
Abstract
Fourteen unprecedented artificial Erythrina alkaloids were isolated from the Erythrina variegata, E. crista-galli and E. arborescens (Fabaceae). The structures of these alkaloids were determined by spectroscopic analyses. Their possible formations were proposed. All isolated compounds showed no cytotoxicity and hypoglycemic activity at cell screening bioassay.
Similar content being viewed by others


1 Introduction
The Erythrina-type alkaloids with 6/5/6/6 spirocycle systems and stable 5S-chiral center are derived from two tyrosine units via oxidative coupling and intramolecular rearrangement. Since the first phytochemical research on Erythrina alkaloid in 1930s [1], the total number now stands at well over 110 alkaloids reported from plants of Erythrina genus [2]. Bioassay screening of these alkaloids showed anxiolytic-like actitivity [3], induced sleep [4], anticonvulsant actitivity [5], neuronal nicotinic acetylcholine receptor antagonism [6], leishmanicidal [7], anticataract [7], and antifeedant activity [8]. Our previous research disclosed natural dimeric [9, 10] and trimeric [11] Erythrina alkaloids, alkaloidal glucosides [10], and complex monomers [12,13,14]. However, during the extraction and separation of Erythrina alkaloids, some artificial products would be produced. Consideration of the NMR spectra characteristics and potential activities of Erythrina alkaloid, we here systematically summarized these artifacts from E. arborescens, E. crista-galli and E. variegata (Fig. 1). Their cytotoxicity against three cancer cells and hypoglycemic activity on 3T3-L1 myoblasts cell were screened. This paper will describe their isolation, structure determination and possible mechanism of formation.

Chemical structures of 14 artificial Erythrina alkaloids
2 Results and Discussion
Alkaloid 1 was obtained as white amorphous powder. Its IR absorption bands at 3441, 1640, 1503, 1480 cm−1 indicated the presence of the hydroxyls and aromatic rings. Moreover, the UV absorptions at 204, 239 and 290 nm indicated a tetrahydroisoquinoline chromophore [15]. These spectra were consistent with the characteristics of an Erythrina alkaloid. Alkaloid 1 had a molecular formula C39H40N2O9 as established by the HRESIMS m/z at 681.2811 [M+H]+, together with the 1H and 13C NMR spectroscopic data. In the 1H NMR spectrum of 1 (Table 1), four aromatic singlet proton (δH 7.08, 7.01, 6.74 × 2), two pairs of olefin [δH 7.16 (dd, J = 10.2, 4.2 Hz), 5.92 (d, J = 10.2 Hz), and 6.56 (dd, J = 10.2, 2.4 Hz), 6.06 (d, J = 10.2 Hz)], two methylenedioxy group (δH 5.96, 5.94, 5.93 and 5.92), and two methoxyl group (δH 3.29 and 3.24) signals indicated that 1 might be an Erythrina alkaloid dimer. In comparison with the reported dimer, erythrivarine A [9], alkaloid 1 had three more signals at δC 208.1, 47.5 and 31.2 in the 13C NMR spectra and 56 mass units higher in molecular weight, which showed an extra 2-oxopropyl group in 1. In the HMBC spectrum, correlations of δH 4.20 (H-8) with δC 53.9 (C-10), δC 136.2 (C-7) and δC 208.1 (C=O) indicated the 2-oxopropyl group at C-8. Further analysis of the 2D NMR revealed that the other parts of compound 1 were consistent with those of erythrivarine A.
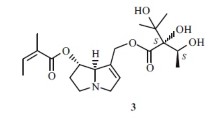
Alkaloids 2 and 3 showed the same molecular formula C40H42N2O9 as established by HRESIMS (m/z 695.2968 [M+H]+ in 2; m/z 695.2970 [M+H]+ in 3). In the 1H and 13C NMR spectra, the chemical shifts of 2 and 3 showed good agreement with those of 1, except those signals around the C-11′ position (C-10′/11′/12′). The C-11′ carbon of 1 was resonated at δ 64.9, however, signals of the same carbon were observed at δ 74.7 and δ 74.5 in 2 and 3, respectively. In addition, an extra methoxyl group (δH 3.54 in 2, δH 3.36 in 3) was observed, which indicated that both 2 and 3 had a methoxyl group at the C-11′ position instead of a hydroxyl group in 1. Chemical shifts of H-10′, H-11′ and H-17′ protons (Table 1) of 2 and 3 implied the configuration of 11′-OCH3 in 2 and 3 were different. In the ROESY spectrum, the NOE correlation of H-3′β/H-4′β and H-11′/H-4′α in 3 suggested its 11′-OCH3 was in β-orientation. The NOE correlation of H-11′/H-4′β in 2 suggested its 11′-OCH3 was in α-orientation.
