
Abstract
This paper reports the results obtained during an intercomparison exercise for the determination of difficult to measure radionuclides in activated reactor pressure vessel (RPV) steel samples. In total, eight laboratories participated analysing 14C, 55Fe and 63Ni activity concentrations in RPV steel. In addition, some laboratories also analysed 60Co activity concentrations. Corresponding activity concentrations were also determined using activation calculations. Robust statistical techniques were utilised for the analysis of the results according to ISO 13528 standard. The results showed good agreement for 55Fe and 63Ni results whereas 14C results had significant differences. 60Co results were in quite good agreement.
Similar content being viewed by others

Introduction
Analysis of difficult to measure (DTM) radionuclides, such as 3H, 14C, 36Cl, 41Ca, 55Fe, 63Ni etc., in radioactive waste is a challenging task not only due to the lengthy radiochemical procedures with several sources of uncertainties but also due to the possible heterogeneity of the studied waste material. In addition, uncertainties can also originate from the sampling technique. For example, volatile radionuclides, such as 3H, can be lost during sampling of contaminated concrete via drilling when the temperature increases and 3H can partly escape as tritiated water vapour or as different tritiated species such as HT, T2 or organic compounds. Even though the contribution of sampling to the overall uncertainty can be significantly larger compared to the uncertainty of the analytical results [1], the validity of analytical results needs to be determined. Most often, a certified reference material is analysed giving validity for the analytical method. However, in case of DTM analyses in radioactive waste, there are no commercial reference materials available for radioanalytical method validation. One way of radiochemical method validation is to attend in an intercalibration exercise in which preferably a real sample is analysed.
This paper presents the results of an intercomparison exercise, which was carried out between eight laboratories on analysis of three corrosion radionuclides (14C, 55Fe and 63Ni) in an activated reactor pressure vessel (RPV) steel. In addition to DTMs, some laboratories also measured 60Co, which is considered as a key nuclide to establish correlation and scaling factors for activation products contained in radioactive waste. The results were analysed according to ISO 13528 standard [2]. The statistical analysis of the measured results was carried out using robust methods and the assigned values were derived from the submitted results. As reported by Leskinen et al. [3], the intercomparison exercise was divided into two phases. First, the participating laboratories submitted preliminary results and they were statistically analysed and discussed. Second, the laboratories were given a chance to correct obvious blunders, add new analysis entries, submit replicate results, and carry out corrective measures, such as efficiency curve corrections, for their final analysis results. The complete procedure is presented in reference [3]. In this paper, the final results are further analysed by comparing them with activation calculation results. The calculated activity concentration results were derived from known chemical composition, irradiation history and cooling time of the RPV steel using a point kinetic code ORIGEN-S [4].
Sample preparation
The studied samples were cut from a 10 × 10 mm pressurised water reactor type RPV steel bar. The samples were prepared as follows; an electric discharge machine was used for cutting of the samples (~ 0.2 mm thin), oxide layer removal was carried out using HCl:H2O:hexamethylenetetramine solution, washing was carried out using ethanol, and the samples were then let to air dry. The samples were weighed, and an identification number was given. The masses of the samples were between 0.1385 and 0.1533 g. Thicknesses of the samples (0.176–0.195 mm) were calculated using the density and the weights of the samples.
Homogeneity and stability verifications
The homogeneity measurements were carried out using an HPGe Be2020 spectrometer (ISOCS Canberra Ltd) connected with Inspector 2000 multichannel analyser using Genie 2000 software and Geometry Composer v.4.4 for efficiency calibrations. RPV steel manufacturer given density and known dimensions of the pieces were used as parameters for the efficiency calibrations. Each sample was positioned on a sample holder in order to maintain constant measurement geometry and the sample holder was placed at 30 mm distance from the detector in order to avoid coincidence counts from 60Co peaks. The measurement time was 15 min until 30,000 counts were collected into the 60Co 1332.5 keV peak. The 60Co results showed relative standard deviation of 1.8% and thus, the samples were considered homogenous. More information on the sample homogeneity studies are presented by Leskinen et al. [3].
The stability of the samples is another important parameter for the intercomparison samples [2]. Since the samples were solid materials and rusting was minimised with the oxide layer removal and dipping into ethanol, the samples were considered to be stable. However, in some cases, the samples had rusted a little, but it was not considered to be a problem, since the masses of the samples were measured at the beginning of the radiochemical analyses by the participants and no significant difference was observed. In total, 20 samples were first analysed for homogeneity and stability and then distributed to the laboratories. Each laboratory received 2–3 samples.
Activation calculations
The studied RPV steel samples had been irradiated with thermal neutron flux of 7.49 × 1019 n cm−2 between December 1980 and July 1985. Therefore, considering the reference date to which all results (measured and calculated) were corrected, the cooling time was approximately 34 years. The flux was semi-empirical based on dosimetric analyses of 59Fe that were performed on the samples in the 1980′s. Material composition was provided by the material owner. However, the complete data is not presented here due to confidentiality reasons. Table 1 lists weight percentages, activation reactions and reaction cross sections of those stable elements that are relevant to this study. Thermal activation cross sections in Table 1 are according to Beckurts et al. [5]. The data provided did not include the weight percentage of Fe and it was calculated by subtracting the combined weight percentages of the elements from 100 wt%. However, Fe and Ni contents were also confirmed experimentally with ICP-OES measurements and they were in good agreement with the data provided [6]. Detailed data on nitrogen impurity was not available. It was reported that nitrogen impurity varies typically from 0.005 to 0.007 wt%. Consequently, the average value of 0.006 wt% was used in the calculations. The initial carbon content was 0.16 wt% which corresponds to a low-carbon steel (< 0.3 wt%). The wt% uncertainties were estimated the same way as in reference [6]. The activation calculation theory, calculation procedures and uncertainties of the calculation results are discussed in depth by Leskinen et al. [6].
