow the unambiguous detection of Bt12 resistance donors.
In addition to QBt.ifa-7DS|Bt12, a minor effect bunt resistance QTL on chromosome 4B (QBt.ifa-4B) was significantly associated with CB in two experiments (Table 2; Fig. 3). However, its effect was not verified in the two independent validation experiments (Table 3; Fig. 3). Interactions of minor resistance QTL with the environment and the genetic background is frequently observed in common and dwarf bunt and was also reported in similar QTL mapping studies. Examples for major and minor genes controlling bunt resistance in a combined action are numerous (Chen et al. 2016; Dumalasova and Bartos 2016; Fofana et al. 2008; Knox et al. 2013; Singh et al. 2016), and although we did not identify an epistatic interaction between QBt.ifa-7DS|Bt12 and QBt.ifa-4B their importance has been highlighted in previous studies (Chen et al. 2016; Knox et al. 2013; Singh et al. 2016).
The bunt differential lines are assumed to be monogenic for a single bunt resistance gene and were used to postulate presence or absence of specific resistance genes in a particular genotype (Goates 1996; Metzger and Hoffmann 1978). Bunt differential line PI119333 is currently used to identify carriers of Bt12 based on concordant reactions to bunt races and resulting virulence patterns (Goates and Bockelman 2012). The observed frequency distribution of RILs for bunt incidence in our mapping population, and the additional minor bunt resistance QTL on 4B inherited from the bunt differential line PI119333 in addition to Bt12 contradict this assumption. Based on race tests, Chen et al. (2016) argued that there is some evidence that the current bunt resistance differential set may in fact not always be monogenic. These suggest that it might be necessary to rethink on the current view that a bunt differential line carries one single major bunt resistance gene, and underlines the necessity to genetically characterize the full set of bunt differential lines currently used for characterizing Bt isolate mixtures and gene postulation.
Lastly it should be noticed that any negative associations between bunt resistance QTL and important agronomic traits are of pivotal interest before a routine application in applied breeding programs can be recommended. Markedly, an overlap of plant height QTL and common bunt QTL were found on chromosome 6D and 4B (Singh et al. 2016). Hence, we also investigated the potential association of plant height, lodging, heading date, mildew and stripe rust severity with CB and DB incidence in the study at hand. However, no significant correlations between any of these agronomic traits and bunt incidence were observed (Table S5), and none of the QTL for these traits overlapped with QBt.ifa-7DS|Bt12 (Table S6).
Summary and conclusion
The knowledge about the genetic architecture of bunt resistance in wheat is currently very scarce, and bunt differential lines are still used to postulate the presence of specific resistance genes in breeding material based on observed disease resistance patterns. Apart from the Bt9 bunt differential line, none of the 16 bunt differential lines has been genetically characterized to date. This study focused on mapping the chromosomal position of bunt resistance gene Bt12 by employing the bunt differential line PI119333 as resistance donor. The QTL QBt.ifa-7DS associated with Bt12 was mapped to chromosome 7DS. Deploying single major bunt resistance genes in elite cultivars has been a successful strategy to combat bunt diseases in the past, most prominently pictured by Bt10 (Laroche et al. 2000). Bt12 is a particularly attractive resistance gene because it is highly effective in controlling CB and DB. The markers that were developed in this study to tag Bt12 will facilitate a marker assisted introgression of Bt12 into regionally adapted germplasm, and the targeted combination with other mapped bunt resistances genes. This opens the way for a knowledge-driven resistance breeding, which is of particular importance for the development of resistant and adapted varieties for organic and low input agriculture.
