
Abstract
Paeonia ludlowii is indigenous to Tibet and has an important ecological and economic value in China. In Tibet, P. ludlowii has been used in folk medicine with relative success. Plant microbial endophytes play an important role in plant growth, health and ecological function. The diversity of endophytic bacteria associated with P. ludlowii remains poorly understood. In this study, the structure of the endophytic bacterial communities associated with different tissues, including fruits, flowers, leaves, stems, and roots, and rhizosphere soils was analyzed with Illumina MiSeq sequencing of bacterial 16S rDNA. A total of 426,240 sequences and 4847 operational taxonomic units (OTUs) were obtained. The OTUs abundance of roots was higher than that of other tissues; however, the OTUs abundance was similar among different deep soil samples. In the plant tissues, Cyanobacteria was the most abundant bacterial phylum, followed by Proteobacteria; however, the most abundant phyla were Proteobacteria and Acidobacteria in soil samples from three different layers. In addition, the diversity and richness of the microorganisms in the soil were very similar to those in roots but higher than those in other tissues of P. ludlowii. Predictive metagenome analysis revealed that endophytic bacteria play critical functional roles in P. ludlowii. This conclusion could facilitate the study of the ecological functions of endophytic bacteria and their interactions with P. ludlowii to analyze the reasons why this important medicinal plant is becoming endangered.
Similar content being viewed by others


Introduction
The Tibetan peony, Paeonia ludlowii, is an endemic species to the Himalayan-Hengduan Mountains; it is renowned as a medicinal plant that reduces inflammation (Liu et al. 2017; Lou et al. 2017) and is only distributed in a small area of southern Tibet in western China (Hao et al. 2014). P. ludlowii, an important rare species with high economic, medicinal and ornamental value (Zhang et al. 1997), is on the verge of extinction as a result of its low germination percentage and long germination period and has been listed in the Endangered Species Red Book of China. Endophyte studies might provide a way to improve seed germination. A large number of articles have documented that endophytes can facilitate interim germination and improve the germination rate, length of coleoptiles and radicles, seedling dry weight, stress resistance and early plant development (Gao and Shi 2018; Li et al. 2017; Hubbard et al. 2014); however, the endophytes of P. ludlowii have not been explored.
Endophytes are nonpathogenic microbes that reside in healthy plant tissues and benefit both the plants and the microbes. Bacterial endophytes play important roles in plant growth, health and ecological function, conferring certain benefits to plants (Lumactud and Fulthorpe 2018). A growing body of literature has reported that bacterial endophytes promote plant growth, improve plant health, enhance plant tolerance to stress and provide many additional benefits (Azevedo et al. 2000; Hardoim et al. 2008; Glick and Stearns, 2011; Mitter et al. 2017). Endophytic bacteria have received increasing research attention because of their potential biological functions in recent years (Compant et al. 2005). Thus, understanding the community and diversity of endophytic bacteria in plant tissues is imperative.
With the development of society, endophytic bacteria of medicinal plants have gained more attention as a consequence of their substantial potential to synthesize numerous novel pharmaceutical compounds, such as antifungal, antibiotic, anticancer, antiviral, and immunosuppressant compounds (Golinska et al. 2015). Therefore, fully understanding the community and diversity of endophytic bacteria may help to exploit the potential of medicinal plants (El-Deeb et al. 2012). It is well known that the application of medicinal plants has a long history in China, and over 500 medicinal plants have been listed in the Chinese Pharmacopeia to date; hence, medicinal plants are a very important and relatively untapped source of pharmaceutical compounds (Golinska et al. 2015). Because of the importance of endophytic bacteria in plants, studying these endophytic bacteria is of immense significance.
In this study, we aimed to investigate the bacterial endophyte diversity and community distribution in P. ludlowii in the Himalayan-Hengduan Mountains and provided a potentially valuable strategy for the propagation of P. ludlowii by analyzing the endophytic bacteria in the different plant tissues and rhizosphere soils of this species.
Materials and methods
Sampling and treatment
P. ludlowii samples were collected from the Himalayan-Hengduan Mountains in MiRui Township (29º32′43.96′′N, 94º38′33.03′′E), Nyingchi City, Tibet, China. The tissues included fruit pods, flowers, leaves, stems, and roots. Rhizosphere soil was also collected (the soil was divided into upper, middle and lower layers at depths of 0–10 cm, 10–20 cm and 20–30 cm, respectively) during the P. ludlowii fruiting stage (June 5, 2018). The plant tissues and soil samples were randomly selected from 10 tree peonies and mixed. All types of samples were taken in triplicate and collected into sterile plastic bags and processed within 24 h. Plant samples were washed with water. Then, each tissue was surface-sterilized with 75% ethanol for 30 s and 10% sodium hypochlorite for 5 min. The samples were then washed with sterile PBS solution. Finally, the success of the sterilization was tested by wiping sterilized tissues across the surface of a Petri plate with Luria–Bertani (LB) medium to ensure the absence of bacterial growth. The soil samples were dried at 105 °C for 24 h.