Methodology for statistical analysis
Statistical analysis of the results was carried out using ISO 13528 standard [2]. Variety of scoring strategies are available, but most often participant’s deviation from an assigned value is compared. Two methods were used for the determination of the assigned value. First, a robust statistical method was utilised for development of assigned values based on the participant’s results. Robust mean and robust standard deviation were calculated using an Algorithm A, which transformed the original data by a process called winsorisation to provide an alternative estimator of mean and standard deviation [2]. This method is robust for outliers, when the expected proportion of outliers is less than 20% [2]. The iterations of the robust mean and standard deviations were continued until there was no change in the 3rd significant figure of the robust means and standard deviations [2]. In this paper, the robust means and standard deviations of 55Fe, 63Ni, 14C and 60Co calculated from participant’s results are referred to as measured assigned values. Second method for determination of the assigned value was carried out by calculating the activity concentrations based on the activation reactions. These results are referred to as calculated assigned values.
Performance assessment was carried out using z score [2]. The participant’s results (noted xi) were assessed against both measured assigned values and calculated assigned values. z score was calculated using Eq. (1). In cases, when the robust standard deviation was large, the uncertainty of the assigned value u(xpt) calculated using Eq. (2) was used as σpt. Selection of the u(xpt) as σpt was used so that the results that were not fit for purpose received an action signal [2]. u(xpt) was used as σpt when robust standard deviation was larger than 20%. The analysis results with z score were marked as acceptable when z ≤ 2.0, a warning signal was given for results with 2.0 < z < 3.0, and results were unacceptable for z ≥ 3.0 [2].
where xi = the value given by a participant i, xpt = the assigned value, σpt = standard deviation for the proficiency assessment
where s* = robust standard deviation of the results, p = number of samples.
Overview of the radiochemical analyses
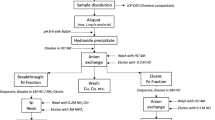
The radiochemical analyses of 55Fe, 63Ni and 14C were based on either internal or published procedures. A general summary of the procedures is presented by Leskinen et al. [3] and detailed procedures in the references [6,7,8,9,10,11,12,13,14]. Due to anonymity, procedures were not linked with specific analytical results. In general, the radiochemical analysis of 55Fe and 63Ni began with complete digestion of the solid material using different acid mixtures with and without heating and sometimes using high pressure (e.g. microwave oven). The participants reported mainly no difficulties in the solubility of the steel samples. However, in the case when no heating was applied, small black particles, which were most likely carbon, remained in the acid digestion solution. After the digestion of the solid samples, separation of Fe and Ni from other elements was carried out using mainly hydroxide precipitation followed by an anion exchange resin (AG 1 × 4 resin by Bio-Rad or Dowex 1 × 4 resin by Dow Chemical Company), which separated Fe and Ni from each other. The eluted Fe fraction from the anion exchange resin was most of the times evaporated to dryness without further purification. The evaporated residues were dissolved and aliquots were taken for the yield and Liquid Scintillation Counting (LSC) measurements. Ni fractions were further purified using Ni-resin by Eichrom Technologies once or twice, since especially 60Co needs to be efficiently removed from the Ni fraction. The eluate was most often evaporated to small volumes and aliquots were taken for the yield and LSC measurements. Fe and Ni yield measurements were carried out mainly using elemental analysis techniques, generally by ICP techniques but also by standard addition.
The analysis of 14C was carried out either using an acid digestion with oxidation of carbon to CO2 or by using a dedicated equipment for volatile radionuclides, namely Oxidizer (Perkin Elmer), Pyroxidiser (Eraly) and Pyrolyser (RADDEC). All the dedicated equipment are furnaces with oxygen as carrier gas in first equipment and air supplemented with oxygen in the last two equipment. In all cases, the released 14C is trapped in a basic solution (generally Carbosorb or NaOH) which is then analysed by LSC. 14C yields were determined using liquid sources.
Overview of the gamma spectrometric analyses
Some participants also carried out analysis of 60Co either in solid form or they dissolved the samples first before measuring the gamma emissions using HPGe gamma detectors. Efficiency calibrations were carried out using calibration sources or Mirion Technologies softwares ISOCS or LabSOCS.
Activation calculation results
Table 2 lists the specific activities of the activated steel samples calculated with ORIGEN-S. Uncertainties of the specific activities were calculated using the law of error propagation in multiplication. The uncertainties arise from the uncertainties in exact original material composition, neutron dose, reaction cross sections and decay time. Cross section uncertainties were assumed according to reference [15]. Uncertainties in material compositions were estimated to be 10% for C, Fe, Co, Ni and 25% for N. Total neutron dose based on the semiempirical dosimetry measurements was obtained directly from the owner of the material and its uncertainty was not available.
Statistical analysis of 55Fe, 63Ni, 14C and 60Co results
In total, 21 55Fe, 15 63Ni, 7 14C and 17 60Co measured data entries were statistically analysed. A summary of the statistically evaluated data, namely standard statistics and robust statistics, is shown in Table 3. In comparison of the Table 3 (measured assigned values) and Table 2 (calculated assigned values) results, it can be calculated that in the case of 55Fe, measured assigned value was 73% of the calculated assigned value. The corresponding percentages for 63Ni was 103%, for 14C 50% and for 60Co 120%. Therefore, the 63Ni results seem to be in an excellent agreement (± 5%), 55Fe and 60Co results in tolerable agreement (± 25%) whereas 14C results exhibit significant difference (± 50%). However, both the calculated and measured assigned values are affected by several parameters and their uncertainties and therefore, further discussion is carried out in the following text.