The HRESIMS of 4 showed the pseudomolecular ion at m/z 370.1651 [M+H]+ (calc. for C21H23NO5, 370.1652). The 13C NMR spectrum of 4 showed a methine at 64.7 ppm, which indicated the existence of hydroxy substituent. The HMBC correlation between δH 7.05 (H-17) with δC 64.7 suggested the hydroxy at C-11. Through detailed comparison of the 1D and 2D NMR spectrum, 4 was basically the same as erythranine [16] except for the 2-oxopropyl substituent (δC 207.9, 47.8 and 30.8). The HMBC spectrum showed correlations from δH 3.98 (H-8) to δC 52.6 (C-10), δC 127.6 (C-7) and δC 207.9 (C=O) disclosed 4 to be 8-(2-oxopropyl)-erythranine.
Alkaloid 5 displayed a hydrogen adduct ion at m/z 354.1709 [M+H]+ (calc. for C21H23NO4, 354.1707). The 1D NMR spectroscopic data of compound 5 were similar to those of 4 except for the following differentiations: in the 1H NMR spectrum, the signal displayed at δH 4.72 in 4 which was assigned to the active hydrogen in the hydroxy was disappeared in compound 5. Correspondingly, the methine signal at δC 64.7 (C-11) in compound 4 was replaced with a methylene (δC 26.0) in 5. Thus, compound 5 was an analogue of 4 without the hydroxy moiety and determined to be 8-(2-oxopropyl)-erythraline.
The HRESIMS of 6 gave a hydrogen adduct ion at m/z 384.1806 [M+H]+, indicative of a molecular formula of C22H25NO5. In comparing with those of 4, the 1H NMR spectrum of 6 gave signal of an addional methyoxyl group (δH 3.55, s, 3H), and its 13C NMR spectrum showed an downfield chemical shift δC 74.8. These findings suggested the C-11 of 6 was substituted by a methoxy rather than a hydroxy. Thus, the structure of 6 was determined to be 8-(2-oxopropyl))-11-methoxy-erythraline.
The molecular formula of 7 was determined to be C21H25NO5 from the HRESIMS m/z at 394.1626 [M+Na]+. Its 1H NMR spectrum showed two aromatic singlet protons (δH 6.84 and 6.76), three conjugate olefin signals (δH 6.60, 6.10 and 5.67), and four methoxy groups (δH 3.28, 3.70, 3.78 and 3.97). The 13C NMR spectrum of 7 showed three methylenes (δC 24.6, 43.3, 44.4), two methines (δC 76.9 and 70.5) and a carbonyl (δC 172.1). These data suggested 7 might be a carbomethoxyl derivative of erysotrine [17]. The HMBC correlations from δH 3.78 (OCH3) and δH 4.33(H-8) to δC 172.1 (C=O) assigned the carbomethoxy at C-8. The molecular formula of 8 was determined to be C20H21NO6 from the HRESIMS m/z at 372.1444 [M+H]+. The 1H and 13C NMR the structural pattern of 8 was identical to that of 5, and the additional carbomethoxyl moiety was identical to that of 7. Accordingly, the structures of 7 and 8 were determined to be 8-carbomethoxyerysotrine and 8-carbomethoxyerythranine, respectively.
Alkaloid 9 showed molecular ion peaks at m/z 370.1652 [M+H]+, suggesting the molecular formulae C21H24NO5. In comparing with compound 5, the 1H and 13C NMR signal of δH 2.13 (CH3) and δC 207.7(CH2COCH3) in 5 were changed to δH 3.61 (OCH3) and δC 172.5(CH2COOCH3) in 9, respectively. The remaining NMR data were almost identical to those of 5. Thus, the structure of 9 was determined to be 8-acetatemethoxyerythraline.