DNA extraction, PCR amplification of the 16S rDNA region and sequencing
Approximately 2 g of sterilized plant tissues were frozen in liquid nitrogen and ground to a fine powder in a sterilized and precooled mortar. Total DNA was extracted using a bacterial DNA extraction kit (Omega, D3350-01, USA). The soil DNA was extracted using a soil DNA isolation kit (Sigma, DNB100-50RXN, USA). Three biological replicates were performed for each tissue and soil sample, and the quantity and quality of DNA were measured with a NanoDrop One spectrophotometer (Thermo Fisher Scientific, USA) and agarose gel electrophoresis (AGE).
The bacterial 16S rRNA gene was amplified using the following primer pairs: forward, 5′- ACTCCTACGGGAGGCAGCA, and reverse, 3′- GGACTACHVGGGTWTCTAAT (Masoud et al. 2011). The 25 μL PCR mixture contained 2.5 μL 10 × reaction buffer, 0.2 μL rTaq DNA polymerase (Invitrogen, USA), 2 μL 5 mmol/L dNTPs, 1 μL 5 μmol/L forward and reverse primers, 10 ng template DNA, and water to a final volume of 25 μL. PCR amplification was performed under the following conditions: initial denaturation at 94 °C for 5 min; 35 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min; a final extension at 72 °C for 10 min; and a final hold at 4 °C until further use. The PCR products were measured on 2% agarose gels containing Gold view and then purified with a DNA gel extraction kit (Tiangen, China). The purified DNA samples were subjected to high-throughput sequencing at the Beijing Genomics Institute (BGI) using Illumina technology. Illumina sequence reads were deposited under the NCBI SRA accession number PRJNA600148.
Sequence processing and analysis
Sequence processing and quality filtering, denoising, trimming, and merging of raw paired-end FASTQ files were performed using an improved dual-indexing approach and FLASH software to obtain clean tags (Fadrosh et al. 2014; Magoc and Salzberg 2011). The chloroplast and mitochondrial DNA was eliminated from further analyses. The obtained clean tags were assigned into operational taxonomic units (OTUs) using USEARCH (v7.0.1090) at 97% identity clustering (Edgar et al. 2013). The observed species index, Chao index, ACE index, Shannon index and Simpson index were determined using R software v3.1.1, which reflected the alpha diversity (Schloss et al. 2009). The Shannon diversity and Chao1 richness were determined and principal coordinate analysis (PCoA) was performed using QIIME (v1.80) (Caporaso et al. 2010). Heatmap and ternary plots were analyzed using R v3.1.1. All data were statistically analyzed using one-way analysis of variance. Tukey’s honestly significant difference test was used to separate means at p = 0.05.
Results
Analysis of sequencing data and bacterial community diversity
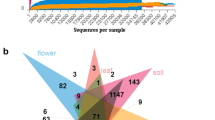
A total of 426,240 high-quality tag sequences were obtained with an average length of 250 bp across fruit, flower, leaf, stem, root, and soil samples after sequence denoising and quality filtering. The numbers of obtained sequences ranged from 43,322 to 59,410, with an average of 53,280 ± 7763 (mean ± SD) sequences across all eight samples. All quality-filtered sequences were clustered into 4847 OTUs at a 97% similarity level. The number of clustered OTUs varied from 43 to 3065. All rarefaction curves gradually saturated with increasing sequencing quantity in all 8 samples that covered the entire group. The results of rarefaction curves showed that OTU abundance was diverse in different tissue and soil samples. The numbers of OTUs were significantly higher in the soil samples than in the tissue samples (upper 3065, middle 3026, deep 2832). The root samples revealed a higher number of OTUs (1217), while fruit, stem and flower tissues showed lower richness, with 43, 46 and 65 OTUs, respectively. The different tissues shared 21 OTUs of the total 1500 OTUs (Fig. 1b), while 1984 OTUs of the total 8923 OTUs were shared among different soil samples (Fig. 1c).