The 55Fe, 63Ni, 14C and 60Co data entries are respectively shown in Figs. 1, 2, 3 and 4 together with measured and calculated assigned values. Some laboratories analysed replicates and thus some sample numbers have 2 to 4 entries. All uncertainties are presented in 2σ except for samples 19 and 20, which were not submitted with uncertainties.

55Fe data entries with 2σ uncertainties in increasing activity concentration order and measured (blue) and calculated (red) assigned values with 2σ uncertainties. (Color figure online)

63Ni data entries with 2σ uncertainties in increasing activity concentration order and measured (blue) and calculated (red) assigned values with 2σ uncertainties. Result of sample number 20 is considered an outlier. (Color figure online)

14C data entries with 2σ uncertainties in increasing activity concentration order and measured (blue) and calculated (red) assigned values with 2σ uncertainties. Acid digestion was used for samples 12, 16, 17 and 18 and combustion method for samples 4, 6 and 13. (Color figure online)

60Co data entries with 2σ uncertainties in increasing activity concentration order and measured (blue) and calculated (red) assigned values with 2σ uncertainties. Samples measured in solution marked with *. (Color figure online)
Table 4 summarises the z score values when the results are compared with the measured assigned values and with the calculated assigned values. In the case of 55Fe, 63Ni and 14C, the uncertainty of the assigned value u(xpt) was used as σpt (see Eqs. 1, 2), since the robust standard deviations were large (30%, 31% and 61%, respectively). The standard deviation of the assigned value was 16% for 60Co, thus it was used in the z score calculations.
The 55Fe results in Fig. 1 and Table 4 show that when measured 55Fe results are compared with the measured assigned values, 57% of the results are within 2σ of the measured assigned value (z ≤ 2.0), 10% in warning signal range (2.0 < z < 3.0) and 33% in unacceptable range (z ≥ 3.0). The z score calculations do not take into consideration the uncertainties of the measured data entries. Therefore, visual assessment of the Fig. 1 shows that when the 2σ uncertainties of the measured data entries are taken into consideration, 76% of the results are within the 2σ uncertainty of the measured assigned value. When the measured 55Fe results are compared with the calculated assigned value, 28% of the results are within 2σ of the measured assigned value (z ≤ 2.0), 48% in warning signal range (2.0 < z < 3.0) and 24% in unacceptable range (z ≥ 3.0). Visual assessment of the Fig. 1 shows that when the 2σ uncertainties of the measured data entries are taken into consideration, 67% of the results are within the 2σ uncertainty of the calculated assigned value. It can be concluded, that the measured assigned value includes higher number of measured 55Fe results within the 2σ uncertainty. Moreover, it can be noticed that when the uncertainties of the measured results are considered, the distributions of the results are comparable with measured and calculated assigned values e.g. the majority of the laboratories (76% and 67%, respectively) obtained satisfactory results for 55Fe determination.
The 63Ni results in Fig. 2 and Table 4 show that when measured 63Ni results are compared with the measured assigned values, 60% of the results are within 2σ of the measured assigned value (z ≤ 2.0) and 40% in unacceptable range (z ≥ 3.0). Visual assessment of the Fig. 1 shows that when the 2σ uncertainties of the measured data entries are taken into consideration, 73% of the results are within the 2σ uncertainty of the measured assigned value. When the measured 63Ni results are compared with the calculated assigned value, 60% of the results are within 2σ of the measured assigned value (z ≤ 2.0), 13% in warning signal range (2.0 < z < 3.0) and 27% in unacceptable range (z ≥ 3.0). Visual assessment of the Fig. 2 shows that when the 2σ uncertainties of the measured data entries are taken into consideration, 73% of the results are within the 2σ uncertainty of the calculated assigned value. It can be concluded, that whatever the type of assigned value is considered with 2σ uncertainty of the measured data entries, the three-fourths of participants obtained satisfactory results for 63Ni determination.
The 14C results in Fig. 3 and Table 4 show that when measured 14C results are compared with the measured assigned values, 43% of the results are within 2σ of the measured assigned value (z ≤ 2.0) and 57% in the warning signal range (2.0 < z < 3.0). Visual assessment of the Fig. 3 shows that when the 2σ uncertainties of the measured data entries are taken into consideration, 71% of the results are within the 2σ uncertainty of the measured assigned value. The measured assigned value for 14C was derived from 7 data entries, which is significantly less than data entries for 55Fe, 63Ni and 60Co (21, 15 and 17, respectively). Additionally, 3/7 of the results were provided by one laboratory and this may have caused a bias. Additionally, the uncertainties of the 14C analysis results had also a wide range (11–41%). When the measured 14C results are compared with the calculated assigned value, 57% of the results are within 2σ of the measured assigned value (z ≤ 2.0), 14% in warning signal range (2.0 < z < 3.0) and 29% in unacceptable range (z ≥ 3.0). Visual assessment of the Fig. 3 shows that when the 2σ uncertainties of the measured data entries are taken into consideration, 57% of the results are within the 2σ uncertainty of the calculated assigned value. It can be concluded, that within the 2σ uncertainties of the measured assigned value includes higher number of results even though the uncertainty of the calculated assigned value is higher than the corresponding measured value. Moreover, it can be noticed that when the uncertainties of the measured results are considered, three-fourths of the 14C results are satisfactory when compared to the measured assigned value whereas only two-thirds of the 14C results are satisfactory compared to the calculated assigned value. However, when the uncertainties raising from the radiochemical analysis and number of data entries are compared with the uncertainties arising from the calculations, an overall conclusion can be made that the results are aligned well enough and at least in same order of magnitude.
