
Abstract
In the northern hemisphere, the spatial structure of many taxa has been shaped by migration patterns after the last glaciation, and phylogeography based on mtDNA variation may reflect the post-glacial demography. The mtDNA lineages are expected to differ in their adaptations to local conditions but little is known about the impact of these conditions on functional genetic variation. Here, we answer this question through an analysis of geographic variation and selection patterns in seven innate immunity genes in free-living bank voles Myodes glareolus from 10 localities across species range assigned to different lineages based on mtDNA. We found clear discrepancies between population structure in mtDNA and each of the studied innate immunity genes. There was no uniform pattern of spatial variation at immunity loci, they differed in the levels of polymorphism, and the results of neutrality tests were not consistent over loci. Each locus comprised a few common haplotypes shared between mitochondrial lineages and studied locations, plus numerous haplotypes unique for each studied site. Our results suggest that the diversity of innate immunity genes cannot be explained solely in terms of demographic processes, and that the observed polymorphism may be attributed to local selection. The strength and direction of selection differed between loci, even within the same gene family, which underlines how crucial it is to take a complex approach while studying the selection patterns acting on immune-related genes.
Similar content being viewed by others


Introduction
In the northern hemisphere, demography and spatial genetic structure of diverse taxa has been shaped by migration patterns after the last glacial maximum that occurred 26–20,000 ya (Hewitt 2000; Schmitt 2007). Forced southward by the expanding glacier, most temperate species survived the glacial maximum in the refugia, and after ice shield retracted, they migrated north recolonizing habitats freed from ice. This pattern is widespread among mammalian taxa (Baca et al. 2017), and studies of isolation within glacial refugia, and the timing and mode of expansion, are important for our understanding of evolutionary processes such as adaptation, speciation, and extinction (Stewart et al. 2010).
Using mitochondrial DNA, numerous species have been divided into lineages corresponding to the glacial refugia (Wielstra et al. 2013; Klütsch et al. 2012; Mäkinen and Merilä 2008). It has been suggested that physiological traits, in particular those associated with geographical variation in the environmental features, may differ between the lineages due to different conditions they experienced while in refugia and during the subsequent migrations (Kotlík et al. 2006). For instance, in yellow-necked mouse (Apodemus flavicollis), frequency of mtDNA haplogroups correlates with winter temperature (Czarnomska et al. 2019). Little is known about functional genetic variation associated with these local adaptations, yet recently it has been shown that bank voles Myodes glareolus originating from two different glacial refugia have different variants in B-globin what has been linked to a resistance against oxidative stress (Kotlík et al. 2014).
Phylogeographic division of species into clades corresponding to glacial refugia is usually based on variation in neutral markers, such mtDNA, although they reflect only a tiny part of the overall genetic variation. The effect of demographic processes on spatial genetic variation may be altered by selection, in particular adaptation to the environmental conditions. Primarily, this mechanism is expected to affect genes whose product can be directly linked to spatially heterogeneous environment. An excellent example for studying these processes are genes encoding components of the immune system. There is a vast evidence that their variation is mostly shaped by the parasite-driven selection (Bernatchez and Landry 2003). The parasite communities, on the other hand, tend to differ between locations (Haukisalmi and Henttonen 2000) constituting spatially heterogeneous selective force (Bonneaud et al. 2006; Kloch et al. 2010; Fumagalli et al. 2010). In mammals and higher vertebrates, the immune system comprises dozens of interacting molecules, yet the mechanisms of the pathogen-driven selection have been comprehensively studied only in the case of major histocompatibility complex (MHC) (e.g., Bernatchez and Landry 2003). Though the MHC plays an important role in antigen-based pathogen recognition, it is not the only nor the main factor responsible for resistance against pathogens (Jepson et al. 1997). In particular, recent studies focused on the components of the innate immunity revealing that the pattern drawn from MHC studies does not necessarily translates into other immunity loci (Kloch et al. 2018).
In the present study, we aimed to investigate the spatial variation at seven immunity loci in bank voles over a large geographic scale in bank voles, M. glareolus, in the context of post-glacial demography. Bank vole M. glareolus is considered one of the key mammal species for genetic studies of post-glacial history (Filipi et al. 2015). Its contemporary populations have been divided into six major lineages corresponding to three refugia on the Mediterranean peninsulas of Iberia, Italy, and the Balkans, two continental named Western and Eastern European, and Carpathian lineage originating from a cryptic glacial refugium in this mountain range (Deffontaine et al. 2005; Kotlík et al. 2006; Wójcik et al. 2010). There is no evidence for severe bottlenecks in bank voles, nor habitat loss, or population fragmentation, thus their genetic variation across species range can be contributed mostly to natural processes related to migration and selection.
Previous studies have shown no congruence between spatial distribution of haplotypes of two immunity loci, namely MHC DRB and TLR2, and mitochondrial lineages (Malé et al. 2012; Morger et al. 2015). However, the selection patterns may differ considerably within the same gene family, toll-like receptors being one of a notable examples (Kloch et al. 2018). Thus, for better understanding how post-glacial demography shapes variation in functional loci, it seems crucial to analyse broader set of genes. To fill this gap, in the present paper we (1) characterized genetic diversity at seven innate immunity loci, (2) analysed selection patterns in the innate immunity loci using sequence- and codon-oriented tests and (3) compared population structure at the innate immunity loci to the structure derived from mtDNA that proved to evolve under neutral conditions (Kotlík et al. 2006; Filipi et al. 2015).
