
Abstract
A common shortcoming of current tissue engineered constructs is the lack of a functional vasculature, limiting their size and functionality. Prevascularization is a possible strategy to introduce vascular networks in these constructs. It includes among others co-culturing target cells with endothelial (precursor) cells that are able to form endothelial networks through vasculogenesis. In this paper, we compared two different prevascularization approaches of bio-artificial skeletal muscle tissue (BAM) in vitro and in vivo. In a one-stage approach, human muscle cells were directly co-cultured with endothelial cells in 3D. In a two-stage approach, a one week old BAM containing differentiated myotubes was coated with a fibrin hydrogel containing endothelial cells. The obtained endothelial networks were longer and better interconnected with the two-stage approach. We evaluated whether prevascularization had a beneficial effect on in vivo perfusion of the BAM and improved myotube survival by implantation on the fascia of the latissimus dorsi muscle of NOD/SCID mice for 5 or 14 d. Also in vivo, the two-stage approach displayed the highest vascular density. At day 14, anastomosis of implanted endothelial networks with the host vasculature was apparent. BAMs without endothelial networks contained longer and thicker myotubes in vitro, but their morphology degraded in vivo. In contrast, maintenance of myotube morphology was well supported in the two-stage prevascularized BAMs. To conclude, a two-stage prevascularization approach for muscle engineering improved the vascular density in the construct and supported myotube maintenance in vivo.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
This article was updated on 11 September 2020 to indicate that the first two authors contributed equally the article.
1. Introduction
Skeletal muscle tissue has a high regenerative capacity that is able to replace damaged muscle tissue upon injury [1, 2]. A central role in muscle regeneration is played by the muscle progenitor cells known as satellite cells. However in cases of volumetric muscle loss, a failure of muscle repair and scar tissue formation is often observed, resulting in functional muscle impairment [2]. The current treatment strategy for localized skeletal muscle repair includes muscle flap transplantation. Therefore, autologous muscle tissue is transplanted from a donor site to the damaged muscle. Unfortunately, this is associated with donor site morbidity and poor flap survival [3]. To limit this problem, microsurgery is used to include vasculature to allow the reconstitution of blood supply in the tissue. In turn, this provides oxygen and nutrients, and removes waste products throughout the transplanted tissue [4, 5].
To avoid the need of donor site tissue and related injuries, tissue engineered skeletal muscle holds promise as an alternative treatment. Skeletal muscle tissue engineering aims to create in vitro muscle tissue that mimics the structure and function of in vivo muscle, and thus offers the potential to repair the damaged muscle area. Similar to muscle flaps, one of the current limitations of avascular tissue engineered constructs is the lack of a blood supply in the initial phase after implantation since passive diffusion is limited to 150–200 µm [4, 6, 7]. In response to the hypoxia that arises when having a tissue engineered graft with wall thickness greater than the diffusion limit, host vessels invade the implanted tissue. However, the rate of spontaneous vascular ingrowth is limited to ∼5 µm h−1 [8], which cannot address the metabolic need in time leading to necrosis in the central area of the graft. As a result, successful use of tissue-engineered constructs in the clinic is limited to thin or avascular tissues, such as skin or cartilage, in which this rate of neovascularization from the host is sufficient. Yet, in other tissue grafts such as muscle, vascularization will be of key importance to achieve successful transplantation. This remains an important hurdle to overcome if the aim is to increase the size of engineered tissue constructs as the lack of oxygen and nutrient supply constrains the size of viable constructs.
Prevascularization is the formation of capillary-like networks before implantation and is aimed to overcome this diffusion limit. As for engineering (immature) blood vessels in skeletal muscle constructs, a few in vitro approaches have been described, such as co-culturing endothelial cells (EC) and myogenic progenitors on different scaffolds and hydrogels, combining endothelial cell sheets with myoblast cell sheets and engineering vasculature using microfabrication techniques [4, 5, 9, 10]. Besides applications in regenerative medicine, such a vascularized muscle model can be used to study mechanisms underlying myogenesis, vasculogenesis and vasculo-myogenic interactions.
This prevascularization is based on de novo blood vessel formation known as vasculogenesis and the advantages of this strategy are fourfold. First, the presence of this preformed endothelial network further enhances host vessel ingrowth upon implantation [11, 12]. Second, having a prevascularized construct bears the advantage that the preformed microvascular network only has to anastomose to the host vasculature after implantation to achieve rapid construct vascularization [13]. Third, this pre-vascularization improves perfusion and survival of the construct upon implantation [11, 12]. Fourth and last, in addition to forming blood vessels, endothelial cells are known for their paracrine activity on developing tissue, including skeletal muscle [14–16]. Although an in vitro prevascularization approach reduces the time needed for complete vascularization, perfusion is not as fast as with in vivo prevascularization using microsurgery to connect the construct to the host vasculature. However, vascularization speed is not the sole factor to consider [16]. Functionality of the newly formed vessels will determine the success of a tissue-engineering strategy as well. More specifically, regression of in vitro formed networks should be avoided [17].
Still, only a limited number of implantation studies focused on pre-vascularized tissue engineered skeletal muscle have been conducted so far. A prevascularized fibrin matrix, obtained using a microsurgically created arteriovenous-loop approach, was injected with expanded primary myoblasts [18]. This prevascularization approach promoted myoblast survival and preservation of the myogenic phenotype, but no differentiation into myotubes occurred. In another approach, a prevascularized construct containing HUVECs (human umbilical vein endothelial cells), C2C12 mouse myoblasts and fibroblasts was implanted, which resulted in enhanced survival and prolonged functionality of the skeletal muscle construct [12, 19]. The used poly-L-lactic acid (PLLA)/polylactic-glycolic acid (PLGA) 3D porous scaffolds however over time release acidic degradation products, which decrease cell survival [20]. Thus, this approach may not represent the most optimal approach for tissue engineering vascularized skeletal muscle. Similarily, an engineered flap composed of human adipose-derived microvascular endothelial cells and mesenchymal stem cells seeded on the same PLLA/PLGA scaffold was used for abdominal wall defect repair [21]. The graft was cultured in vitro until a small capillary net was formed. This was folded around an exposed femoral artery and vein of the recipient rat. After 21 d, the grafts with the arteries and veins were transferred to an abdominal full-thickness wall defect. This resulted in a highly vascularized, well-integrated muscle flap. However, further reports on functional muscle regeneration using this approach are missing.
Another disadvantage in the above mentioned prevascularization studies is the use of solid scaffolds. The skeletal muscle tissue-engineering approach used in our lab is based on previous work of the Vandenburgh lab and uses hydrogels, allowing cells to self-organize and migrate [22, 23]. In addition, a hydrogel better mimicks the physiological environment of developing skeletal muscle which has been shown to influence the proliferation of muscle progenitor cells [24]. In this approach, cells isolated from a human muscle biopsy are expanded to several millions of muscle cells (myoblasts and a minority of fibroblasts). These are cast in silicone molds in the presence of a natural hydrogel. After one week, this results in the formation of a bio-artificial muscle (BAM) containing multinucleated myotubes, well-aligned in the direction of the attachment points. The BAM is limited in size (2 cm long, ±1 mm thick) due to the absence of a vascular network. To increase the size of the BAM, which would expand its applications and utility, we already succeeded in prevascularizing a BAM in vitro in a previous study [10]. This was done by seeding HUVECs together with muscle cells in a fibrin hydrogel followed by one week of tissue contraction and cell differentiation. This is a direct 3D co-culture approach, defined as a one-stage prevascularization approach in this manuscript (BAM-1s). Despite detailed in vitro analysis, our previous study lacked an in vivo part in wich the functionality of the preformed endothelial networks was tested. In this follow-up study we aim to (i) further improve the prevascularization with minimal loss of myotube formation in the BAM based on a novel two-stage BAM prevascularization approach (BAM-2s) and (ii) assess in vivo behaviour of prevascularized BAMs in a mouse model.