Majority of 60Co measurements were carried out in solid form. However, some measurements were carried out in the acid digested solutions (marked with * in Fig. 4 and in Table 4). The results have a consistent trend and no major difference between measurements in solid and solution can be observed. The 60Co results in Fig. 4 and Table 4 show that when measured 60Co results are compared with the measured assigned values, 100% of the results are within 2σ of the measured assigned value (z ≤ 2.0). When the measured 60Co results are compared with the calculated assigned value, 59% of the results are within 2σ of the measured assigned value (z ≤ 2.0), 12% in warning signal range (2.0 < z < 3.0) and 29% in unacceptable range (z ≥ 3.0). Visual assessment of the Fig. 4 shows that when the 2σ uncertainties of the measured data entries are taken into consideration, 82% of the results are within the 2σ uncertainty of the calculated assigned value. It can be concluded that when the 2σ uncertainties of the measured data entries are considered, all of the 60Co results are satisfactory when compared to the measured assigned value and only three-fourths when compared to the calculated assigned value.
Discussion
Critical considerations in the radiochemical analysis of 55Fe
55Fe decays via electron capture to 55Mn with the emission of Auger electrons and low-energy X-rays, namely 5.888 keV, 5.899 keV and 6.490 keV. Low-energy X-rays can be measured using low-energy gamma spectrometers, gas flow proportional counters, and LSC. If the low-energy gamma spectrometry is the measurement technique of choice, the 55Fe analysis results have been reported to be comparable with LSC results, but the analysis is affected by self-attenuation, which depends on geometrical parameters and chemical composition of the precipitated sample [16]. However, the most common measurement technique is LSC, which can detect both the K-Auger electrons and the K X-ray emissions. However, due to the low energies, the measurement of 55Fe is challenged by several phenomena related to the LSC measurement technique. First, the LSC samples need to be dark-adapted long enough time in order to eliminate the chemiluminescence, which exhibits signal in the lower channels of the LSC spectrum similar to 55Fe emissions. Normally, chemiluminescence decays away within 1 h of sample preparation [17], but sometimes cooling of the sample to approximately 10 °C at least 1 h and use of a coincidence technique has been required to overcome the effects of the chemiluminescence [18]. Second, the measurement of low energy 55Fe is highly affected by quenching, which has an especially significant effect on the low energy emissions. Since the slope of the counting efficiency curve is steep in the beginning of the graph, even small changes in the quenching can result in a significant increase or decrease in the counting efficiency resulting in a bias in the measurement results. In the analysis of 55Fe, the yellow colour of Fe(III) is a very efficient colour quenching agent. In order to overcome the colour quenching effect, reduction of Fe(III) to Fe(II) has been carried out using reducing agents such as ascorbic acid [19]. However, the reduction was not permanent and the colour returned into the LSC samples after some days [19]. Another solution is to form a stable iron complex such as a colourless Fe-phosphate, which can be achieved by dissolving the purified Fe fraction into dilute phosphoric acid [20]. Third, selection of the scintillation cocktail has an effect on the tolerance for acids and hence, measurable amount of Fe [20]. The tolerance of acids, namely 1 M HCl and 1 M HNO3 by different scintillation cocktails and their effect on the counting efficiency have been studied by Warwick [21]. Majority of the participants in the intercomparison exercise diluted the 55Fe fractions into 1 M H3PO4 [3]. However, further details in the colours of the 55Fe fractions or counting efficiencies were not submitted. Nevertheless, after discussions on the preliminary results, two participants recalculated the 55Fe results using improved quenching analysis resulting in consistent results compared with the majority of the submitted results [3]. The improved quenching analysis included a preparation of a new 55Fe quenching curve and calculation of activity results against it and recalculation of the activity results using a Hidex provided CoreF function for conversion of TDCR (triple to double count rate) to counting efficiency [3]. In the first case, the activity results decreased on average 38% and in the second case, on average 41%. These results emphasise the importance of correct quenching correction in the analysis of 55Fe via LSC.
In addition to challenges in the LSC measurement technique, purification of 55Fe from interfering radionuclides needs to be carefully carried out. Special care is also needed in the evaporation of the Fe fraction in HCl solution to near dryness since prolonged heating converts the Fe (III) chloride residue to a much more persistent oxide [20]. 55Fe purification via TRU resin treatment or hydroxide precipitation followed by ion exchange chromatography [3] seemed to eliminate all the interfering radionuclides from the Fe fraction since none of the participants expressed difficulties in the 55Fe sample preparation.
Critical considerations in the radiochemical analysis of 63Ni
63Ni is a pure beta emitter with a maximum energy of 66.98 keV. Careful radiochemical purification is required prior to LSC detection in order to eliminate the interfering radionuclides. 60Co is a problematic interference especially in the analysis of 63Ni for two reasons. First, as the radiochemical behaviour of these two metals is very similar, Co reduces the radiochemical yield of Ni by competing with Ni for available sorption sites in extraction chromatographic Ni resin. Steel samples may contain both active and inactive isotopes of Co and Ni, especially if stable Co and Ni carriers have been added to the sample in the beginning of the analysis, leading easily to the exceeded binding capacity of the extraction chromatography resin. Second, 60Co is disturbing the activity measurements of 63Ni due to overlapping beta energies, the endpoint beta energies being 318 keV for 60Co and 67 keV for 63Ni.