a Rarefaction curves for bacterial endophyte OTUs in different tissues and soils of P. ludlowii. b Venn diagrams showing the number of shared and unique OTUs in different tissue and soil samples
Among the types of tissue, the highest richness and diversity of the bacterial community were found in roots, and the richness and diversity of the bacterial community were the lowest in stems (Table 1). In the rhizosphere soils, the upper soil and middle soil had the same diversity, whereas the richness of the bacterial community in the upper soil was higher than that in the middle soil, and the lowest richness and diversity were observed in the deep soil (Table 1).
Microbial taxonomic analysis at the phylum and class levels
The tag numbers of each taxonomic rank from phylum to species or each OTU in different samples were summarized with QIIME. The classification of sequences also demonstrated bacterial community differences in tissues and rhizosphere soils at the phylum level. In this study, the sequences were clustered into 43 bacterial phyla. The bacterial composition varied among the different tissues; for example, the number of bacterial phyla was higher in roots (28 phyla) than in fruits (8 phyla), while the phyla were similar in different rhizosphere soil samples from the deepest layer. It was obvious that Cyanobacteria was the most dominant phylum, accounting for 70.3–88.0% of all bacterial sequences in the different tissues, followed by Proteobacteria (11.9–19.6%). The abundance of each phylum varied in the different tissues. Actinobacteria, Verrucomicrobia, Bacteroidetes, Acidobacteria, and Firmicutes were also predominant phyla (> 1% relative abundance) in the root tissues, contributing to 1.2–3.4% of the relative abundance, whereas the abundance of these five phyla was less than 1% in other tissues. These results demonstrated that the relative abundance of bacterial communities in the roots was much higher than that in other tissues.
The abundance of bacterial communities was significantly higher in the rhizosphere soil than in the tissues. Proteobacteria and Acidobacteria were the dominant phyla, accounting for 21.2–31.1% and 20.9–23.6% of all bacterial sequences, respectively, followed by Actinobacteria (5.7–15.4%), Bacteroidetes (4.0–9.0%), Crenarchaeota (1.3–5.7%), Firmicutes (2.2–3.1%), Gemmatimonadetes (1.8–2.6%), Planctomycetes (5.6–8.0%), and Verrucomicrobia (9.9–13.6%) (Fig. 2a). At the class level, species in Cyanobacteria and Alphaproteobacteria showed virtually absolute dominance, representing 70.3–88.0% and 11.9–19.5% of the relative abundance, respectively. However, Actinobacteria (2.7%), Bacilli (1.1%), Gammaproteobacteria (2.1%), Saprospirae (1.6%), and Spartobacteria (1.2%) were also predominant (> 1% relative abundance) in the root tissues, whereas their abundance was less than 1% in other tissues. In the rhizosphere soil, Alphaproteobacteria, Acidobacteria-6, Betaproteobacteria, Chloracidobacteria, Gammaproteobacteria, and Spartobacteria were the predominant groups, comprising approximately 48.2% of the relative abundance at the class level (Fig. 2b), and the abundances of the classes exceeding 1% in every tissue are shown in Table 2. These results showed that the relative abundance of bacterial communities in the rhizosphere soil was significantly higher than that in the plant tissues.

The relative abundances of bacterial communities at (a) the phylum level and (b) the class level in the different samples
Bacterial community composition analysis at the family and genus levels
The heatmap analysis at the family level revealed that bacterial communities were classified into 40 families, which were significantly different between the plant tissues and rhizosphere soils (Fig. 3). The overall bacterial composition of the families differed significantly in the different samples. Chitinophagaceae was the most dominant family in the different plant tissues, accounting for 0.02–1.6% of the relative abundance. However, most of the sequencing data from the plant tissues, accounting for 76.4–88.0% of the total relative abundance, were not clustered into families, which resulted in a lower relative abundance in plant tissues, with the exception of the root tissues. These results demonstrated that the distribution of bacterial communities was much higher in root tissues than in other tissues. In the rhizosphere soil, the distribution and composition of bacterial communities were similar; the dominant families were Chthoniobacteraceae and Pseudomonadaceae, accounting for 6.6–9.0% and 3.4–6.8% of the relative abundance, respectively, followed by Chitinophagaceae (1.8–4.9%), Comamonadaceae (1.4–2.6%), Hyphomicrobiaceae (1.2–1.3%), Nitrososphaeraceae (1.3–5.7%), Sinobacteraceae (1.1–1.7%), and Sphingomonadaceae (1.6–2.4%).