2. Methods
2.1. Cell culture media
The medium that was employed for the isolation and proliferation of myogenic progenitors (skeletal muscle growth medium, SkGM) was composed of high glucose Dulbecco's Modified Eagle Medium (DMEM, Gibco), 10% fetal bovine serum (FBS, Thermo Fisher), 50 μg ml−1 gentamicin (Life Technologies), and 1% Ultroser solution (Pall corporation). Differentiation medium (SkFM) composed of DMEM with high glucose containing 10 ng ml−1 hEGF, 10 µg ml−1 insulin, 50 µg ml−1 BSA and 50 µg ml−1 gentamicin was used to promote formation of multinucleated myotubes.
2.2. Cell culture
Green Fluorescent Protein (GFP)-labeled HUVECs (GFP-HUVECs, Angio-proteomie) were seeded in cell culture flasks which were precoated for 1 h at 37 °C with gelatin (0.1% gelatin, Millipore) and cultured in endothelial growth medium (EGM-2 with bullet kit, Lonza). HUVECs were split at 90% confluence and used in the experiment at passage 7.
Human skeletal muscle cells were isolated from a fresh human muscle tissue biopsy, of an adult male aged 60, obtained from the Human Body Donation programme of KU Leuven University as described in [25]. The donor provided written consent (for research and educational purpose) prior to death. Briefly, isolated tissue was cut in strips of approximately 2 mm × 10 mm using sterile forceps and scalpel after removing excess connective tissue and fat. Before isolation of cells, muscle strips were pinned under tension into a sylgard coated 6-well plate to maintain cell survival while avoiding muscle atrophy. Two days after pinning the strips, enzymatic digestion was performed by incubating the muscle strips at 37 °C for 1 h in DMEM high glucose with Glutamax and pyruvate supplemented with 0.1% collagenase, type II (Sigma) and 4 mg ml−1 dispase II (Roche Diagnostics). After the incubation, isolated cells were collected by filtering through a 100 μm cell strainer (Falcon) and fragments were incubated again to digest the whole tissue. Isolated cells were pooled, centrifuged for 5 min at 200 × g and resuspended in SkGM. Muscle cells were split at 60%–70% confluence and used in experiments at 12 doublings. In the isolated muscle cell population, myoblast percentage and fusion index were determined as described before [10, 25].
Muscle cells that were used for tissue engineering of BAMs, and subsequently for implantation, expressed the far-red fluorescent protein miRFP670 [26]. The expression of miRFP670 was obtained by transducing 5 × 105muscle cells in 3.15 ml SkGM for 48 h with 350 µl 1.21 × 106 pg p24 ml−1 CMV-miRFP670 cDNA-containing HIV1-derived retroviral particles. Retroviral vectors were produced as previously described by the Leuven Viral Vector Core (Full description in [27] and with improvements in [28]). The transfer plasmid pCH-CMV-miRFP670-3xflag-IRES-blasti was made by cloning the miRFP670 sequence (RefSeq KX421097.1) into a pCH-CMV-eGFP-3xflag-Ires-blasti plasmid. All cloning steps were verified by sequencing. This resulted in the expression of triple flag-tagged miRFP670 under the control of a cytomegalovirus (CMV) promotor and with simultaneous expression of a blasticidin resistance cDNA. Lentiviral vector particle number was estimated by quantifying the surface protein p24 with enzyme linked immunosorbent assay (ELISA) (HIV-1 core profile ELISA, DuPont), which was determined to be 1.21 × 106 pg p24 ml−1.
2.3. Tissue-engineering of BAMs
Three different types of BAMs were used: (i) BAMs containing only muscle cells (2×106 muscle cells construct−1) cultured for 2 weeks in SkGM/SkFM (termed BAM), (ii) the one-stage prevascularized BAM with muscle cells and endothelial cells (1.4×106 muscle cells and 0.6 × 106 HUVECs construct−1) cultured for 1 week in EGM-2 (termed BAM-1s) and (iii) the two-stage prevascularized BAM with initially only muscle cells (2×106 muscle cells construct−1) cultured for 1 week in SkGM/SkFM followed by an additional embedding step in a fibrin hydrogel containing 2 × 106 GFP labeled HUVECs (termed BAM-2s) (schematic representation in figure 1). In previous experiments, the initial seeding density of in total 2 × 106 cells has been set based on the obtained aligment and dense formation of myofibers [10]. For the BAM-1s, this same initial cell density appeared to result in best myofiber formation and the total amount of cells was divided over the two cell types. The ratio of cells used was described to be the best trade-off between maximal myogenesis while having interspersed endothelial networks in [10]. Finally, for the BAM-2s the initial amount of cells (muscle cells) was kept constant (2×106) as this allows the direct comparison with the BAM in terms of myofibers while endothelial cell numbers were increased to 2 × 106 HUVECs. The latter was optimized separately in terms of maximal endothelial network formation extent.

Figure 1. Schematic representation of tissue engineering strategies. (top) BAM represents bio-artificial muscle in a fibrin hydrogel containing aligned myotubes, differentiated for 2 weeks in SkGM and SkFM medium. (middle) BAM-1s is a prevascularization strategy based on co-culture of endothelial and muscle cells in a fibrin hydrogel, cultured for 1 week in EGM medium which results in aligned myotubes with interspersed endothelial networks. (bottom) BAM-2s is a prevascularization strategy by embedding a one-week old BAM in a new fibrin hydrogel containing endothelial cells (EC). Culturing this construct for an additional week in EGM medium, results in a myotube bundle surrounded by an endothelial coat containing an endothelial network.
Download figure:
Standard image High-resolution imageTissue-engineering of BAMs was described in [10, 25]. Briefly, cells were mixed with 500 µl thrombin (4 U ml−1, Bio Phar Laboratories LLC). The mixture was cast into 25-mm-long silicone rubber molds with end attachment sites spaced 20 mm. Then, to form a fibrin hydrogel (1 mg ml−1) containing the cells, 500 µl fibrinogen (2 mg ml−1, Merck Chemicals) was added and the cell-gel mix was mixed. Following 2 h incubation at 37 °C, SkGM (for BAM) or EGM-2 medium (for BAM-1s) supplemented with fibrinolysis inhibitors aprotinin (92.5 µg ml−1 Carl Roth) and tranexamic acid (400 µM, Sigma) was added. For BAM, the medium was switched to differentiation medium (SkFM) two days after casting. To obtain the third type of BAM, BAM-2s, 1 week old BAMs were embedded in 1 ml fibrin (created similar as above) hydrogel containing 2 × 106 HUVECs and cultured for another week in EGM-2. Medium was replaced every 2 d and BAMs were kept in culture for 7 or 14 d prior to thickness measurement and fixation.
2.4. Thickness measurements
Cross-sectional thickness of BAMs in culture and post-implantation was measured with a sterile micrometer at day 7 or day 14 after casting.
2.5. In vivo implantation of BAMs
All animal experiments were evaluated and approved by the Animal Ethics Committee of the KU Leuven (P099/2017) and were performed according to international guidelines. BAMs were implanted subcutaneously on the fascia of the latissimus dorsi muscle for a period of 14 d in 6–7 weeks old non-obese diabetic severe combined immunodeficiency (NOD/SCID) mice (Janvier) (n = 12 for BAM, n = 8 for BAM-1s and n = 4 for BAM-2s). Imbalance in numbers is related to the surgical procedure in which a lower number of animals with the BAM-2s constructs survived. The higher number of the BAM as compared to the BAM-1s and BAM-2s is because the BAM condition was used as a reference in each animal. All animals were anesthetized and maintained with isoflurane (0.5%–2%, 3–5 l min−1) and oxygen while being kept warm on a heating pad during surgery and recovery. The fascia of the latissimus dorsi muscle was scraped with a sterile scalpel until minor bleeding was observed. Both sides of the BAM were secured to the muscle with non-resorbable polypropylene suture (Ethicon) parallel to the spine. After implantation, meloxicam (2 mg kg−1; Meloxidyl CEVA) and buprenorphine (0.1 mg kg−1; Vetergesic) (for 2 d) were administered subcutaneously. Animals were euthanized 5 or 14 d later. Construct and underlying host tissue were explanted to capture both construct maintenance and host vessel ingrowth as well as construct integration into the host respectively. Host muscle without direct contact with the implanted constructs (gastrocnemius) was explanted each time and served as a negative control for all graft-specific stainings. Explants were washed in PBS for 5 min and fixed using 4% formaldehyde solution (w/v) for 6 h at room temperature (RT).