Stable Ni and Co carriers are often added before radiochemical separation of 63Ni from 60Co, for enabling quantitative precipitation of 63Ni from the solution and confirming the complete separation of 63Ni from 60Co during the separation procedure. On the other hand, addition of Co carrier in analysis of 63Ni is concluded to be preferable only after the extraction chromatography separation step, due to fore mentioned competition between Co and Ni in Ni resin easily leading to the binding capacity of the resin being exceeded [17, 22]. The use of Co and Ni carriers, i.e. milligram amounts of these metals, can be even seen visually while using the separation method by Hazan and Korkisch [13]: blue Co and green Ni belts are seen in the resin column after elution of Fe, but without using carriers there is only a yellow column.
One of the laboratories reported a problem with 60Co in the LSC spectrum of purified 63Ni fraction, when Ni had been separated with anion exchange in acetone and HCl media, as described in reference [13]. Although this separation method was previously found to be successful for purifying 63Ni from 60Co in activated pressure vessel steel, it was not adequately efficient with these intercomparison samples for an unknown reason. The presence of 60Co in 63Ni fraction after anion exchange separation was observed both in gamma and LSC spectra (Fig. 5). As seen in Fig. 5, the 63Ni beta spectrum peaking at channel region 250–300 is overlapped by 60Co beta spectrum peaking at channel region 500–550. Due to high amount of 60Co compared to 63Ni in the LSC spectrum, it was impossible to determine the activity of 63Ni correctly at this purification stage.

LSC spectrum of 63Ni after the first separation by ion exchange using the method by Hazan and Korkisch [13]. The 63Ni beta spectrum peaks at channel region 250–300 and 60Co beta spectrum peaks at channel region 500–550. The measurement time was 360 min
Further purification of the Ni fraction containing 63Ni and excessive amounts of 60Co was carried out with Ni resin. As seen in Fig. 6, after extraction chromatography separation there was only 20% of the 60Co activity left, compared to the same sample earlier after anion exchange only (Fig. 5). Now it was possible to measure the activity of 63Ni with LSC, although there was still a small visible effect from 60Co interfering with the beta spectrum of 63Ni. If a second Ni resin separation cycle would have been performed for these samples, then it might have been possible to achieve a negligible activity level of 60Co in the 63Ni fractions [23].

LSC spectrum of 63Ni after ion exchange [13] and Ni resin separation. The 63Ni beta spectrum peaks at channel region 250–300 and 60Co beta spectrum peaks at channel region 500–550. The measurement time was 480 min
The purified 63Ni samples, which first suffered from excessive 60Co interference (Fig. 5) and later had only moderate amounts of 60Co in the LSC spectra (Fig. 6), were measured by a gamma spectrometer in order to remove the effect of 60Co in the LSC spectra of the 63Ni fractions. The activity of 60Co, obtained by gamma spectrometry, was subtracted from the total beta peaks of 63Ni and 60Co in the LSC spectrum. In order to perform this correction for obtaining pure 63Ni beta activity, the counting efficiency of the liquid scintillation counter for the both radionuclides 63Ni and 60Co was determined, as the efficiency value is different for these two beta energies. In this particular case, the counting efficiency of the Quantulus 1220 liquid scintillation counter was 71% for 63Ni and 99% for 60Co, respectively, using 63Ni and 45Ca standard solutions for determining the counting efficiency. Due to availability reasons 45Ca was used instead of 60Co in efficiency determination for 60Co, but the beta energies of 60Co (318 keV) and 45Ca (257 keV) are adequately similar enabling the use of 45Ca standard in this context. 60Co standard has been used for determination of 45Ca inversely in a corresponding way [24].
Critical considerations in the radiochemical analysis of 14C
14C is a pure beta emitter which decays by emitting particles with the maximum energy of 156 keV and can be analysed using LSC. In the case of irradiated steel samples, 14C is mainly produced from neutron capture of trace level nitrogen (see Table 1). For this type of matrix, Mibus et al. [25] highlighted, that there is a considerable uncertainty present whether 14C produced by activation of 14N will be present in the same chemical form as the carbon or nitrogen (e.g. iron carbide or nitride forms) present in the steel at the time of their production. Additionally, there is a very limited experimental information available on the speciation of 14C and its release mechanisms [25, 26]. Many papers deal with the measurements of 14C in environmental or biological samples [10] but a very few in nuclear waste [8, 14]. The major difficulty of 14C determination in radioactive waste deals with its complete extraction from the matrix, especially for steel samples [6, 8, 14, 27,28,29] and the elimination of volatile interfering radionuclides such as 3H, 129I, 99Tc or 35S before LSC analysis. In agreement with the literature, two methods (e.g. acid digestion and furnaces) were applied during the intercomparison exercise. Both methods relied on the quantitative liberation of 14C from the solid matrix, its conversion to CO2 and trapping in Carbosorb®, Carbotrap® or NaOH solutions. The trapping solutions were mixed with liquid scintillation cocktail and measured using LSC. Whatever the implemented procedure, no laboratory observed interfering radionuclides in the LSC spectra. However, significant chemiluminescence was observed in NaOH mixtures with liquid scintillation cocktail and therefore, the samples were kept in dark over 24 h before LSC measurements.
In order to provide reliable results, quantitative conversion of carbon to CO2 needs to be guaranteed. In acid digestion, this can be achieved by using effective oxidising acids. In this study, acid mixtures of H2SO4 + HNO3 and HNO3 + HCl + HClO4 were used. In the latter case, HClO4 was added in a second step after aqua regia medium in order to optimise the oxidation of carbon to CO2 [6]. It was highlighted that fresh HClO4 was needed, otherwise the carbon was not efficiently oxidised. As no commercial activated steel samples are available, the 14C recovery yield for a steel was determined experimentally and assumed to behave similarly with the standard liquid sources (approximately 100%). This hypothesis may induce a bias since liquid source containing 14C may not have the same behaviour as carbon contained in solid matrix i.e. steel. In order to overcome this difficulty in yield determination, 14C analysis can be carried out using both of the above methods and if the results are similar, it can be concluded that analysis result (including yield correction) is reliable. Even though this approach is widely used in internal method validation, intercomparison exercise with several laboratories and data entries are required for more reliable method validation. For example, samples 12 and 13 were analysed by one laboratory using both acid digestion and a furnace giving comparable results. However, these results differ to some extent from the other data entries.