References
Appels R et al (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361:eaar7191. https://doi.org/10.1126/science.aar7191
Bhatta M, Morgounov A, Belamkar V, Yorgancilar A, Baenziger PS (2018) Genome-wide association study reveals favorable alleles associated with common bunt resistance in synthetic hexaploid wheat. Euphytica 214:200. https://doi.org/10.1007/s10681-018-2282-4
Bokore F, Cuthbert R, Knox RE, Pozniak C, N'Diaye A, Sharpe A, Ruan Y (2017) Molecular mapping of common bunt resistance in a 'Vesper' x 'Lilian' population Canadian. J Plant Pathol 39:90–90. https://doi.org/10.1007/s00122-019-03403-3
Broman KW, Sen S (2009) A guide to QTL mapping with R/qtl: online complements. Springer, Dordrecht Heidelberg London New York. https://doi.org/10.1007/978-0-387-92125-9
Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890. https://doi.org/10.1093/bioinformatics/btg112
Chen JL, Guttieri MJ, Zhang JL, Hole D, Souza E, Goates B (2016) A novel QTL associated with dwarf bunt resistance in Idaho 444 winter wheat. Theor Appl Genet 129:2313–2322. https://doi.org/10.1007/s00122-016-2783-2
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Dumalasova V, Bartos P (2016) Reaction of wheat to common bunt and dwarf bunt and reaction of triticale to dwarf bunt. Czech J Genet Plant Breed 52:108–113. https://doi.org/10.17221/48/2016-cjgpb
Dumalasova V, Simmonds J, Bartos P, Snape J (2012) Location of genes for common bunt resistance in the European winter wheat cv Trintella. Euphytica 186:257–264. https://doi.org/10.1007/s10681-012-0671-7
Fofana B, Humphreys DG, Cloutier S, McCartney CA, Somers DJ (2008) Mapping quantitative trait loci controlling common bunt resistance in a doubled haploid population derived from the spring wheat cross RL4452 x AC Domain. Mol Breed 21:317–325. https://doi.org/10.1007/s11032-007-9131-9
Gilmour AR, Gogel BJ, Cullis BR, Welham SJ, Thompson R (2015) ASReml user guide release 4.1 functional specification. VSN International Ltd, Hemel Hempstead. http://www.vsni.co.uk
Goates BJ (1996) Common Bunt and Dwarf Bunt. In: Wilcoxson RD, Saari EE (eds) Bunt and smut diseases of wheat: concepts and methods of disease management. CIMMYT, Mexico, pp 12–25
Goates BJ (2012) Identification of new pathogenic races of common bunt and dwarf bunt fungi, and evaluation of known races using an expanded set of differential wheat lines. Plant Dis 96:361–369. https://doi.org/10.1094/pdis-04-11-0339
Goates BJ, Bockelman HE (2012) identification of new sources of high levels of resistance to dwarf bunt and common bunt among winter wheat landraces in the usda-ars national small grains collection. Crop Sci 52:2595–2605. https://doi.org/10.2135/cropsci2012.01.0060
Ishikawa G, Saito M, Tanaka T, Katayose Y, Kanamori H, Kurita K, Nakamura T (2018) An efficient approach for the development of genome-specific markers in allohexaploid wheat (Triticum aestivum L.) and its application in the construction of high-density linkage maps of the D genome. DNA Res 25:317–326. https://doi.org/10.1093/dnares/dsy004
Knox RE, Campbell HL, Depauw RM, Gaudet D, Puchalski B, Clarke FC (2013) DNA markers for resistance to common bunt in 'McKenzie' wheat. Can J Plant Pathol 35:328–337. https://doi.org/10.1080/07060661.2013.763292
Laroche A, Demeke T, Gaudet DA, Puchalski B, Frick M, McKenzie R (2000) Development of a PCR marker for rapid identification of the Bt-10 gene for common bunt resistance in wheat. Genome 43:217–223. https://doi.org/10.1139/gen-43-2-217
Matanguihan JB, Murphy KM, Jones SS (2011) Control of common bunt in organic wheat. Plant Dis 95:92–103. https://doi.org/10.1094/pdis-09-10-0620
Menzies JG, Knox RE, Popovic Z, Procunier JD (2006) Common bunt resistance gene Bt10 located on wheat chromosome 6D. Can J Plant Sci 86:1409–1412. https://doi.org/10.4141/p06-106
Metzger RJ, Hoffmann JA (1978) New races of common bunt useful to determine resistance of wheat to dwarf bunt. Crop Sci 18:49–51. https://doi.org/10.2135/cropsci1978.0011183X001800010013x
Mourad AMI, Sallam A, Belamkar V, Mahdy E, Bakheit B, Abo El-Wafaa A, Stephen Baenziger P (2018) Genetic architecture of common bunt resistance in winter wheat using genome-wide association study. BMC Plant Biol 18:280. https://doi.org/10.1186/s12870-018-1435-x