Heatmap analysis of the bacterial composition at the family level for all sample types. The clustering indicates the similarity of certain species among different samples. The heatmap colors from red to green represent the relative abundances from low to high
The sequences were classified into 19 different genera, and the heatmap analysis revealed that Rhodanobacter was a genus specific to root tissues (Fig. 4). The distribution of each genus was significantly different across all sample types; for example, there were only three genera and 4 genera in fruit and flower tissues, respectively. DA101 and Pseudomonas were highly abundant in the rhizosphere soil, followed by Candidatus Nitrososphaera and Rhodoplanes. These results showed that the relative abundance of bacterial communities was much higher in the soil than in the plant tissues; nevertheless, the relative abundance of bacterial groups in the root tissues was the highest among the plant tissues, followed by leaves. The clustering analysis indicated that there were more similarities in the plant tissues, except for the roots, and the species compositions of the roots and soil were more similar at the family and genus levels (Figs. 3, 4).

Heat map analysis of the bacterial composition at the genus level for all sample types. The clustering indicates the similarity of certain species among different samples. The heat map colors from red to green represent the relative abundances from low to high
Comparative analysis of the bacterial communities in the different samples
To display the differences in OTUs composition in the different samples, PCA was used to illustrate that the bacterial communities in different tissues and soils formed individual clusters (Fig. 5a). The PCA indicated that the community structures were similar in fruit, flower, stem and leaf tissues; in contrast, there were significant differences in community structures among the root tissues and soils, which showed that the roots and soils had their own unique bacterial community structures (Fig. 5a). The results of complete linkage clustering (CLC) tree analysis were similar to those of the PCA (Fig. 5b). In the plant tissues, the roots and other tissues were separated into two different clusters, whereas the root and soil samples were classified into the same group (Fig. 5b). These results indicated that the microbiota in the root samples were largely different from those in other samples.

Beta diversity analysis; a principal coordinate analysis (PCoA) based on the relative abundance of bacterial OTUs. b The complete linkage clustering (CLC) of the bacterial communities in different samples based on unweighted UniFrac distances
Predictive metagenome analysis
The PICRUSt approach was used to perform functional classification with the KEGG Orthology (KO) database, and 6 of the level 1 KO groups were found according to their predicted metagenomes, which were involved in cellular processes, environmental information processing, genetic information processing, metabolism, organismal systems and, to a lesser extent, human diseases. A total of 81 levels 3 KO groups were found according to the predicted metagenomes. The major gene families were those relating to ABC transporters, DNA repair and recombination proteins, general function prediction, purine metabolism, peptidases, photosynthesis proteins, ribosomes and transporters (Fig. 6). The relative abundance was similar in the whole tissue and soil samples (Fig. 6).

Gene profiles of the bacterial community in P. ludlowii tissues and rhizosphere soil predicted using PICRUSt
Discussion
The data presented here aimed to explore the endophytic bacterial diversity and rhizosphere soil bacterial diversity of the endangered medicinal plant P. ludlowii. Most previous studies on plants with Illumina amplicon sequencing have demonstrated that endophytic bacteria play an important role in plant growth (Badri et al. 2009; Müller et al. 2016; Bulgarelli et al. 2012; Weyens et al. 2009). To our knowledge, this is the first report of Illumina amplicon sequencing being used to study the endophytic bacterial community structure in the endangered medicinal plant P. ludlowii.
The sequence analysis revealed that endophytic bacteria were clustered into 43 phyla; Cyanobacteria was the most dominant phylum, accounting for 70.3–88.0% of all bacterial sequences in the different tissues, followed by Proteobacteria (11.9–19.6%). Cyanobacteria are the oldest photoautotrophs with nitrogen fixation capabilities and synthesize a large variety of metabolic compounds that exhibit biomaterial and biofertilizer production capabilities, including phycobiliproteins (PBPs), which may be an important target in biotechnology and biomedical research (Gonzalez et al. 2019; Rastogi and Sinha 2009; Rastogi et al. 2017). Cyanobacteria are well-known bacteria with an immense amount of pharmacologically active functions, such as antibiotic, anti-inflammatory, antioxidant and anti-Alzheimer's disease properties (Chaubey et al. 2019; Sonani et al. 2014; Singh et al. 2014). P. ludlowii has anti-inflammatory and antioxidative functions, while Cyanophyta is the dominant phylum in the endophytic bacterial community of P. ludlowii; therefore, the medicinal value of P. ludlowii may be associated with Cyanobacteria, and the pharmacological characteristics of Cyanobacteria may contribute to the high-quality medicinal value of P. ludlowii.