The molecular formulas of compounds 10 and 11 were deduced to be C21H23NO6 and C22H25NO6 from the HRESIMS at m/z 386.1599 [M+H]+ and 400.1758 [M+H]+, respectively. The 1H and 13C NMR data of both 10 and 11 are very similar to those of 9 except that the methylene signal was replaced by an oxymethine signal at the C-11 position. Further, in the 13C NMR, the signal for C-11 appeared at 64.8 and 74.7 ppm for compounds 10 and 11, similar to that of 4 and 6, respectively. Thus, 10 was identified as 8-acetatemethoxyerythranine. 11 had an extra methoxy and was identified as 8-acetatemethoxy-10β-methoxyerythraline.
The molecular formula 12 was established as C22H27NO5 based on the HRESIMS m/z = 386.1964 [M+H]+. From the 1H and 13C NMR data, the structure of 12 was very similar to 9 except for the replacement of methylenedioxy group by two methoxys at C-15 and C-16. This was confirmed from the HMBC and HSQC spectra. Alkaloid 12 was thus identified as 8-acetatemethoxyerysotrine.
The HRESIMS m/z at 394.1628 [M+Na]+ of 13 assigned the molecular formula to be C21H25NO5, 58 mass units higher than that of erysotrine. Its 13C NMR spectrum gave an additional methylene (δC 34.9) and a carbonyl (δC 172.3) signals, indicating the existence of an acetyl group. The HMBC correlations from δH 2.51 (CH2CO) and δH 4.14 (H-8) to δC 172.3 (C=O) suggested that the acetyl group was located at C-8. Accordingly, the structure of 13 was determined to be 8-acetylerythsotrine. The molecular formula of 14 was dirermined to be C20H21NO5 by the HRESIMS m/z at 378.1313 [M+Na]+. The 1D NMR spectrum gave signals similar to that of erythraline expect for the replacement of a methylene by an acetyl group (δC 35.4 and δC 172.3). In the HMBC spectrum, the correlations from δH 4.07 (H-8) to δC 172.3 (COOH) and δH 2.74 (CH2COOH) to δC 65.4 (C-8) and δC 172.3 (COOH) confirmed that 14 was an 8-acetyl derivative of erythraline.
The configurations of H-8 for compound 1–14 were determined to be β based on ROESY experiments with correlations of H-3β/H-4eq, H-4eq/H-10ax and H-10eq/H-8 (Fig. 2). Further, together with 5S-configuration in all Erythrina alkaloids [18], so absolute configuration of alkaloids 1–14 could be determined.

Selected NOE interaction of alkaloid 9
Since N-containing compounds were main candicates of anticancer and hypoglycemic drugs, so alkaloids 1–14 were evaluated for their cytotoxicity against human A-549 lung cancer, SGC-7901 gastric cancer, and HeLa cell lines using the MTT method. In addition, their hypoglycemic activity on 3T3-L1 myoblasts cell were screened. Unfortunately, none of them showed positive activity. Alkaloids 1–14 possessed acetonyl, acetyl methyl, acetate, or methyl formate groups, which indicated they were artificial products. Without considering the artifitial units, these alkaloids are known. Duing the extraction and isolation, methanol, acetone, petroleum ether, especial ethyl acetate, were used as solvents. Accordingly, acetone and residual of acetic acid, methyl acetate and methyl formate in above solvents would become reaction reagents. Alkaloids 1–6 and 9–14 were formed firstly through an iminium immediate by oxidation, then by nucleophilic attack from carbanion of acetone, acetic acid, and methyl acetate in base condition. On the other hand, the iminium immediate could be tautomerized to inmine and attacked to methyl formate, generating the carbomethoxy substitued products (7–8) (Fig. 3).

Possible formation of two typically artificial Erythrina alkaloids
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were measured with a Jasco p-1020 digital polarimeter. UV spectra were recorded on a Shimadzu 2401PC spectrophotometer. IR spectra were obtained on a Bruker Tensor 27 infrared spectrophotometer with KBr pellets. 1H, 13C and 2D NMR spectra were obtained on Bruker AV-600, AVANCE III-500 and 400 MHz spectrometers with SiMe4 as an internal standard. Chemical shifts (δ) were expressed in ppm with reference to the solvent signals. MS data were recorded on an UPLC-IT-TOF MS. Column chromatography (CC) was performed on either silica gel (200–300 mesh, Qingdao Marine Chemical Co., Ltd., Qingdao, China) or RP-18 silica gel (20–45 μm, Fuji Silysia Chemical Ltd., Japan). Fractions were monitored by TLC on silica gel plates (GF254, Qingdao Marine Chemical Co., Ltd., Qingdao, China), and spots were visualized with Dragendorff’s reagent spray. MPLC was performed using a Buchi pump system coupled with RP-18 silica gel-packed glass columns (15 × 230 and 26 × 460 mm, respectively). HPLC was performed using Waters 1525 pumps coupled with analytical or preparative Sunfire C18 columns (4.6 × 150 and 19 × 250 mm, respectively). The HPLC system employed a Waters 2998 photodiode array detector and a Waters fraction collector III.