The second method applied to extract 14C was the implementation of oxidative thermal desorption in furnace systems working either with an oxygen/nitrogen flow or air flow followed by mixture of oxygen and air in the final phase of the analysis. Use of oxygen gas enhances the oxidation process. After precise weighing, the samples (approx. 0.14 g) were introduced in the furnaces. Depending on the device, catalysts such as Pt-aluminia, were added in the tube assembly to finalise the oxidation of 14C into CO2. As Warwick et al. [29] underlined, the temperature applied during the pyro-combustion plays an essential role to extract quantitatively the volatile radionuclides whatever the analysed sample. As an example, depending on its speciation and the studied matrix, the tritium release temperature varied from 100 °C up to 900 °C respectively for steel and graphite. Typical fusion temperatures for steel samples are around 1500 °C, which is not achievable by the furnaces mentioned in this study, with maximum temperatures of 1100 °C and 950 °C, respectively. The differences in 14C values observed in this study could be due to the differences of temperatures that can be reached by commercial furnaces. One can also assume that the 14C recovery yield assumed to be close to 100% as for liquid standards might be overestimated in the case of pyrolysis-combustion and might induce an underestimation of the 14C activity concentrations. Actually, as reported in [14], the quantity of sample and the time–temperature profile also influence greatly the extraction yield. In the case of concretes, the granulometry of the particles also has an impact: the recovery yield can vary from 60% up to 85% [14]. The same effects might be observed for steel samples.
The present intercomparison exercise shows that further work is needed in order to determine 14C activity in activated steels with a good accuracy. The ideal way to obtain accurate 14C measurements is to have a complete acid digestion and quantitative oxidation of carbon to CO2 or to implement furnace analysis using small sample sizes or samples with a low granulometry and to achieve temperature higher than 1100 °C.
Critical considerations in the analysis of 60Co
60Co decays with β− sending out X-rays, betas and gammas. The two distinctive gamma energies, namely 1173 keV and 1332 keV, are easily detected via gamma spectrometry and thus, 60Co is considered to be an easy to measure radionuclide. In this study, even though the statistical analysis carried out for the measured 60Co results showed good results, there was 50% difference between the lowest and highest data point. The difference is large when considering that 60Co is considered to be easy to measure. However, in order to obtain reliable results, a few requirements need to be fulfilled. First set of requirements include the detector, which needs to be properly maintained and calibrated. This is one of the main principles of quality control in analytical laboratories and thus it is considered to be self-evident in this study.
Secondly, the measurement geometry needs to be suitable for the sample size and activity level. For example, depending on the activity level, significant amount of coincidence can be detected in the 60Co gamma spectrum. Without coincidence correction, the analysed results are underestimated. The coincidence effect is clear with short source-to-detector distances and with big detectors and thus the effect can be minimised by increasing distance between the detector and sample. In this study, coincidence corrections were carried out when needed.
Thirdly, self-absorption of the sample matrix needs to be considered. A traditional way is to dissolve a solid material prior to the gamma measurement in order to reduce the uncertainty rising from self-absorption. In this study, however, the samples were thin (less than 0.2 mm) and the measured gamma energies were high and therefore the self-absorption was considered to be negligible. Some measurements were carried out in liquid form, which can be an additional benefit since the sample sizes were bigger. The bigger size may be advantageous if the sample is measured near or on the detector, because the efficiency on the detector can change rapidly (e.g. heterogeneity in the detector crystal) and thus even small changes in the geometry can have an effect on the results. In both cases (e.g. measurement in solid and liquid form), the measurement geometry needs to be kept constant. This can be carried out by using a sample holder.
Fourth parameter to consider is the efficiency calibration, which is the most likely reason behind the discrepancies in the 60Co results. The traditional way to measure gamma radionuclides from a solid sample is to dissolve the sample into a known volume, measure it and calculate the activity concentration using efficiency calibrations, which has been carried out using a standard solution with known activities and geometries. Another way is to measure the sample from such a distance that the sample can be considered to be a point source and thus the efficiency calibration can be made by using simple point source. In this study, the laboratories also used LabSOCS or ISOCS calibrations in which the efficiency calibration is carried out using modelling. When the modelling tools are used, sample parameters (e.g. density, size, distance from detector) are submitted in the modelling program, which calculates the efficiency curve. Therefore, experimentally determined calibration curves are not needed and the sample can be measured as such.
As a summary, the 60Co results show more variation than expected in an intercalibration for gamma radionuclides. However, the main objective of this intercalibration was the analysis of difficult to measure radionuclides, which means that the submitted 60Co results may have not been analysed with best effort. It can be concluded, though, that even if analysis of gamma emitters is expected to be a straightforward process, reliable results require appropriate attention and care.
Critical considerations in the activation calculations
The uncertainties in the calculation model arise from the uncertainties of the input parameters. First parameter is the neutron flux. In this study, the neutron flux was provided by the material owner as calculated and semiempirical fluences. Calculated fluence used information from irradiation source, which was in this case a nuclear reactor. Semiempirical calculations were explained in the Activation calculation section. Semiempirical flux results were considered to be more realistic and thus they were used in the activation calculations of this study. However, out of interest, when calculated flux was used instead of semiempirical flux, the activation results were 23% lower for 55Fe, 15% lower for 63Ni, 8% lower for 14C and 14% lower for 60Co.