2.6. Whole mount immunofluorescence staining
To assess in vitro BAM formation, the 3 types of BAM (BAM, BAM-1s, BAM-2s) were washed (3 × 5 min in PBS) and then removed from the attachment sites. Then, they were pinned on Styrofoam to preserve their original shape during fixation in 4% formaldehyde for 1 h at room temperature (RT) and stored at 4 °C in PBS. Explanted constructs from the in vivo implantation study were fixed for 6 h at RT in 4% formaldehyde and stored at 4 °C in PBS in the dark. For tropomyosin immunohistochemical staining, samples were fixed a second time, in methanol (−20 °C, 20 min) immediately prior to staining. Next, the samples were permeabilized in blocking buffer containing 0.2% Triton-X-100 (Sigma) and 1% bovine serum albumin (BSA, Sigma) in PBS for 3 h at RT. Subsequently, the samples were incubated overnight at 4 °C with a monoclonal mouse antibody against tropomyosin (Sigma, T9283, 1:100 in blocking buffer) or with polyclonal rabbit antibody against collagen IV (Abcam, ab6586, 1:400 in blocking buffer). To stain capillaries, the explanted BAM constructs were incubated overnight at 4 °C with a polyclonal rabbit antibody against CD 31 (Invitrogen, PA16301, 1:30 in blocking buffer). Next, the human BAMs and explants were washed and incubated with a polyclonal goat anti-mouse secondary antibody (Alexa Fluor 633, A-11 059 or A-21 063, Invitrogen, 1:200), a polyclonal goat anti-rabbit secondary antibody (Alexa Fluor 633, A21070, Thermo Fisher) or a polyclonal donkey anti-rabbit secondary antibody (Alexa Fluor 488, A-21 206, Life Technologies) for 3 h in the dark, followed by incubation with DAPI (Life Technologies, 0.1 µg ml−1 in PBS) for 1 h at RT. Samples were stored in PBS in the dark until visualized.
2.7. Confocal imaging
Confocal imaging and data analysis of in vitro BAMs and in vivo BAMs were performed as described in [10]. Briefly, BAMs and explanted tissue were visualized by confocal microscopy (Zeiss LSM710). Per BAM, 5 z-stacks were acquired with Plan-Apochromat 25x/0.8, WD 0.57 mm objective every 5 µm. Since explanted BAMs at day 14 were surrounded by host tissue, the distance to the construct exceeded the working distance of the objective. Therefore, explanted tissue was embedded in liquid 4% agarose in distilled water, cooled until the agarose solidified and cut in 200 µm sections using a vibratome (Microm HM 650 V, VWR) to allow visualization within the whole explanted tissue (in collaboration with Prof. Veerle Baekelandt, KU Leuven). More detailed images were acquired with Plan-Apochromat 63x/1.2, WD 0.49 mm. Three dimensional reconstructions of stacked images were generated with Imaris 9.2.0 software (Bitplane).
2.8. Histology
4% formaldehyde fixed BAMs and explanted constructs were dehydrated through an alcohol series and embedded in paraffin. Tissue was sectioned at a thickness of 5 µm with a microtome (Leica RM2125 RTS). Tissue slices were stained with Martius Scarlet Blue (MSB) staining [29] to evaluate the extracellular matrix deposition in BAMs before and after implantation. Murine muscle was used as positive control.
2.9. Data analysis
Myotube analysis and endothelial network analysis in in vitro constructs and after implantation were performed as previously reported in Gholobova et al [10]. For myotube formation 20 µm z-projections were manually analyzed for different parameters of myotube formation: myotube alignment, length, density and diameter. Myotube alignment was represented as the mean of the standard deviation of the angles of the myotubes. A lower number thus indicates better alignment. For endothelial network analysis, 60 µm z-projections were automatically analyzed by a customized version of the 'Angiogenesis Analyzer', an ImageJ plugin created by Gilles Carpentier [10, 30]. Moreover, a customized algorithm was developed that was able to detect non-perfused and perfused vessels. This tool is able to measure several parameters of endothelial networks of which we used: total length of endothelial network (master segments, branches and isolated segments), % master segments (length of master segments, longest elements giving origin to branches, divided by total network length), % branching length (length of interconnected segments and branches, elements delimited by two junctions, or a junction and one extremity, divided by total network length) and % isolated segments (length of isolated segments, binary lines which are not branched, divided by total network length).
2.10. Detection of anastomosis
Two different approaches were used to evaluate anastomosis and perfusion of the implanted endothelial networks with host vessels; in the first approach 1.25 μg g−1 body mass rhodamine-labelled Ulex europaeus agglutinine-I (UEA-I; Vectorlabs) was injected intravenously in the tail vein, circulation was allowed for 2 min, whereafter the animal was euthanized by cervical dislocation. UEA-I specifically targets human endothelial cells. Presence of UEA-I positive human endothelial networks was confirmed with confocal imaging of explanted constructs. In the second approach, human GFP labeled endothelial networks in the explanted constructs were examined for the presence of auto-fluorescent red blood cells by confocal microscopy.
2.11. Quantitative real-time PCR
RT-qPCR was performed to determine the expression of the different myosin heavy chain isoforms (MYH1, MYH3, MYH8) and myogenin (MyoG) in the three types of human BAMs to reflect the developmental state of the myotubes in the tissue-engineered constructs. To normalize for the amount of cells, three reference genes were used: GAPDH, HSP90AB1 and RPL13A. RT-qPCR was performed as described previously [31]. Briefly, BAMs were washed with PBS and lyzed by sonication. After centrifugation (5 min, 2600 g), high quality total RNA was isolated from supernatant using the PureLink RNA Mini Kit (Ambion #12 183 018A) as evaluated by A260/A280. 2 µg of isolated RNA was reverse transcribed with the qScript cDNA SuperMix (Quantabio #95 048-100). PCR was performed with GeneAmp® PCR System 9700 (Applied Biosystems). Gene-specific primers (table 1) were designed using NCBI/Primer-Blast and Primer3plus. Primer efficiency was determined by serial dilution of cDNA during RT-qPCR and verified to be >90%. RT-qPCR was performed with the LightCycler® 480 Instrument (Roche) using Perfecta Sybr Green Supermix (Quantabio #95 054-500). All samples, 10 µl reaction volume, were run in technical duplicates. 40 cycles were run with a DNA dissociation step (95 °C, 15 s), a primer annealing step and an amplification step (60 °C, 45 s). Melting curve analysis was performed to verify the amplification product. Relative mRNA expression was statistically analyzed with relative expression software tool (REST 2009, Qiagen). The REST software calculates whether gene expression is significantly different between the sample and control group using bootstrap randomization [32].