3.2 Plant Material
Flowers of E. variegata Linn and E. crista-galli Linn were collected in February and April, respectively, 2014 in Simao of Yunnan Province, People’s Republic of China. Leaves and flowers of Erythrina arborescens Roxb. Hort. Beng were collected in October 2014 in Jianshui of Yunnan Province. These plant samples were identified by Dr. Chun-Xia Zeng. The voucher specimens (Cai20140207, Cai20140407, Cai20141003 and Cai20141004) have been deposited in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, the Chinese Academy of Sciences.
3.3 Extraction and Isolation
The dried and powdered flowers of E. variegata (10.0 kg) were extracted with 90% MeOH (25 L) for three times. The extracts were concentrated under reduced pressure, and then dissolved in 2% acetic acid to adjust pH to 2–3 and then partitioned twice with EtOAc. The aqueous layers were basified with NH3·H2O to adjust pH to 8–9 and then extracted with EtOAc to give a crude alkaloid fraction (110 g). The crude alkaloid was subjected to column chromatography (CC) over silica gel with gradient CHCl3-Acetone (1:0 to 1:1) to afford seven fractions (Fr. I–Fr. VII). Fr. I (6.1 g) was divided into 2 subfractions (Fr. I-1–Fr. I-2) by using RP-MPLC eluting with MeOH-H2O (50–100%). Fr. I-2 was isolated by preparative C18 HPLC column with a gradient of MeOH–H2O (60:40–70:30, v/v) to obtain 9 (35 mg) and 11 (41 mg). Fr. II (4.5 g) was separated using C18 MPLC column with a gradient of MeOH–H2O (40:60–90:40, v/v) to afford 10 (35 mg) and 8 (13 mg). Fr. IV (8.5 g) was fractionated by C18 MPLC column with a gradient of MeOH–H2O (10:90–90:10, v/v) to give six subfractions (Fr. IV-1–Fr. IV-6). Fr. IV-2 was subjected to a preparative C18 HPLC column with a gradient of MeOH–H2O (50:50–60:40, v/v) to afford 6 (20 mg) and 5 (32 mg) Fr. IV-4 was subjected to a preparative C18 HPLC column with a gradient of MeOH–H2O (70:30–80:20, v/v) to give 1 (5 mg). Fr. V (12 g) was chromatographed on a C18 MPLC column eluted with a gradient of MeOH–H2O (10:80–100:0, v/v) to give six subfractions (Fr. V-1–Fr.V-6). 2 (29 mg) and 3 (21 mg) was obtained from Fr.V-5 using a preparative C18 HPLC column with a gradient of MeOH–H2O (65:35–25:75, v/v).
Flowers of E. crista-galli (11 kg) were powdered and extracted with 90% MeOH (25 L) for three times. The extract was concentrated in vacuo to give a brown residue. The crude alkaloid (90 g) were obtained using the same acid–base treatment method described above, and then subjected to column chromatography (CC) over silica gel and eluted with gradient CHCl3-Acetone (1:0 to 1:1) to afford four fractions (Fr. I–Fr. IV). Fr. I (12.1 g) was further chromatographed on a C18 MPLC column eluted with a gradient of MeOH–H2O (50:50–100:0, v/v) to give the two subfractions (Fr. I-1–Fr. I-2). Alkaloid 12 (51 mg) was obtained from Fr. I-1 using a column chromatography (CC) over silica gel and eluted with petroleum ether-acetone (4:1).