Proteobacteria and Acidobacteria are common microbial communities in other plants and soils (Manter et al. 2010; Bulgarelli et al. 2012; Jackson et al. 2013; Lin et al. 2019; Juan et al. 2019; Yaoben et al. 2019). Similarities were evaluated in this study. Proteobacteria and Acidobacteria were the predominant phyla in the rhizosphere soil, accounting for 21.2–31.1% and 20.9–23.6% of all bacterial sequences, respectively, while the abundance of Proteobacteria decreased with increasing soil depth. In contrast, Acidobacteria increased with increasing soil depth. It has been reported that Proteobacteria plays an important role in natural processes and has potential for application in treating wastewater, increasing tolerance to pollutants and improving the soil environment (Jeon et al. 2003; Yaoben et al. 2019; Kragelund et al. 2007). The dominant taxa in the upper soil supported the upper soil as the main site of soil organic matter degradation and soil improvement versus the other soil layers. Proteobacteria were more ubiquitous in all samples of P. ludlowii, and their potential function may be to regulate the immune system of P. ludlowii and improve the viability of P. ludlowii in extreme environments.
The sequences were classified into 19 different genera among all tissues and soils; however, the abundance of the bacterial communities was significantly different in all samples. Pseudomonas was the dominant genus in rhizosphere soil; however, its relative abundance decreased with soil depth, and several prior documents have shown that Pseudomonas is commonly distributed in plants and is known to have beneficial effects on plant growth and nutrient availability in addition to showing biocontrol activity against pests (Chen et al. 2014; Lee et al. 2019; Sousa et al. 2016). Nevertheless, Pseudomonas was less abundant in the roots, leaves, flowers and fruits and was even absent from the stems, which may be the reason that P. ludlowii is endangered. P. ludlowii may not provide a superior colonization environment for Pseudomonas, resulting in the lower distribution of this taxa in P. ludlowii, reducing the defensive abilities of the plant, affecting the absorption and utilization of nutrients, and eventually reducing the plant growth rate.
Rhizosphere microorganisms could contribute to plant health, growth and productivity (Cheng et al. 2020; Mendes et al. 2013). However, some important microbial groups, such as Pseudomonas and Bacillus, decreased with soil depth. Previous studies revealed that Bacillus potentially manipulates the host’s redox status and contributes to overcoming a critical period in development and seedling establishment due to its high catalase activities and superoxide contents (Pitzschke 2016). The dominant taxa in the upper soil supported the upper soil as a site of significant activity versus the other soil layers. The results indicated that the upper soil might play more vital roles than the middle and deep soil in improving seed germination, plant growth, and nutrient availability.
Endophytic bacteria are mainly derived from rhizosphere soil bacteria. In this study, the bacterial structure of plant tissues was significantly different, and the results were consistent with those of other studies showing that different plant tissues harbored different bacterial communities (Liu et al. 2015; Ren et al. 2019; Mûller et al. 2016). The structure of the bacterial community was similar among different deep soils; however, the bacterial structures of the roots and soil were more similar compared with those of other tissues (stem, leaf, flower and fruit), and the bacterial structure of plant tissues and soils had different levels of overlap. This indicated that endophytic bacteria might come from rhizosphere soil and be transferred from roots to other plant tissues due to the relationship between the roots and soil. Many previous studies have shown that endophytic bacteria mainly come from rhizosphere soil and are affected by soil microbes (Mûller et al. 2016; Bertollo 2001; McInory and Kloepper 1994).
The abundance of gene families in tissues and soil communities and the functional classification schemes of the KO and Clusters of Orthologs Groups (COG) databases were determined by phylogenetic investigation of communities by reconstruction of their unobserved states (PICRUSt). PICRUSt analysis showed that the gene families belonging to cellular processes, environmental information processing, genetic information processing, metabolism and organismal systems were detected among the samples. Among them, those related to metabolism were markedly most abundant. Metabolism, including energy, fatty acid, histidine, methane, nitrogen, propanoate and tryptophan metabolism, is widespread and plays extensive roles in prokaryotes. The analysis of level 3 KO groups showed that transporters were most abundant across all samples; most transporters were present in cell membranes and involved in detoxification processes, organ growth, nutrition, development and response to abiotic and biotic stresses (Do et al. 2018). The functional abundance of DNA repair and recombination proteins, ABC transporters, general function prediction, ribosomes and the two-component system was similar between the different tissues and soils, whereas the functional abundance of peptidases, photosynthesis and photosynthesis proteins was much higher in plant tissues than in soil. Overall, the functional abundance of gene families was similar, and the results indicated that there were relationships between endophytic bacteria and soil microorganisms. Thus, it is possible that P. ludlowii has high adaptability to local environmental conditions and has formed mutualistic associations with bacteria.