Table 1. List of primers used for RT-qPCR.
| Gene | Orientation | Primer sequence (5'—3') | Amplicon size (bp) |
|---|---|---|---|
| MYH1 | Forward | GGG AGA CCT AAA ATT GGC TCA A | 106 |
| Reverse | TTG CAG ACC GCT CAT TTC AAA | ||
| MYH3 | Forward | CTT GTG GGC GGA GGT CTG G | 119 |
| Reverse | AGC TAT GCC GAA CAC TTC CAT | ||
| MYH8 | Forward | ACA TTA CTG GCT GGC TGG AC | 143 |
| Reverse | ACC TTT CTT CGC GCT GCT AT | ||
| MyoG | Forward | GTG TGT AAG AGG AAG TCG GTG TC | 90 |
| Reverse | GAA GGC CTC ATT CAC CTT CTT | ||
| GAPDH | Forward | TCA AGA AGG TGG TGA AGC AGG | 168 |
| Reverse | ACC AGG AAA TGA GCT TGA CAA A | ||
| HSP90AB1 | Forward | AGA AAT TGC CCA ACT CAT GTC C | 75 |
| Reverse | ATC AAC TCC CGA AGG AAA ATC TC | ||
| RPL13A | Forward | CCT GGA GGA GAA GAG GAA AGA GA | 126 |
| Reverse | TTG AGG ACC TCT GTG TAT TTG TCA A |
2.12. Statistics
Number of replicates refers to number of analyzed images for microscopic analyses or to number of BAMs for thickness analyses. D'Agostino & Pearson normality test and Bartlett's test were used to verify normality of the data and equality of variances, respectively. Normally distributed data with equal variances were analyzed by an unpaired student t-test when two groups were compared. For comparing several normally distributed groups, a one-way ANOVA was used with a Bonferroni multiple comparison post test. For groups that were not normally distributed and/or had unequal variances, a non-parametric Mann-Whitney test was used for comparison between two groups while Kruskal-Wallis test followed by a Dunn's post test was performed for multiple comparisons. All values of analyzed images and relative expression data were displayed in Whisker boxplots and thickness analyses as mean ± standard deviation. * p < 0.05, ** p < 0.01, *** p < 0.001.
3. Results
3.1. In vitro BAM formation
First, cells isolated from a human muscle biopsy were expanded and characterized. This muscle cell population, previously described in [31] contained 82 ± 5% desmin-positive myoblasts (n = 6) at 12 doublings. Myoblasts had a fusion index of 80 ± 15% (n = 6) as determined by tropomyosin staining, forming myotubes with 10 ± 4 nuclei per myotube (figure S1 (available online at stacks.iop.org/BF/12/035021/mmedia)). To make bio-artificial muscle with only muscle cells (BAM), 2 × 106 muscle cells were mixed in a fibrin extracellular matrix (1 mg ml−1) and cultured in skeletal muscle growth medium (SkGM) for 2 d, followed by skeletal muscle differentiation medium (SkFM) for 12 d. The cell-fibrin hydrogel mix contracted around two attachment points and formed a 2 cm long muscle bundle (schematic representation in figure 1, macroscopic image in figures 2(a) and (d)), containing aligned multinucleated myotubes (figure 3(a)) with a thickness of 1.25 ± 0.2 mm (n = 21).

Figure 2. Implantation of BAM, BAM-1s and BAM-2s constructs. Macroscopic images of BAM, BAM-1s and BAM-2s at day 0 of implantation (a) and (d), at day 5 (b) and (e) and 14 (c) and (f) of post-implantation. BAM constructs were sutured on the left fascia of the latissimus dorsi muscle, BAM-1s or BAM-2s on the right fascia of the latissimus dorsi muscle. (g), (h) and (i) show enlarged images of explanted constructs with surrounding host tissue at day 14 post-implantation. Scale bars represent 0.5 cm (a)–(f) and 0.2 cm (g)–(i).
Download figure:
Standard image High-resolution image
Figure 3. In vitro myotube formation and prevascularization. Fluorescent confocal images of in vitro tissue-engineered skeletal muscle (BAM) with only muscle cells (BAM), a one-stage co-culture BAM with GFP labeled HUVECs (green) (BAM-1s) and a two-stage co-culture BAM with an endothelial coat (BAM-2s). The 3 types of BAMs show presence of aligned multinucleated (DAPI, blue) myotubes (red) (a), (b) and (d). In BAM-1s, endothelial networks (green) are interspersed between the aligned myotubes (b). These endothelial networks are positive for collagen IV (c), in red). In BAM-2s, interconnected endothelial networks are located in the endothelial coat surrounding the myotube bundle (d), (e) and are negative for collagen IV (f). Scale bars represent 100 µm (a), (b), (d) and (e)) and 50 µm (c) and (f).
Download figure:
Standard image High-resolution image3.2. In vitro myotube formation in prevascularized BAMs
Two different prevascularization strategies of BAMs were evaluated. The first approach was previously evaluated in vitro and described in [10]. This co-culture strategy, here termed the 1-stage prevascularization (or BAM-1s) was made by co-culturing muscle cells with endothelial cells (HUVECs) in a 70:30 ratio and a total number of 2 × 106 cells in fibrin hydrogel (schematic representation in figure 1, macroscopic image in figure 2(a) right side). As we described previously, culturing HUVECs in skeletal muscle media results in poor HUVEC survival, therefore we cultured BAM-1s in EGM-2, an optimized medium for endothelial cells [10]. After one week in culture, the cell—fibrin hydrogel contracted to a muscle bundle with interspersed endothelial network and a total thickness of 1.34 ± 0.15 mm (n = 16) (figure 2(a) right side). No significant difference in BAM-1s thickness was present in comparison to BAM. However, as reported before [10], myotube formation in BAM-1s is compromised in the EGM medium resulting in decreased myotube diameter, length and density compared to BAM (compare figure 3(a) with 3(b) and figures 4(a), (c) and (d). No difference in myotube alignment was observed (figure 4(b)). RT-qPCR analysis of pre-implantation BAM, BAM-1s and BAM-2s for myosin heavy chain (MYH) isoform expression showed a presence of less mature myotubes (figure 5) in BAM-1s compared to BAM reflected by a 2.5 and 4.7-fold lower expression level of respectively the adult isoform MYH1 and the perinatal isoform MYH8. Also, a respectively 3-fold and 1.4-fold lower expression level was observed for the embryonic isoform MYH3 and myogenin, indicating a lower myogenic differentiation in BAM-1s. Earlier reports on the in vitro evaluation of this co-culture BAM-1s also described a decrease in myotube diameter and density compared to BAM [10].
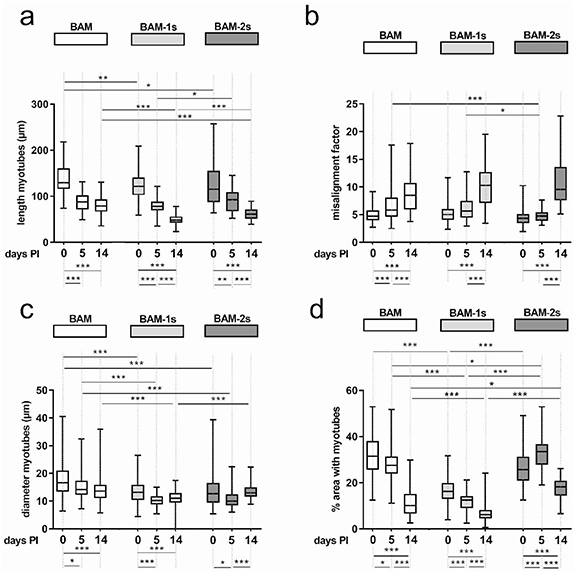
Figure 4. Morphological analysis of myotubes in BAMs in vitro and in vivo. Myotube formation and morphology was quantified with the following parameters: myotube length (a), alignment (b), diameter (c) and myotube density (d). These parameters were determined in BAM, BAM-1s and BAM-2s at 0, 5 and 14 d post implantation (PI). Myotube analysis was performed based on tropomyosin staining for all conditions. Statistical significances for comparisons in a BAM condition between different time points are indicated below the graphs and for comparisons between different conditions at a specific time point are indicated above the graphs. For multiple comparisons a nonparametric Kruskal-Wallis test with Dunn's post test corrected for multiple testing was used. * p < 0.05, ** p < 0.01, *** p < 0.001.