Piepho H-P, Möhring J (2007) Computing heritability and selection response from unbalanced plant breeding trials. Genetics 177:1881–1888. https://doi.org/10.1534/genetics.107.074229
R Core Team (2016) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org/
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci U S A 81:8014–8018. https://doi.org/10.1073/pnas.81.24.8014
Sen S, Churchill GA (2001) A statistical framework for quantitative trait mapping. Genetics 159:371–387
Singh A, Knox RE, DePauw RM, Singh AK, Cuthbert RD, Kumar S, Campbell HL (2016) Genetic mapping of common bunt resistance and plant height QTL in wheat. Theor Appl Genet 129:243–256. https://doi.org/10.1007/s00122-015-2624-8
Steffan PM, Torp AM, Borgen A, Backes G, Rasmussen SK (2017) Mapping of common bunt resistance gene Bt9 in wheat. Theor Appl Genet 130:1031–1040. https://doi.org/10.1007/s00122-017-2868-6
Taylor J, Butler D (2017) R Package ASMap: efficient genetic linkage map construction and diagnosis. J Stat Softw. https://doi.org/10.18637/jss.v079.i06
van Ooijen JW (1992) Accuracy of mapping quantitative trait loci in autogamous species. Theor Appl Genet 84:803–811. https://doi.org/10.1007/bf00227388
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78. https://doi.org/10.1093/jhered/93.1.77
Wang R et al (2019) Identification and assessment of two major QTLs for dwarf bunt resistance in winter wheat line ‘IDO835’. Theor Appl Genet 132:2755–2766. https://doi.org/10.1007/s00122-019-03385-2
Wang S, Knox RE, DePauw RM, Clarke FR, Clarke JM, Thomas JB (2009) Markers to a common bunt resistance gene derived from 'Blizzard' wheat (Triticum aestivum L.) and mapped to chromosome arm 1BS. Theor Appl Genet 119:541–553. https://doi.org/10.1007/s00122-009-1063-9
Wang S et al (2014) Characterization of polyploid wheat genomic diversity using a high-density 90000 single nucleotide polymorphism array. Plant Biotechnol J 12:787–796. https://doi.org/10.1111/pbi.12183
Wilkinson PA et al (2016) CerealsDB 3.0: expansion of resources and data integration BMC. Bioinformatics 17:256. https://doi.org/10.1186/s12859-016-1139-x
Zou J et al (2017) Mapping of QTLs associated with resistance to common bunt, tan spot, leaf rust, and stripe rust in a spring wheat population. Mol Breeding 37:144. https://doi.org/10.1007/s11032-017-0746-1
Acknowledgments
Open access funding provided by University of Natural Resources and Life Sciences Vienna (BOKU). We thank Herbert Huss and AGES for collecting and providing CB inoculum. We acknowledge Matthias Fidesser for excellent technical support and supervising the CB field trials. We thank Ricarda Pernold for assisting in the CB scorings in 2018, as well as Michael Haag and Mathias Schneider for assisting in the CB scorings in 2016. We are grateful to Verena Strasser for her work on KASP genotyping. We thank Jianli Chen and Rui Wang (University of Idahoo, Aberdeen) for genotyping the parental lines for marker polymorphism with their KASP markers. This work was financed by the Austrian Federal Ministry of Agriculture, Forestry, Environment and Water Management within the framework of the ERA-Net project Core Organic 2 (project number 100898).
Author information
Authors and Affiliations
Contributions
AEM performed the study and wrote the manuscript. BE, JH and BP helped with the collection of phenotypic data for CB trials and genotyping of populations. MB designed and performed KASP assays and edited the manuscript. SM helped with the statistical analysis of data. DH conducted the DB field trial. HB initiated and guided the study and obtained funding. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical standard
The authors declare that the experiments conducted for this publication comply with the current laws of Austria.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Muellner, A.E., Eshonkulov, B., Hagenguth, J. et al. Genetic mapping of the common and dwarf bunt resistance gene Bt12 descending from the wheat landrace PI119333. Euphytica 216, 83 (2020). https://doi.org/10.1007/s10681-020-02614-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-020-02614-w