Second parameter affecting the calculation model is the material composition, which is not always known in detail enough and conservative assumptions need to be made regarding the initial concentrations of the elements which can be activated. In many cases, trace level impurities, such as Co in steel, are a significant source of activity and thus initial Co concentration should be reliably determined. In this study, the amounts of Fe, Ni, Co, C, and N were reported as weight percentages (Table 1). The amounts of Fe and Ni were 96.8 wt% and 1.6 wt%, respectively. The provided Co, C and N amounts were below 0.1 wt% (0.09, 0.16 and 0.006, respectively) and such low levels of elements require sensitive measurement techniques. For example, 0.2 g of studied sample would contain only tens to hundreds of nanograms of Co, C and N. In this study, the approximation of the trace level results is not known and thus they may be one source of uncertainty in the calculated results.
Third parameter affecting the calculation model is the cross section. The cross sections of the studied nuclides are based on empirical research. In activation calculations, thermal neutrons are of interest. If fast neutron reactions would be taken into account, they would only have a minor impact on the results. For example the absorption cross section thermal neutron reaction of 59Co(n,γ)60Co is 18.7 barns, whereas the cross section for fast neutron reaction 63Cu(n,\(\alpha\))60Co is around the order of millibarns. Additionally, irradiation history did not take into account maintenance periods during irradiation and thus, the fluence was assumed to be accumulated in a single burnup period.
Fourth parameter affecting the calculation model is the decay or cooling time, which should include maintenance periods when the irradiation source was not in operation and after the sample was taken from the irradiation position. The uncertainty of cooling time was estimated to be 1 month. Depending on nuclide half-lives, at maximum this can have an effect of around 2% for 55Fe, whereas for long-lived nuclides 14C and 63Ni the effect is below 0.1%.
Considerations on uncertainty estimations
Majority of the provided uncertainties of the 14C, 55Fe and 63Ni results included both measurement uncertainties (e.g. LSC and yield measurements) and uncertainties in the radiochemical analysis (e.g. weight, volume etc.). 55Fe and 63Ni results for samples 19–20 did not include uncertainty estimations. Uncertainties in the radiochemical analysis of 55Fe and 63Ni in samples 9–11 were considered to be low, when compared to the LSC uncertainties and therefore a global 15% uncertainty based on the LSC uncertainty was used.
In the case of the 60Co results, the uncertainty estimations for samples 1–2, 14–15 and 16–18 included only uncertainties in the gamma spectrometry measurements whereas all the other 60Co results included also other uncertainties such as uncertainties arising from weight measurements.
In general, significant variations can be seen in the reported uncertainties. In Figs. 1, 2, 3 and 4, the 2σ uncertainties varied from 9 to 23% for 55Fe, from 5 to 23% for 63Ni, from 11 to 41% for 14C, and from 1 to 18% for 60Co. Therefore, it can be concluded that open discussion on the uncertainty determinations are needed in order to produce representative analysis result and thus more effort should be given to it, especially when DTM radionuclides are analysed.
Conclusions
In conclusion, beta-emitter radionuclides are difficult to be measured accurately in nuclear wastes and particularly in some matrices such as steels and concretes. However, their determinations are very important for nuclear waste management in order to obtain accurate results for scaling factors and to make comparison with calculated activities. Intercomparison exercises implemented to validate the accuracy of radiochemical procedures are quite common in liquid matrix but scarcer in solid matrix. The present intercomparison exercise focused on 55Fe, 63Ni and 14C measurements in activated steel. It underlined the importance of comparing the results to others, especially for laboratories which have a little experience. No major difficulty was observed for 55Fe analysis for the majority of the participants. Concerning 63Ni determination, it can be inferred that caution has to be taken towards 60Co, which was not totally removed with some purification methods and can induce an overestimation of 63Ni activity concentration. The intercomparison exercise also emphasized that further work has to be undertaken to obtain accurate 14C measurements due to the uncertainties related to the quantitative extraction of 14C from the solid matrix. 60Co is an easy to measure radionuclide since it can be quantified using gamma spectrometry. However, the results showed that even with easy to measure radionuclides, careful analysis of the measurement data is required.
The overall benefit for the participating laboratories was the validation of the radiochemical methods for 55Fe, 63Ni and 14C analysis since the results were statistically analysed by comparing them to peer provided results (measured assigned value) and modelled results (calculated assigned value). Additionally, the participants benefited from the possibility to submit initially preliminary results and then final results after discussion with the peers. Therefore, the intercomparison exercise improved the quality of the analytical results, and it had a positive impact on the development of the quality assurance in the participating laboratories on a long run. The consortium will carry out a similar intercomparison exercise for the analysis of activated concrete in 2020.