Download figure:
Standard image High-resolution image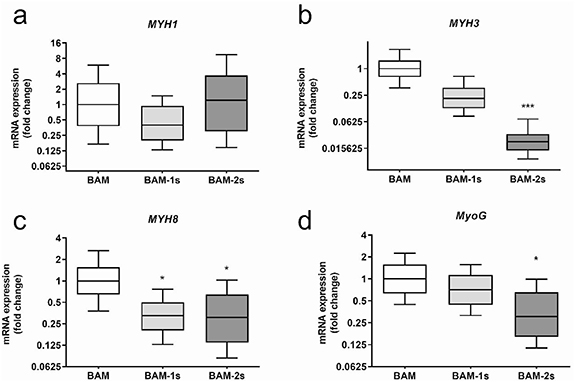
Figure 5. Myogenic gene expression. Relative expression of MYH isoforms and myogenin in pre-implantation BAMs, direct co-culture BAMs (BAM-1s) and BAMs with an endothelial coat with HUVECs (BAM-2s). Expression levels of these genes were quantified by RT-qPCR as the fold changes relative to gene expression in BAMs with only muscle cells (represented with Whisker box plots). To compare the three conditions, a nonparametric Kruskal-Wallis test with Dunn's post test corrected for multiple testing was used. *p < 0.05, **p < 0.01, ***p < 0.001.
Download figure:
Standard image High-resolution imageTo circumvent this compromised myotube formation, we also explored a two-stage prevascularization approach (BAM-2s). First, 2 × 106 muscle cells were mixed with a fibrin hydrogel (1 mg ml−1) to engineer a one-week old BAM as described before [10]. Then, we embedded the BAM in a fibrin hydrogel containing 2 × 106 HUVEC cells, further referred to as the endothelial coat. In each tissue-engineering stage, a 2 × 106 cell ml−1 ratio was used, as was done with BAM and BAM-1s. This construct was cultured for another week in EGM-2 to stimulate endothelial network formation (schematic representation in figure 1, macroscopic image in figure 2(d) right side). Endothelial cells in this coat layer spontaneously formed endothelial networks, and this layer was 117 ± 12 µm thick (n = 6) one week after casting (figure 3(e)). The advantage of this procedure is that differentiation of each cell type occurred in its own specialized medium, resulting in better survival and morphology of each cell type. The resulting BAM-2s was 1.5 ± 0.15 mm thick (n = 16, figure 2(d)), which was significantly thicker than BAM and BAM-1s. After 14 d in culture, we visualized myotube formation using confocal microscopy (figure 3(d)). Myotube density was significantly higher in BAM-2s compared to BAM-1s and similar to BAM (figure 4(d)). Myotube length and diameter in vitro was highest in BAM (figures 4(a) and (c)). No differences were observed for myotube alignment between the 3 different BAM types (figure 4(b)). Regarding MYH isoform expression in BAM-2s, no difference with BAM was observed for MYH1. A lower expression of MYH3, MYH8 and myogenin was observed, respectively 45, 3.2 and 3.3-fold lower than in myotubes in BAM (figure 5). There were no significant differences in MYH or myogenin expression in BAM-2s versus BAM-1s (figure 5).
3.3. In vitro endothelial network formation in BAM-1s and BAM-2s
Endothelial networks were formed through self-assembly and self-organization in BAM-1s and BAM-2s fibrin constructs (figures 3(b), (d) and (e)). In BAM-1s, endothelial networks were interspersed between aligned myotubes. In BAM-2s, the endothelial networks (figure 3(e)) penetrated the myotube bundle occasionally in the transition area between the BAM and the endothelial coat (figure 3(d)). When comparing endothelial networks in BAM-1s and BAM-2s, a higher network complexity could be observed in BAM-2s (figures 3(b), (e) and 6). For all measured parameters, BAM-2s outperformed BAM-1s (figure 6). This means that in BAM-2s, the endothelial networks were longer, more branched with less isolated segments not participating in a network compared to BAM-1s. Although the endothelial network characteristics were better in BAM-2s than BAM-1s, they were negative for collagen IV, a basement membrane protein important in vascular maturity and stabilization [33] (figure 3(f)). Networks in BAM-1s were positive for collagen IV (figure 3(c)) but they regressed after one week in vitro, a clear indication of the poor stability of the network (figure S2).

Figure 6. Morphological analysis of endothelial networks in vitro and in vivo in BAMs. Endothelial network formation was quantified with the following parameters: (a) total length/100 µm2, (b) % master segments (frequently branching long segments), (c) % branching and (d) % isolated segments. These parameters were determined in BAM, BAM-1s and BAM-2s at 0, 5 and 14 d post implantation (PI). Network analysis was performed based on GFP fluorescence for BAM-1s and BAM-2s (containing GFP-labeled HUVECs) and CD31-staining for BAM. Statistical significances for comparisons in a BAM condition between different time points, are indicated below the graphs and for comparisons between different conditions at a specific time point are indicated above the graphs. For multiple comparisons a nonparametric Kruskal-Wallis test with Dunn's post test corrected for multiple testing was used. * p < 0.05, ** p < 0.01, *** p < 0.001.
Download figure:
Standard image High-resolution image3.4. In vivo implantation and explantation
Next, we evaluated whether different strategies of prevascularization affected the constructs in terms of (i) myotube survival, (ii) myotube maturation and (iii) functionality of the endothelial networks in vivo. Therefore, we implanted the three BAM types on the fascia of the latissimus dorsi muscle on the back of NOD/SCID mice. The prevascularized constructs (BAM-1s and BAM-2s) were implanted on the right fascia of the latissimus dorsi muscle, while BAMs (without prevascular networks) were implanted on the left fascia of the latissimus dorsi (figures 2(a) and (d)) as a reference. Both on day 5 and day 14 post implantation (PI), differences in vascularization between constructs could be observed macroscopically (figures 2(b) and (e), (c) and (f), (h) and (i)). While the appearance of the muscle cell only BAM construct did not change (white), BAM-1s and BAM-2s were red, pointing to the presence of blood (figures 2(b), (c) and (e), (f); right side). In contrast to day 5, the different types of BAM were integrated with host tissue at day 14. Therefore, BAM constructs were explanted together with surrounding host tissue at day 14. To visualize myotube characteristics throughout the whole construct at day 14 PI, the tissue was cut into 200 µm sections with a vibratome.
3.5. In vivo myotube maintenance
Myotube survival and maturation in vivo were evaluated morphologically at 5 and 14 d after implantation. Myotubes were still present at day 14 of implantation in all conditions (figure 7). However, compared to pre-implantation, a reduction in myotube length in all conditions was shown at day 5 which was even more prominent at day 14 (figure 4(a)). At this time point, myotube length was decreased by 40% (BAM), 61% (BAM-1s) and 47% (BAM-2s). Similarly, myotube alignment decreased over 14 d, depicted in figure 4(b) as an increase in misalignment. On the other hand, there was an overall decrease from day 0 to day 14 PI in myotube diameter for BAM and BAM-1s conditions, while this was not the case for BAM-2s. Finally, myotube density decreased in all conditions from day 0 to day 14 PI although this effect was least prominent in the BAM-2s condition (67.8% (BAM), 61% (BAM-1s) and 33,4% (BAM-2s), figures 4(d) and 7(d), (e) and (f)).