Crude alkaloid extract (85.2 g) and (62.5 g) were obtained from the leaves (15.8 kg) and flowers (6.5 kg) of Erythrina arborescens, respectively. The crude alkaloid of leaves was divided into nine fractions (Fr. I–Fr. IX). Fr. V (7.1 g) was fractionated by C18 MPLC column with a gradient of MeOH–H2O (20:80–80:20, v/v) to give three subfractions (Fr. V-1–Fr. V-3). Fr. V-3 was subjected to a preparative C18 HPLC column with a gradient of MeOH–H2O (60:40–70:30, v/v) to afford 4 (1 mg) and 14 (5 mg). The crude alkaloid of flowers was divided into seven fractions (Fr. I–Fr. VII). 7 was obtained from Fr. II by C18 MPLC column with a gradient of MeOH–H2O (40:60–100:0, v/v) and then purified by preparative C18 HPLC column with a gradient MeOH–H2O (50:50–60:40, v/v). Fr. IV (4.5 g) was chromatographed on a C18 MPLC column eluted with a gradient of MeOH–H2O (20:80–70:30, v/v) to give four subfractions (Fr. IV-1–Fr. IV-4). Fr. IV-3 was further purified by a preparative C18 HPLC column with a gradient of MeCN–H2O (25:75–35:65, v/v) to afford 13 (20 mg).
8α-(2-oxopropyl)-erythrivarine A (1): white powder; \([\alpha]^{20}_{\text D}\) − 121.2 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 204 (4.26), 239 (3.88) and 290 (3.46) nm; IR (KBr) νmax 3441, 2924, 1640, 1503, 1480, 1226, 1100, 1041 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Table 1; positive HRESIMS m/z 681.2811 [M+H] + (calcd. for C39H41N2O9, 681.2110).
8α-(2-oxopropyl)-11′-O-methyl-erythrivarine A (2): white powder; \([\alpha]^{20}_{\text D}\) −114.1 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 203 (4.12), 238 (3.66) and 289 (3.43) nm; IR (KBr) νmax 3441, 1639, 1490, 1234, 1101, 1040 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Table 1; positive HRESIMS m/z 695.2968 [M+H]+ (calcd. for C40H43N2O9, 695.2969).
8α-(2-oxopropyl)-11′-epi-O-methyl-erythrivarine A (3): white powder; \([\alpha]^{20}_{\text D}\) −165.2 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 204 (4.22), 238 (3.88) and 289 (3.46) nm; IR (KBr) νmax 3441, 2924, 1629, 1503, 1482, 1234, 1100, 1040 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Table 1; positive HRESIMS m/z 695.2970 [M+H] + (calcd. for C40H43N2O9, 695.2969).
8α-(2-oxopropyl)-erythrinine (4): white powder; \([\alpha]^{20}_{\text D}\) + 133.0 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 204 (4.71), 239 (3.63) and 289 (3.32) nm; IR (KBr) νmax 2930, 1630, 1503, 1489 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Tables 2 and 4; positive HRESIMS m/z 370.1651 [M+H]+ (calcd. for C21H24NO5, 370.1652).
8α-(2-oxopropyl)-erythraline (5): white powder; \([\alpha]^{23}_{\text D}\) + 80.5 (c 0.10, MeOH); UV(MeOH) λmax (log ε) 201 (4.11), 238 (2.37), 289(1.29) nm; IR (KBr) νmax 1712, 1480, 1423 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Tables 2 and 4; positive HRESIMS m/z 354.1709 [M+H]+ (calcd. for C21H23NO4, 354.1707).
8α-(2-oxopropyl)-11-methoxy-erythraline (6): white powder; \([\alpha]^{20}_{\text D}\) + 95.3 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 204 (4.76), 238 (3.74) and 288 (3.48) nm; IR (KBr) νmax 1631, 1504, 1483 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Tables 2 and 4; positive HRESIMS m/z 384.1806 [M+H]+ (calcd. for C22H26NO5, 384.1805).
8α-carbomethoxyerysotrine (7): white powder; \([\alpha]^{22}_{\text D}\) + 74.0 (c 0.21, CH3OH); UV (CH3OH) λmax (log ε) 203 (3.79), 225 (3.45) and 277 (3.05) nm; 1H (400 Hz) and 13C (125 Hz) NMR data (acetone-d6), Tables 2 and 4; Positive ESIMS m/z 394 [M+Na]+., HRESIMS m/z. 394.1626 [M+Na]+; (calcd. for C21H25NO5Na, 394.1625).