References
Petersen L, Minkkinen P, Esbensen KH (2005) Representative sampling for reliable data analysis: theory of Sampling. Chemom Intell Lab Syst 77:261–277
International Standard ISO 13528:2015(E) (2015) Statistical methods for use in proficiency testing by interlaboratory comparison. ISO, Geneva
Leskinen A, Tanhua-Tyrkkö M, Kekki T, Salminen Paatero S, Zhang W, Hou X, Stenberg Bruzell F, Suutari T, Kangas S, Rautio S, Wendel C, Bourgeaux-Goget M, Stordal S, Isdahl I, Fichet P, Gautier C, Brennetot R, Lambrot G, Laporte E (2020) Intercomparison exercise in analysis of DTM in decommissioning waste. NKS-429, NKS-B, Roskilde, Denmark
Gauld IC, Radulescu G, Ilas G, Murphy BD, Williams ML (2011) Isotopic depletion and decay methods and analysis capabilities in SCALE. Nucl Technol 174:169–195
Beckurts KH, Wirtz K (1964) Neutron physics, appendix I 407–416. Springer, Berlin
Leskinen A, Salminen-Paatero S, Räty A, Tanhua-Tyrkkö M, Iso-Markku T, Puukko E (2020) Determination of 14C, 55Fe, 63Ni and gamma emitters in activated RPV steel samples—a comparison between calculations and experimental analysis. J Radioanal Nucl Chem 323:399–413
Gautier C, Colin C, Garcia C (2015) A comparative study using liquid scintillation counting to determine 63Ni in low and intermediate level radioactive waste. J Radioanal Nucl Chem 308:261–270
Hou X (2005) Rapid analysis of 14C and 3H in graphite and concrete for decommissioning of nuclear reactor. Appl Radiat Isot 62:871–882
Hou X, Østergaard LF, Nielsen SP (2005) Determination of 63Ni and 55Fe in nuclear waste samples using radiochemical separation and liquid scintillation counting. Anal Chim Acta 535(1–2):297–307
Hou XL, Østergaard LF, Nielsen SP (2005) Determination of 63Ni and 55Fe in nuclear waste and environmental samples. Anal Chim Acta 535:297–307
Hou XL, Østergaard LF, Nielsen SP (2007) Determination of 36Cl in nuclear waste from reactor decommissioning. Anal Chem 79:3126–3134
Hou X (2018) Analytical procedure for simultaneous determination of 63Ni and 55Fe. NKS-B RadWorkshop
Hazan I, Korkisch J (1965) Anion-exchange separation of iron, cobalt and nickel. Anal Chim Acta 32:46–51
Brennetot R, Giuliani M, Guégan S, Fichet P, Chiri L, Deloffre P, Masset A, Mougel C, Bachelet F (2017) 3H measurement in radioactive wastes: EFFICIENCY of the pyrolysis method to extract tritium from aqueous effluent, oil and concrete. Fusion Sci Technol 71:397–402
NRG Petten, nuclear reaction program TALYS. https://www.talys.eu/home/. Accessed 5Aug 2019
Taddei MHT, Macacini JF, Vicente R, Marumo JT, Sakata SK, Terremoto LAA (2013) A comparative study using liquid scintillation counting and X-ray spectrometry to determine 55Fe in radioactive wastes. J Radioanal Nucl Chem 295:2267–2272
Warwick PE, Croudace IW (2006) Isolation and quantification of 55Fe and 63Ni in reactor effluents using extraction chromatography and liquid scintillation analysis. Anal Chim Acta 567:277–285
Geckeis H, Hentschel D, Jensen D, Görtzen A, Kerner N (1997) Determination of 55Fe and 63Ni using semi-preparative ion chromatography—a feasibility study. J Anal Chem 357:864–869
König W, Schupfner R, Schüttelkopf H (1995) A fast and very sensitive LSC procedure to determine 55Fe in steel and concrete. J Radioanal Nucl Chem 193(1):119–125
Warwick PE, Croudace IW, Bains MED (1998) An optimised method for the measurement of 55Fe using liquid scintillation analysis. Radioact Radiochem 9(2):19–25
Warwick PE (1999) The determination of pure beta-emitters and their behaviour in a salt-marsh environment. Ph.D. thesis, University of Southampton, School of Ocean and Earth Science, pp 72–76
Lee C, Martin JE, Griffin HC (1997) Optimization of measurement of 63Ni in reactor waste samples using 65Ni as a tracer. Appl Radiat Isot 48:639–642
Eriksson S, Vesterlund A, Olsson M, Ramebäck H (2013) Reducing measurement uncertainty in 63Ni measurements in reactor coolant water high in 60Co activities. J Radioanal Nucl Chem 296:775–779
Ponge-Ferreira CRP, Koskinas MF, Dias MS (2004) Standardization of 45Ca radioactive solution by tracing method. Braz J Phys 34(3A):939–941
Mibus J, Diomidis N, Wieland E, Swanton SW (2018) Release and speciation of 14C during the corrosion of activated steel in deep geological repositories for the disposal of radioactive waste. Radiocarbon 60:1657–1670
https://www.projectcast.eu/. Accessed 10 Feb 2020
Schumann D, Stowasser T, Volmert B, Gunther-Leopold I, Linder H, Wieland E (2014) Determination of the 14C content in activated steel components from a neutron spallation source and a nuclear power plant. Anal Chem 86:5448–5454
Berg JF, Fonnesbeck JE (2001) Determination of 14C in activated metal waste. Anal Chimica Acta 447:191–197
Warwick PE, Kim D, Croudace IW, Oh J (2010) Effective desorption of tritium from diverse solid matrices and its application to routine analysis of decommissioning materials. Anal Chimica Acta 676:93–102
Acknowledgements
Open access funding provided by Technical Research Centre of Finland (VTT). The authors would like to thank the Nordic Nuclear Research NKS-B programme (www.nks.org) for funding the DTM Decom project in which the intercomparison exercise was carried out. The authors would also like to thank the other participating laboratories, namely Technical University of Denmark, Cyclife Sweden AB, Fortum Power and Heat Oy, IFE Kjeller and IFE Halden for provision of data and collaboration. National fundings were given by Finnish Research Programme on Nuclear Waste Management KYT 2022. The authors would also like to thank Fortum Power and Heat Oy for their collaborative actions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leskinen, A., Salminen-Paatero, S., Gautier, C. et al. Intercomparison exercise on difficult to measure radionuclides in activated steel: statistical analysis of radioanalytical results and activation calculations. J Radioanal Nucl Chem 324, 1303–1316 (2020). https://doi.org/10.1007/s10967-020-07181-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-020-07181-x