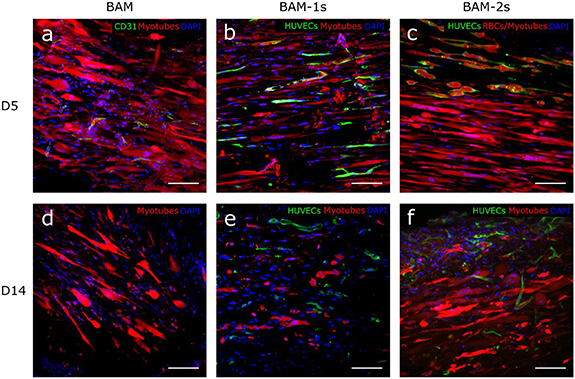
Figure 7. Confocal images of myotube and endothelial network in BAMs 5 and 14 d post-implantation. Fluorescent confocal images of musce cell only BAMs explanted after 5 (a) or 14 d (d) in vivo showing myotubes (red), nuclei (DAPI, blue) and endothelial cells (CD31, green). Images with the same setup are shown for BAM-1s explanted after 5 (b) or 14 (e) days and BAM-2s explanted after 5 (c) or 14 (f) days. For BAM-1s and BAM-2s, endothelial cells are labeled with GFP. Scale bars represent 100 µm.
Download figure:
Standard image High-resolution image3.6. In vivo vascularization and anastomosis
In vivo vascularization of BAMs was assessed 5 and 14 d after implantation. GFP labeled endothelial vessels, which originated from the HUVECs, were present in BAM-1s and BAM-2s at day 5 and 14 (figures 6, 7 and 8). Ingrowth of host blood vessels in muscle cell only BAM was visualized by CD31-staining. There was a higher vascular density, determined by total network length per 100 µm2, in the prevascularized BAMs compared to muscle cell only BAM (figures 6(a) and 7). After 5 d in vivo, the location of the endothelial vessels remained similar as to the situation in vitro for BAM-1s and BAM-2s: while interspersed between myotubes in BAM-1s, endothelial vessels in BAM-2s surrounded the myotube bundle (figures 7(b) and (c)). However, at day 14, vascular structures derived from HUVECs were also observed between myotubes in BAM-2s (figure 7(f)). Although there was a general decrease in the vascular density in both prevascularized constructs, pointing to degeneration of part of the network (figure 6(a)), the remaining network was more interconnected (figures 6(c) and (d)). To assess the functionality of the networks, a rhodamin-labelled human EC specific agglutinin (UEA-I) was injected intravenously. Detection of this dye within the BAMs points to a functional network which is anastomosed with the host blood vessels [5]. The latter could also be verified by the presence of red blood cells in the GFP labeled networks. At day 5, in both BAM-1s and BAM-2s, the majority of endothelial vessels (>90%) contained red blood cells, while only sparse presence of UEA-I-positive endothelial networks was observed (figures 8(b) and (c)). This pointed to early anastomosis with little perfusion. At day 14, a part of the GFP-labeled endothelial vessels in BAM-1s and BAM-2s were UEA-I positive (figures 8(e) and (f), S3, supplementary Videos S1 and S2) and contained red blood cells (figures 8(b) and (c), supplementary Video S3), showing functionality of implanted networks .
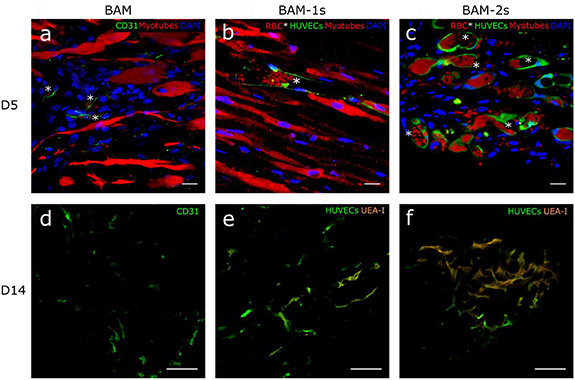
Figure 8. In vivo perfusion of prevascularized endothelial networks at 5 and 14 d post-implantation. Fluorescent confocal images of implanted BAM at day 5 and day 14 with only muscle cells (BAM) and CD 31 positive capillaries (green) (a) and (d), of a one-stage co-culture BAM with GFP labeled HUVECs (BAM-1s) (green) (b) and (e) and a two-stage co-culture BAM with an endothelial coat containing GFP labeled HUVECs (green) (BAM-2s) (c) and (f). At day 5 post-implantation prevascularized BAMs contain GFP labeled endothelial vessels (green) that are perfused with red blood cells (red dots, asterisks (b) and (c)). At day 14 GFP labeled endothelial vessels (green) are UEA-I positive (orange) (e) and (f), showing perfusion of the newly formed blood vessels due to anastomosis between host vessels and implanted networks. Muscle cell only BAMs contain some CD31 positive capillaries but no perfusion of the construct with UEA-I at day 14 was observed (d). Scale bars represent 20 µm (a)–(c) and 100 µm (d)–(f).
Download figure:
Standard image High-resolution image3.7. Extracellular matrix remodeling after implantation
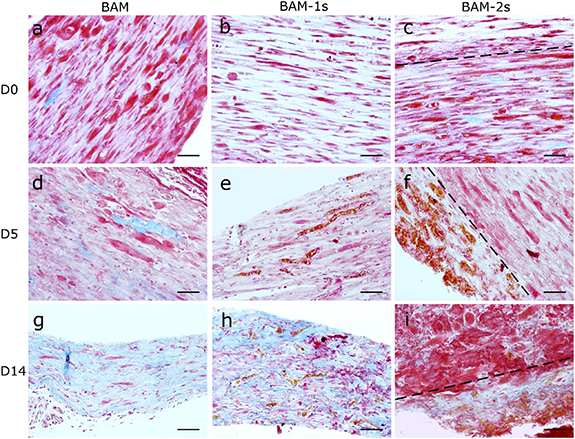
Extracellular matrix (ECM) remodeling in vivo was evaluated by Martius Scarlet Blue (MSB) which stains muscle tissue red, fibrin pink/purple, collagen blue and red blood cells orange. The ECM of in vitro cultured BAMs mainly consists of fibrin (figures 9(a)–(c)) with limited deposition of collagen. No significant ECM remodeling had occurred by day 5 PI (figures 9(d)–(f)). The presence of myotubes as seen with confocal microscopy (figure 7) was confirmed with MSB staining (figure 9). The presence of blood vessels in BAM-1s and BAM-2s constructs was also confirmed with MSB staining; red blood cells stained in orange were clearly detectable and located within the BAM-1s or surrounding the BAM-2s (figures 9(e), (f), (h) and (i)). After 14 d in vivo, fibrin ECM in BAM and BAM-1s was remodeled into a collagen ECM (figures 9(g) and (h)). In BAM-2s, a difference could be observed between the inner part of the construct containing mainly myotubes with traces of fibrin and collagen versus the endothelial coat which showed primarily collagen as ECM (figure 9(i)).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. Extracellular matrix remodeling in BAMs. Martius Scarlet Blue (MSB staining) of muscle cell-only BAM, one-stage co-culture BAM (BAM-1s) and two-stage co-culture BAM (BAM-2s) at day 0 of implantation and day 5 and 14 post-implantation. The extracellular matrix (ECM) of in vitro BAMs consist mainly of fibrin (light pink) (a)–(c) with some collagen deposition (blue). Aligned myotubes are shown in dark pink/red. At day 5 post-implantation (d)–(f), the ECM still consists mainly of fibrin, while at day 14 post-implantation, fibrin is being remodeled to collagen. Red blood cells are stained in orange and are present in capillaries at day 5 and day 14 in BAM-1s and BAM-2s (e)–(f), (h)–(i)). Scale bars represent 20 µm, dashed line in (c), (f) and (i) indicates the border between the endothelial coat and the muscle bundle.
Download figure:
Standard image High-resolution image{kind=link}
4. Discussion
At present, the use of muscle flaps to treat volumetric muscle loss has significant drawbacks [2]. Tissue engineered skeletal muscle holds promise for an alternative treatment. The tissue-engineering approach used in this paper further builds on a muscle tissue engineering approach, called bio-artificial muscle (BAM) containing aligned myotubes [23, 34]. Still, many challenges towards clinical as well as in vitro testing applications remain. One of them is the limited size of the constructs, imposed by the limit of passive diffusion of nutrients and gases. Prevascularization is being explored as a method to allow in vitro perfusion and to enhance in vivo survival and engraftment of tissue-engineered constructs [12, 35, 36]. In this paper, we have evaluated two prevascularization strategies of tissue-engineered bio-artificial muscle (BAM) to tackle the limitation.