8α-carbomethoxyerythrinine (8): white powder; \([\alpha]^{20}_{\text D}\) + 118.3 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 205 (4.69), 239 (3.79) and 289 (3.51) nm; IR (KBr) νmax 2923, 1630, 1503, 1488 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Tables 2 and 4; positive HRESIMS m/z 372.1444 [M+H]+ (calcd. for C20H22NO6, 372.1443).
8α-acetatemethoxyerythraline (9): white powder; \([\alpha]^{20}_{\text D}\) + 171 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 204 (4.78), 239 (3.88) and 290 (3.52) nm; IR (KBr) νmax 1628, 1501, 1489 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Tables 3 and 4; positive HRESIMS m/z 370.1652 [M+H]+ (calcd. for C21H24NO5, 370.1653).
8α-acetatemethoxyerythrinine (10): white powder; \([\alpha]^{20}_{\text D}\) + 89.4 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 204 (4.65), 238 (3.68) and 290 (3.33) nm; IR (KBr) νmax 2924, 1628, 1488 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Tables 3 and 4; positive HRESIMS m/z 386.1599 [M+H]+ (calcd. for C21H24NO6, 386.1598).
8α-acetatemethoxy-11β-methoxyerythraline (11): white powder; \([\alpha]^{20}_{\text D}\) + 154.2 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 204 (4.62), 238 (3.74) and 289 (3.41) nm; IR (KBr) νmax 1630, 1503, 1484 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Tables 3 and 4; positive HRESIMS m/z 400.1758 [M+H]+ (calcd. for C22H26NO6, 400.1757).
8α-acetatemethoxyerysotrine (12): white powder; \([\alpha]^{20}_{\text D}\) + 113.3 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 204 (4.72), 239 (3.66) and 288 (3.51) nm; IR (KBr) νmax 1628, 1503, 1488 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Tables 3 and 4; positive HRESIMS m/z 386.1964 [M+H]+ (calcd. for C22H28NO5, 386.1965).
8α-acetylerythsotrine (13): white powder; C21H25NO5; \([\alpha]^{22}_{\text D}\) + 5.2 (c 0.18, CH3OH); UV (CH3OH) λmax (log ε) 203 (3.90), 229 (3.56) and 282 (2.93) nm; 1H (400 Hz) and 13C (125 Hz) NMR data (acetone-d6), Tables 3 and 4; Positive ESIMS m/z 394 [M+Na]+., HRESIMS m/z. 394.1628 [M+Na]+; (calcd. for C21H25NO5Na, 394.1625).
8α-acetylerythraline (14): white powder; \([\alpha]^{20}_{\text D}\) + 105 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 204 (4.71), 249 (3.72) and 289 (3.62) nm; IR (KBr) νmax 3341, 1639, 1503, 1442 cm−1; 1H (600 MHz) and 13C NMR (150 MHz) data (acetone-d6), see Tables 3 and 4; positive HRESIMS m/z 378.1313 [M+H]+ (calcd. for C20H22NO5, 378.1314).
3.4 Cytotoxicity
The human A-549 lung cancer, SGC-7901 gastric cancer, and HeLa cell lines were used in the cytotoxic assay. These cells were grown in DMEM media (HyClone, USA) supplemented with 10% fetal bovine serum (HyClone, USA) at 37 °C in 5% CO2. The cytotoxicity of all alkaloids were determined based on the MTT method in 96-well microplates. In short, 100 µL adherent cells were seeded into each well and incubated for 12 h before the addition of the test alkaloids/drug. At the same time, the suspended cells were seeded at an initial density of 1 × 105 cells/mL just before the addition of the alkaloids/drug. Each tumor cell line was exposed to a test compound at concentration 20 μM in DMSO in triplicate for 48 h, with camptothecin as the positive control. After treatment, cell viability was assessed.