References
Badri DV, Weir TL, van der Lelie D, Vivanco JM (2009) Rhizosphere chemical dialogues:335 plant-microbe interactions. Curr Opin Biotechnol 20(6):642–650. https://doi.org/10.1016/j.copbio.2009.09.014
Bertollo P (2001) Assessing landscape health: a case study from northeastern Italy. Environ Manag 27:349–365. https://doi.org/10.1007/s002670010154
Bulgarelli D, Rott M, Schlaeppi K, Loren EV, van Themaat N, Ahmadinejad FA, Rauf P, Huettel B, Reinhardt R, Schmelzer E, Peplies J, Gloeckner FO, Amann R, Eickhorst T, Schulze-Lefert P (2012) Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488(7409):91–95
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336. https://doi.org/10.1038/nmeth.f.303
Chaubey MG, Patel SN, Rastogi RP, Srivastava PL, Singh AK, Madamwar D, Singh NK (2019) Therapeutic potential of cyanobacterial pigment protein phycoerythrin: in silico and in vitro study of BACE1 interaction and in vivo Aβ reduction. Int J Biol Macromol 134:368–378. https://doi.org/10.1016/j.ijbiomac.2019.05.06
Chen B, Shen J, Zhang X, Pan F, Yang X, Feng Y (2014) The endophytic bacterium, Sphingomonas SaMR12, improves the potential for zinc phytoremediation by its host, Sedum alfredii. PLoS ONE. https://doi.org/10.1371/journal.pone.0106826
Cheng Z, Lei S, Li Y, Huang W, Ma R, Xiong J, Zhang T, Jin L, Haq HU, Xu X, Tian B (2020) Revealing the variation and stability of bacterial communities in tomato rhizosphere microbiota. Microorganisms. https://doi.org/10.3390/microorganisms8020170
Compant S, Duffy B, Nowak J, Clement C, Barka EA (2005) Use of plant growth-promoting bacteria for biocontrol of plant diseases: principles, mechanisms of action, and future prospects. Appl Environ Microbiol 71(9):4951–4959
De Azevedo JL, Maccheroni W, Pereira JO, Araujo WL (2000) Endophytic microorganisms: a review on insect control and recent advances on tropical plants. Electron J Biotechnol 3(1):15–16
El-Deeb B, Fayez K, Gherbawy YA (2012) Isolation and characterization of endophytic bacteria from Plectranthus tenuiflorus medicinal plant in Saudi Arabia desert and their antimicrobial activities. J Plant Interact 8:56–64. https://doi.org/10.1080/17429145.2012.680077
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. https://doi.org/10.1038/nmeth.2604
Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, Ravel J (2014) An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. https://doi.org/10.1186/2049-2618-2-6
Fei R, Wei D, Dong-Hui Y (2019) Endophytic bacterial communities of Jingbai Pear trees in north China analyzed with Illumina sequencing of 16S rDNA. Arch Microbiol 201:199–208. https://doi.org/10.1007/s00203-018-1597-9
Gao T, Shi XY (2018) Preparation of a synthetic seed for the common reed harboring an endophytic bacterium promoting seedling growth under cadmium stress. Environ Sci Pollut Res Int 25:8871–8879. https://doi.org/10.1007/s11356-018-1200-6
Glick BR, Stearns JC (2011) Making phytoremediation work better: maximizing a plant's growth potential in the midst of adversity. Int J Phytoremediation. https://doi.org/10.1080/15226514.2011.568533
Gonzalez A, Riley KW, Harwood TV, Zuniga EG, Risser DD (2019) A tripartite hierarchical sigma factor cascade promotes hormogonium development in the filamentous cyanobacterium nostoc punctiforme. mSphere. https://doi.org/10.1128/mSphere.00231-19
Hao H, He Z, Li H, Shi L, Tang Y (2014) Effect of root length on epicotyl dormancy release in seeds of Paeonia ludlowii, Tibetan peony. Ann Bot 113(3):443–452. https://doi.org/10.1093/aob/mct273
Hardoim PR, van Overbeek Leo S, Dirk EJ (2008) Properties of bacterial endophytes and their proposed role in plant growth. Trends Microbiol 16:463–471. https://doi.org/10.1016/j.tim.2008.07.008
Hubbard M, Germida JJ, Vujanovic V (2014) Fungal endophytes enhance wheat heat and drought tolerance in terms of grain yield and second-generation seed viability. J Appl Microbiol 116:109–122. https://doi.org/10.1111/jam.12311