The first approach was previously evaluated in vitro [10]. In this one-stage prevascularization approach (BAM-1s) a direct co-culture of endothelial cells and muscle cells resulted in aligned multinucleated myotubes with interspersed endothelial networks. The natural hydrogel fibrin was used as a scaffold, because of its pro-angiogenic and stiffness characteristics [37]. As reported before [10] myotube formation was suboptimal in the endothelial growth medium (EGM) in which BAM-1s was cultured when compared to BAMs cultured in skeletal muscle cell-specific media (SkGM/SkFM). The impaired myotube formation in EGM was also described by others [12] but is unavoidable in the one-stage approach since EC survival is severly compromised in SkGM/SkFM. In this follow-up study, the goal was to test the functionality of the preformed endothelial networks after implantation in vivo.
To avoid the reported suboptimal myotube formation in BAM-1s, we established another prevascularization strategy, consisting of two stages, termed BAM-2s. In the first stage, muscle cell-only BAMs were cultured for one week in SkGM/SkFM, allowing for optimal myotube formation. In a second stage, an endothelial cell coat was added in which endothelial networks formed during the second week. Then, the BAM-2s was cultured in EGM, supporting survival and differentiation of the ECs. This two-stage strategy indeed resulted in less compromised myotube formation, while supporting endothelial network formation even stronger than in the one-stage approach. Also these BAM-2s were tested in an in vivo settting in this study.
We are aware that there are multiple differences between the one-stage and two-stage prevascularization protocols since both strategies have been optimized separately to produce the best possible co-culture BAM. For example, the prolonged culture time of the muscle cells in the two-stage approach resulted in better myotube formation, as also reported elsewhere [38]. However, applying the same culture time in the one-stage approach was detrimental for the ECs since culturing BAM-1s longer than 1 week resulted in endothelial network degradation (figure 2(s)).
Vascularization is key for muscle development and function. Therefore, we assessed the myogenic gene expression in the three types of BAMs before implantation using quantitative real-time PCR. Myosin heavy chain isoforms reflect the developmental state of muscle fibers [39]. Relative expression levels of MYH1, MYH3, MYH8 and myogenin were analyzed to assess the influence of our vascularization strategy on muscle development. Myogenin expression is indicative for early myogenic differentiation and myofiber formation [31] while MYH3, MYH8 and MYH1 are expressed in embryonic, perinatal and adult stages, respectively. Analysis showed that the significantly higher MYH8, MYH3 and/or myogenin levels indicate a more active myofiber formation in BAM compared to BAM-1s or BAM-2s. This is not surprising given the optimal conditions for myofiber formation provided by the skeletal muscle fusion medium in this condition. No significant difference in adult isoform MYH1 expression was observed between the three conditions pointing out that pre-implantation, the endothelial networks were not able to promote muscle fiber maturation. The BAM-2s approach was explored with the intention to improve myogenesis and/or vasculogenesis compared to the BAM-1s strategy. Although this was successful, the BAM-2s still underperformed compared to the BAM regarding myogenesis, as shown by the image analysis and qPCR data. This can be explained by the need to maintain the BAM-2s in EGM for one week. Myogenic cell lineages are typically cultured in a high-glucose basal medium with at least 10% FBS while the EGM-2 medium only contains 5% FBS. The negative impact on myogenic cell lineages when defining co-culture media has been described by us and others [10, 12, 40]. Still, the BAM-2s provided the best trade-off between vasculogenesis and myogenesis in a tissue engineered muscle construct.
Characterization of endothelial networks in vitro indicated that the most interconnected networks with the highest vascular density were observed in BAM-2s. Indeed, an increased endothelial cell density in the fibrin hydrogel was already described to result in an enhanced number of branches and total vessel length [13], and this is consistent with the high endothelial cell density in the coat of the BAM-2s approach. Also, our previous study showed improved endothelial network formation in vitro when a higher density of endothelial cells was used [10]. A well-organized endothelial network in vitro is important for the in vivo vascularization and tissue integration in the host tissues as shown before [5, 19]. One week after implantation, the extent of vascularization in the co-culture flaps was greater than in the myotube only flaps. Vascularization and efficient perfusion are also challenges in cardiac tissue engineering. Perfusable microvascular constructs containing patterned microchannels with endothelial cells integrated with the coronary vasculature of infarcted rat hearts to a greater degree than non-perfusable constructs at 5 d post-implantation [41].
Besides analysis of morphological parameters, collagen IV deposition was assessed. Collagen IV is the main component of the vascular basement membrane matrix. Basement membrane matrix assembly is a critical step in vessel maturation and vessel stability as observed in in vitro and developmental studies [33, 42–44]. Endothelial networks in BAM-1s were surrounded by a thin layer that contained collagen IV, suggesting the presence of a basement membrane, in contrast to endothelial networks in BAM-2s. In BAM-1s, endothelial cells were mixed with muscle cells containing both myoblasts and fibroblasts [10], in contrast to BAM-2s where only endothelial cells were present in the coat region. Several groups have successfully shown that addition of mesenchymal-derived cells, such as fibroblasts, pericytes and/or smooth muscle cells improved endothelial network maturation and in vitro/in vivo network stabilization in tissue engineered constructs [45–49]. These cells are able to differentiate into supportive mural cells around newly formed endothelial networks in co-culture settings with endothelial cells [47, 50, 51]. Pericytes for example are known for their role in stimulating basement membrane matrix deposition by endothelial cells [33]. Optimizing fibroblast—endothelial cell ratios and/or evaluation of other mesenchymal-derived cells as mural cells may still improve prevascularization outcome in the two-stage approach. In this study we have used HUVECs as endothelial cell source. HUVECs are easy to isolate, abundant and well characterized. Furthermore, they are capable of endothelial network formation in vitro [5, 10, 12]. Other examples of endothelial cell sources able to form endothelial networks are microvascular endothelial cells [52], embryonic heart endothelial cells [53], pluripotent cell derived endothelial cells [54] and blood outgrowth endothelial cells [55] and are worthwhile for further investigation in prevascularization strategies.
To evaluate whether prevascularization improves in vivo myotube survival and tissue perfusion we implanted BAMs with and without endothelial networks on the fascia of the latissimus dorsi muscle of NOD/SCID mice for 5 or 14 d. In all conditions, myotubes were still observed at day 14 of implantation in the construct. However, myotube length, density and alignment decreased in all conditions in vivo. Myotube length was decreased by 40% (BAM), 61% (BAM-1s) and 47% (BAM-2s) over 14 d while myotube density was decreased by 67.8% (BAM), 61% (BAM-1s) and 33,4% (BAM-2s). This deterioration in myotube characteristics over time could be caused by the initial lack of perfusion. The detrimental effects of this initial hypoxia state on the myotubes are not completely counteracted, as also reported elsewhere [56]. In addition, the supply of nutrients and oxygen in the first days in vivo was limited compared to the in vitro situation where constructs were completely submerged in a nutrient-rich medium. Finally, the decrease may also be explained by the occurrence of xenogeneic rejection. Although NOD/SCID mice lack functional T and B cells, have impaired macrophage function and lack the complement system [57], a low level of natural killer (NK) cell activity remains present [58–60]. Macrophage, neutrophil and mast cell invasion may also result in rejection, as seen with implantation of human mesenchymal stem cells on poly( -caprolactone) in a NOD/SCID model [60]. Immunodeficient animal models are also deprived of pro-regenerative effects of the immune response during skeletal muscle repair [61–63]. Inflammatory cells, such as macrophages and neutrophils, are recruited and start producing growth factors and cytokines important for satellite cell activation, proliferation and differentiation and capillary ingrowth to injured muscle [2]. For clinical application, use of human autologous muscle and endothelial cells would eliminate xenogeneic responses. In our study, the overall decrease in myotube density was least present for the BAM-2s which we hypothesize to be due to (i) the improved myotube characteristics before implantation compared to the BAM-1s and (ii) the pre-vascularization which was absent in the BAM. An implantation time exceeding 14 d may even improve myotube maturation as reported previously [64]. As the ultimate aim was to develop a muscle tissue engineering strategy with a prevascularization approach balancing between optimal vasculogenesis and myogenesis, the comparison between BAM-1s vs BAM-2s is crucial. We can conclude that for three out of four measured parameters for myotube characteristics, the two-stage prevascularization approach outperformed the one-stage approach.