3.5 Hypoglycemic Activity
3T3-L1 myoblasts cells were purchased from American Type Culture Collection (Manassas, VA). Cells were maintained in DMEM supplemented with 10% FBS or CS (for 3T3-L1 cells), 100 units/ml penicillin and 100 mg/ml streptomycin in 10 cm diameter dishes in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. Cells were maintained in continuous passages by trypsinization of subconfluent cultures and fed fresh medium every 48 h. For differentiation, L6 myoblasts were transferred to DMEM with 2% FCS in tissue culture plates for 5-6 days, 3T3-L1 cells were exposed to 0.5 mM IBMX, 1 mM dexamethasone, 1 mM rosiglitazone and 1 mg/mL insulin for 3 days, and 1 mg/ml insulin for the other day. For glucose uptake assay, cells were serum starved for 4 h in 96-well plates, followed by incubated with insulin and alkaloids 1–14 for 24 h. Finally, the supernatants of cultured cells were collected and subjected to glucose assay using a commercially kit. The quantified values were normalized based on the results of the MTS assay.
References
K. Folkers, R.T. Major, J. Am. Chem. Soc. 59, 1580–1581 (1937)
A.F. Parsons, M.J. Palframan, Alkaloids 68, 39–81 (2010). https://doi.org/10.1016/S1099-4831(10)06802-1
O. Flausino, L.D. Santos, H. Verli, A.M. Pereira, V.D. Bolzani, R.L. Nunes-de-Souza, J. Nat. Prod. 70, 48–53 (2007)
M. Ozawa, K. Honda, I. Nakai, A. Kishida, A. Ohsaki, Bioorg. Med. Chem. Lett. 18, 3992–3994 (2008)
S.A. Faggion, A.O.S. Cunha, H.A. Fachim, A.S. Gavin, W.F. dos Santos, A.M.S. Pereira, R.O. Beleboni, Epilepsy Behav. 20, 441–446 (2011)
F. Crestey, A.A. Jensen, M. Borch, J.T. Andreasen, J. Andersen, T. Balle, J.L. Kristensen, J. Med. Chem. 56, 9673–9682 (2013)
D.R. Callejon, T.B. Riul, L.G.P. Feitosa, T. Guaratini, D.B. Silva, A. Adhikari, R.L. Shrestha, L.M.M. Marques, M.D. Baruffi, J.L.C. Lopes, N.P. Lopes, Molecules 19, 5692–5703 (2014)
W.W. Cornelius, T. Akeng’a, G.O. Obiero, K.P. Lutta, Rec. Nat. Prod. 3, 96–103 (2009)
B.J. Zhang, M.F. Bao, C.X. Zeng, X.H. Zhong, L. Ni, Y. Zeng, X.H. Cai, Org. Lett. 16, 6400–6403 (2014)
B.J. Zhang, W.N. Xiao, J. Chen, M.F. Bao, J. Schinnerl, Q. Wang, X.H. Cai, Tetrahedron (2019). https://doi.org/10.1016/j.tet.2019.130515
B.J. Zhang, B. Wu, M.F. Bao, L. Ni, X.H. Cai, RSC Adv. 6, 87863–87868 (2016)
J. Wu, B.J. Zhang, W.N. Xiao, M.F. Bao, X.H. Cai, RSC Adv. 7, 51245–51251 (2017)
J. Wu, B.J. Zhang, M.F. Bao, X.H. Cai, Nat. Prod. Res. 33, 2004–2010 (2019)
H.E. Zhao, J. Wu, F.Q. Xu, M.F. Bao, C.S. Jin, X.H. Cai, Nat. Prod. Res. 33, 1298–1303 (2019)
K. Ito, H. Furukawa, H. Tanaka, J. Chem. Soc. Chem. Commun. 17, 1076–1077 (1970)
K. Ito, H. Furukawa, H. Tanaka, Yakugaku Zasshi 93, 1215–1217 (1973)
A.S. Chawla, S. Chunchatprasert, A.H. Jackson, Org. Magn. Reson. 21, 39–41 (1983)
Y. Tsuda, T. Sano, Alkaloids 48, 249–337 (1996). https://doi.org/10.1016/S1099-4831(08)60032-2
Acknowledgements
This project is supported in part by the National Natural Science Foundation of China (31872677). The authors are grateful to Dr. Chun-Xia Zeng for identification of plant samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest.
Additional information
Dedicated to Professor Han-Dong Sun on the occasion of his 80th birthday.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, BJ., Wu, J., Bao, MF. et al. Artificial Erythrina Alkaloids from Three Erythrina Plants, E. variegata, E. crista-galli and E. arborescens. Nat. Prod. Bioprospect. 10, 57–66 (2020). https://doi.org/10.1007/s13659-020-00235-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-020-00235-3