Jackson CR, Randolph KC, Osborn SL, Tyler HL (2013) Culture dependent and independent analysis of bacterial communities associated with commercial salad leaf vegetables. BMC Microbiol. https://doi.org/10.1186/1471-2180-13-274
Jeon CO, Lee DS, Park JM (2003) Microbial communities in activated sludge performing enhanced biological phosphorus removal in a sequencing batch reactor. Water Res 37:2195–2205. https://doi.org/10.1016/S0043-1354(02)00587-0
Juan H, Chunni Y, Jialiang L, Wenzhu G, Prasad SR, Chong C, Jun X (2019) Feasibility study of vertical flow constructed wetland for tertiary treatment of nanosilver wastewater and temporal-spatial distribution of pollutants and microbial community. J Environ Manag 245:28–36. https://doi.org/10.1016/j.jenvman.2019.04.128
Kragelund C, Caterina L, Borger A, Thelen K, Eikelboom D, Tandoi V, Kong Y, Waarde J, Krooneman J, Rossetti S, Thomsen TR (2007) Identity, abundance and ecophysiology of filamentous Chloroflexi species present in activated sludge treatment plants. FEMS Microbiol Ecol 59:671–682
Kumar SN, Hasan SS, Jitendra K, Isha R, Pathan AA, Asha P, Shazi S, Samudrala G, Datta M (2014) Crystal structure and interaction of phycocyanin with β-secretase: a putative therapy for Alzheimer's disease. CNS Neurol Disord Drug Targets 13:691–698. https://doi.org/10.2174/1871527313666140228114456
Li XZ, Song ML, Yao X, Chai Q, Simpson WR, Li CJ, Nan ZB (2017) The effect of seed-borne fungi and endophyte on seed germination and biomass of Elymus sibiricus. Front Microbiol 8:2488. https://doi.org/10.3389/fmicb.2017.02488
Liu Y, Li YH, Yao S, Wang H et al (2015) Diversity and distribution of endophytic bacterial community in the Noni (Morinda citrifolia L.) plant. Afr J Microbiol Res 24(9):1649–1657
Liu MH, Lin AH, Ko HK, Perng DW, Lee TS, Kou YR (2017) Prevention of bleomycin-induced pulmonary inflammation and fibrosis in mice by paeonol. Front Physiol 8:193. https://doi.org/10.3389/fphys.2017.00193
Lumactud R, Fulthorpe RR (2018) Endophytic bacterial community structure and function of herbaceous plants from petroleum hydrocarbon contaminated and non-contaminated sites. Front Microbiol 9:1926. https://doi.org/10.3389/fmicb.2018.01926
Lee SA, Kim Y, Kim JM, Chu B, Joa JH, Sang MK, Song J, Weon HY (2019) A preliminary examination of bacterial, archaeal, and fungal communities inhabiting different rhizocompartments of tomato plants under real-world environments. Sci Rep 9:9300. https://doi.org/10.1038/s41598-019-45660-8
Lin C, Kai F, Jie Z, Zhi-Ping Y, Xing-Fan D, Guang-Hui D, Han-Bo Z (2019) Enrichment of soil rare bacteria in root by an invasive plant Ageratina adenophora. Sci Total Environ 683:202–209. https://doi.org/10.1016/j.scitotenv.2019.05.220
Magoc T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27(21):2957–2963. https://doi.org/10.1093/bioinformatics/btr507
Manter DK, Delgado JA, Holm DG, Stong RA (2010) Pyrosequencing reveals a highly diverse and cultivar-specific bacterial endophyte community in potato roots. Microb Ecol 60:157–166. https://doi.org/10.1007/s00248-010-9658-x
Masoud W, Takamiya M, Vogensen FK, Lillevang S et al (2011) Characterization of bacterial populations in Danish raw milk cheeses made with different starter cultures by denaturating gradient gel electrophoresis and pyrosequencing. Int Dairy J 21:142–148. https://doi.org/10.1016/j.idairyj.2010.10.00
McInory JA, Kloepper JW (1994) Studies on indigenous endophytic bacteria of sweet corn and cotton. In: Gara FO, Dowling DN, Boesten B (eds) Molecular ecology of rhizosphere microorganisms. VCH Verlagsgesellschaft, Federal Republic of Germany, Weinheim. https://doi.org/10.1002/9783527615810.ch2
Mendes R, Garbeva P, Raaijmakers JM (2013) The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol Rev 37(5):634–663. https://doi.org/10.1111/1574-6976.12028
Mitter EK, de Freitas JR, Germida JJ (2017) Bacterial root microbiome of plants growing in oil sands reclamation covers. Front Microbiol 8:849. https://doi.org/10.3389/fmicb.2017.00849
Müller DB, Christine V, Yang B, Vorholt JA (2016) The plant microbiota: systems-level insights and perspectives. Annu Rev Genet 50:211–234. https://doi.org/10.1146/annurev-genet-120215-034952