-caprolactone) in a NOD/SCID model [60]. Immunodeficient animal models are also deprived of pro-regenerative effects of the immune response during skeletal muscle repair [61–63]. Inflammatory cells, such as macrophages and neutrophils, are recruited and start producing growth factors and cytokines important for satellite cell activation, proliferation and differentiation and capillary ingrowth to injured muscle [2]. For clinical application, use of human autologous muscle and endothelial cells would eliminate xenogeneic responses. In our study, the overall decrease in myotube density was least present for the BAM-2s which we hypothesize to be due to (i) the improved myotube characteristics before implantation compared to the BAM-1s and (ii) the pre-vascularization which was absent in the BAM. An implantation time exceeding 14 d may even improve myotube maturation as reported previously [64]. As the ultimate aim was to develop a muscle tissue engineering strategy with a prevascularization approach balancing between optimal vasculogenesis and myogenesis, the comparison between BAM-1s vs BAM-2s is crucial. We can conclude that for three out of four measured parameters for myotube characteristics, the two-stage prevascularization approach outperformed the one-stage approach.
Next to examining the effects of prevascularization on myotube maintenance in vivo, we also evaluated the effect on in vivo vascularization. Prevascularized BAMs showed a significantly higher vascular density in vivo compared to BAMs with only muscle cells, as reported previously by others in tissue-engineered constructs [12, 19]. In this study, already at day 5 of implantation anastomosis of the implanted networks with host vessels was observed by the presence of red blood cells in the human endothelial networks. Anastomosis was further confirmed by perfusion of a fluorescent dye injected in the tail vein, which colocalized with the transplanted endothelial cells. Similar to our findings, evidence of anastomosis of endothelial networks in a fibrin gel (without muscle cells) at day 5 PI was found [13]. This anastomosis speed may not be sufficient for oxygen-sensitive tissues such as cardiomyocytes causing ischemia that leads to cell death [65]. Improving the structure and maturation of in vitro networks and optimizing the mesenchymal-derived cell/endothelial cell ratio as proposed before [19, 35, 47, 49], addition of exogenous growth factors (e.g. VEGF, FGF and PDGF) to promote in vivo vascularization [66] and optimizing fibrin concentration, type and ultrastructure [37, 67] may further reduce anastomosis time and increase vascularization. Despite anastomosis of HUVEC endothelial vessels, shown by red blood cell presence, no perfusion of intravenously injected agglutinin was observed at day 5, suggesting a restricted perfusion. It has been observed with laser speckle imaging that thrombi occurred early after anastomosis of implanted endothelial vessels with host vessels [68]. By day 7 however, functional perfused vessels were reported [68]. In our experiments, onset of full perfusion as demonstrated with fluorescent agglutinin occurred between day 5 and 14 after implantation and showed functionality of the endothelial networks. No differences were found in endothelial network parameters between the one-stage and the two-stage approach constructs.
Fibrin degradation is controlled in vitro by modulating the fibrinolysis through fibrinolysis inhibitors. However, in vivo fibrinolysis occurs, which results in the rapid degradation of fibrin ECM. Nevertheless, fibrin BAMs maintained structural integrity over 14 d and fibrin ECM was remodeled to collagen as shown by MSB staining in constructs explanted after 14 d. This is in line with previous in vivo studies in which fibrin degradation was observed over two weeks in acellular fibrin clots [69] and collagen deposition was shown in tissue engineered skeletal muscle with a fibrin ECM [64]. Recent work from our group showed that replacement of the degrading fibrin ECM through deposition of autologous collagen enabled the maintenance of the BAM without fibrinolysis inhibition, while increasing the visco-elastic properties of the BAMs [70]. This ECM remodeling can be attributed primarily to fibroblasts that make up 30% of the cell population in the primary human skeletal muscle cells used for making the BAMs. Furthermore, since electrical stimulation has been demonstrated to increase collagen deposition as well, maturating myotubes may also contribute to collagen deposition [71]. The same can be said about ECs which have been shown to locally degrade or deposit matrix proteins upon sprouting [72]. The differences in ECM we observed between the inner myotube part versus the outer endothelial coat of the BAM-2s may be explained by the presence of the vasculature. Taken together, many cell types may contribute to collagen deposition and the results obtained in this work are a combined effect of the presence of fibroblasts and the increased vascularization and myogenesis in and around the implants. ECM remodeling should be controlled, since too extensive collagen deposition might result in fibrotic scarring. This would ultimately interfere with the engraftment and graft function. In this context, the elimination of an inflammatory trigger and resolution of inflammation are important for preventing fibrosis [73]. Furthermore, characterizing the presence of invading immune cells could help understand the evolution of ECM remodeling and engraftment [74, 75]. From our results, no sign of fibrotic tissue was observed around implanted BAMs on histological sections at day 14. Together with the high vascular density and perfusion at day 14 this proves successful engraftment of BAM-1s and BAM-2s as opposed to the muscle cell only BAM, which was not perfused.
In conclusion, we tested two prevascularization strategies of bio-artificial skeletal muscle and evaluated construct survival and engraftment in the host tissue after implantation in vivo. The two-stage prevascularization approach resulted in an endothelial network that became functional in vivo and supported best myotube survival. Although further improvements can still be done, this strategy opens perspectives for engineering larger skeletal muscle constructs.
Acknowledgments
The authors are grateful to Stephanie De Vleeschauwer, Erna Dewil and Marleen Lox, Aernout Luttun, Sander Craps, Greetje Vande Velde, Veerle Baekelandt and Joris Van Asselberghs for technical assistance, Rik Gijsbers for miRFP670 cDNA-containing HIV1-derived retroviral vector, Evie Vereecke for human muscle samples, Laurens Willaert and Rune Peyskens for myotube morphological analysis and Sigrid Vanryckeghem for administrative support. This work was funded by Fonds Wetenschappelijk Onderzoek—Vlaanderen (1.5.298.17 N and mandate to Lisanne Terrie); and AFM-Telethon trampoline grant (19802).
Conflict of Interest
The authors confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Data availability
The raw data required to reproduce these findings are available to download from https://data.mendeley.com/datasets/6p47mbpr9f/draft?a=e80a2562-a984-4dad-8adb-6bd3060ecac5 and https://data.mendeley.com/datasets/8gkvvy8j9h/draft?a =1194a3ca-b02e-4627-b1a5-5283b8698986. The processed data required to reproduce these findings are available to download from https://data.mendeley.com/datasets/595jwcnnkm/draft?a=8ff32c59-4c2f-4312-8c91-18c654ed6a6f.
Author contributions
DG and LTh contributed to conception and designed the project. DG performed in vitro experiments. DG, KM, LH, LTe, JD were responsible for in vivo experiments. DG, LD, LTe performed data collection. DG, Lte, MG and GC performed data analysis and GC performed customized software development. DG and MG performed interpretation and statistical analysis. DG and LTe wrote the manuscript with input from all authors. LTh supervised the project. All authors read and approved the manuscript.