Patrycja G, Magdalena W, Gauravi A, Dnyaneshwar R, Hanna D, Mahendra R (2015) Endophytic actinobacteria of medicinal plants: diversity and bioactivity. Antonie Van Leeuwenhoek 108:267–289. https://doi.org/10.1007/s10482-015-0502-7
Pitzschke A (2016) Developmental peculiarities and seed-borne endophytes in quinoa: omnipresent robust bacilli contribute to plant fitness. Front Microbiol. https://doi.org/10.3389/fmicb.2016.00002
Rastogi RP, Sinha RP (2009) Biotechnological and industrial significance of cyanobacterial secondary metabolites. Biotechnol Adv 27:521–539. https://doi.org/10.1016/j.biotechadv.2009.04.009
Rastogi RP, Madamwar D, Pandey A (2017) Algal green chemistry: recent progress in biotechnology. Elsevier, Amsterdam
Schloss PD, Westcott SL, Thomas R, Hall JR, Martin H, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Blaz S, Thallinger GG, Van Horn David J, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. https://doi.org/10.1128/AEM.01541-09
Sonani RR, Singh NK, Kumar J, Thakar D, Madamwar D (2014) Concurrent purification and antioxidant activity of phycobiliproteins from Lyngbya sp. A09DM: an antioxidant and antiaging potential of phycoerythrin in Caenorhabditis elegans. Process Biochem 49:1757–1766. https://doi.org/10.1016/j.procbio.2014.022
Sousa J, Olivares FL (2016) Plant growth promotion by streptomycetes: ecophysiology, mechanisms and applications. Chem Biol Technol Agric. https://doi.org/10.1186/s40538-016-0073-5
Thi DTH, Enrico M, Youngsook L (2018) Functions of ABC transporters in plant growth and development. Curr Opin Plant Biol 41:32–38. https://doi.org/10.1016/j.pbi.2017.08.003
Weyens N, van der Lelie D, Taghavi S, Vangronsveld J (2009) Phytoremediation: plant-endophyte partnerships take the challenge. Curr Opin Biotechnol 20(2):248–254. https://doi.org/10.1016/j.copblo.2009.02.012
Yaoben L, Yanmei Ye, Yiming Hu, Haokun S (2019) The variation in microbial community structure under different heavy metal contamination levels in paddy soils. Ecotoxicol Environ Saf 180:557–564. https://doi.org/10.1016/j.ecoenv.2019.05.057
Yiting L, Chenggui W, Qian T, Wenhao Z, Zhenhua F, Xingfang Yu, Xiaoshan G, Jianshun W (2017) Paeonol inhibits IL-1β-induced inflammation via PI3K/Akt/NF-κB pathways. In vivo and vitro studies. Inflammation 40:1698–1706. https://doi.org/10.1007/s10753-017-0611-8
Zhang XB, Ni AL, Zhang WM, Wang CC (1997) Advances in researches on seed dormancy of officinal plants. Chin Tradit Herb Drugs 6:376–378
Acknowledgements
This research was supported by the Tibet Department of Science and Technology General Scientific Research Project (Grant No. XZ2017ZRG-22), the coordinated innovation program for the research and development of Tibet characteristic resources (Grant No. 300201), The Forth National Survey of Traditional Chinese Medicine Resources, Chinese or Tibet Medicinal Resources Investigation in Tibet Autonomous Region (State Administration of Chinese Traditional Medicine)(Grant No. 20190411–542222), and Research and Formulating of Standards for the Construction of China Wild Traditional Chinese Medicine Resources Protection Base (Tibet region)( Grant No. 2060302–1702-13)
Author information
Authors and Affiliations
Contributions
YZL, EHZ, and QLL designed experiments and collected samples; YZL, EHZ and XY performed the experiments; FY, LY, HC and MSH performed the statistical analysis; EHZ wrote the first draft of the manuscript; KTZ and XZL modified the manuscript; all authors read and approved the submitted version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Communicated by Erko Stackebrandt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lu, Y., Zhang, E., Hong, M. et al. Analysis of endophytic and rhizosphere bacterial diversity and function in the endangered plant Paeonia ludlowii. Arch Microbiol 202, 1717–1728 (2020). https://doi.org/10.1007/s00203-020-01882-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-020-01882-3