Materials and methods
Material and genotyping
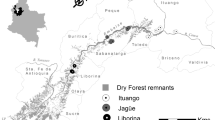
Genetic polymorphism in 7 innate immunity genes (TLR1, 2, 4, 5, 9, TNFa, TNFb) and in mitochondrial gene coding cytochrome b (hereafter cytB) was characterized in 69 voles originating from 10 sites (populations) across Europe (Fig. 1, Table S1). In the current work, we used samples provided by members of the scientific community (researchers are listed in the Acknowledgements), and collected as part of other studies. The tissues were obtained either from biopsies taken from anaesthetised animals (Germany, Sweden, Russia) or animals killed in accordance with the European ethical normative (Directive 2010/63/EU) through anaesthetic overdose and/or cervical dislocation (France, Poland, Slovakia, Serbia). Relevant permission from ethical committees are provided in the Supplementary Materials.

Location of the studied sites along with assignment of samples to the mitochondrial lineages: yellow—Western clade, green—Carpathian clade, blue—Balkan clade, red—Eastern clade. The background map originates from Wikimedia Commons, under Creative Commons licence, modified
From each site we analysed from 3 to 10 individuals (Table S1), which—given the length of analysed amplicons—is an accepted number for studies covering a wide geographical area providing accurate estimates of population genetic parameters (Pluzhnikov and Donnelly 1996; Moeller et al. 2007). Loci were amplified using primers designed and optimized for the bank vole, the PCR details and primer sequences with relevant references are given in Table S2. Resulting amplicons were pooled for each individual, purified twice using CleanUp kit (Aabiot). Library was prepared using Nextera XT DNA kit and sequenced in a single run on Illumina MiSeq using MiSeq Reagent Kit (300 cycles) producing 2 × 150 bp reads. Reads were processed as described in Kloch et al. (2018).
Neutral phylogeny in mitochondrial cytochrome B
First, to ensure that cytB is not under selection and thus allow for inferring neutral population structure, in DnaSP v.6 (Rozas et al. 2017) we performed a series of neutrality tests that confirmed this assumption (Tajima D = − 1.09ns, Fu and Li D = − 2.22ns, F = − 2.12ns, McDonald–Kreitman NI = 1.164ns, G = 0.047ns). Maximum likelihood phylogenetic trees for cytB were inferred using Mega v. 7.0 (Kumar et al. 2016). The model of nucleotide evolution was determined using corrected Akaike’s Information Criteria in jModelTest version 2.1.10 (Darriba et al. 2012). The robustness of the tree was tested with 1000 bootstrap replications, using the Tien Shan red-backed vole (M. centralis) sequence as an outgroup. In the analysis, we included two mitochondrial haplotypes from each lineage described by Kotlík et al. (2006) to assure that the obtained phylogeny is concordant with previously described division of European bank voles into mitochondrial lineages.
Phylogeography and population structure
The population structure of innate immunity loci was analysed using discriminant analysis of principal components (DAPC) implemented in the R package adegenet (Jombart et al. 2010; R Core Team 2018). First, using the clustering algorithm based on k-means, we sought the best partitioning of the observed variation with no assumptions about group membership of the samples. To identify the optimal number of clusters, we used a plot of the BIC (Bayesian Information Criterion) score of a given partition against the number of clusters included in this partition, as implemented in the find.clusters function. Next, we used DAPC to investigate variation between actual populations (sampled sites).
To visualise the genealogical relationships between haplotypes we constructed minimum spanning networks using Popart (Leigh and Bryant 2015).
Tests of selection acting on innate immunity loci
We focused on functional variation of the studied innate immunity loci, and from this part of the analysis we excluded non-coding parts of the sequenced amplicons in TNFa and TNFb. We also discarded a pseudogene detected in TLR5. The pseudogenes had a 2 bp deletion in position 224 resulting in premature stop codon in position 379, and all individuals with the pseudogene were heterozygotes.
Using DnaSP v.6 we calculated basic measures of sequence diversity: the number of variable sites (S), haplotype diversity (Hd), average number of nucleotide differences (k), and nucleotide diversity per site (π). Separately for each gene, we tested for departures from neutrality using three tests suitable for single loci (Nielsen 2001): Tajima’s D (Tajima’s (1989), Fay and Wu’s H (Fay and Wu 2000), and the Ewens–Watterson test (Ewens 1972; Watterson 1978). Tajima’s D allows an excess of intermediate or low frequency alleles to be detected, while Fay and Wu’s measures the abundance of high-frequency derived alleles in relation to intermediate frequency variants. The Ewens–Watterson test, in turn, compares observed homozygosity to the homozygosity expected given the observed number of alleles. Since all these tests are sensitive to demography and background selection, we also computed three compound tests proposed by Zeng et al. (2006) which are powerful to detect positive selection and robust against demographic assumptions Zeng et al. (2006, 2007). The calculations were performed in the program dh (available from the website http://zeng-lab.group.shef.ac.uk) using a mouse sequence as an outgroup and with default 10,000 coalescent simulations.
Since selection may affect particular parts of a protein differently, we aimed to detect sites under selection based on the dN/dS ratio at each codon (Kosakovsky Pond and Frost 2005) using the DataMonkey server (Delport et al. 2010). First, we tested for possible recombination events that might have affected the diversity of the studied loci using Genetic Algorithm Recombination Detection (GARD) (Kosakovsky Pond et al. 2006), and the identified recombination effects were accounted for in the models. Next, we ran two phylogenetically controlled models of selection: (1) MEME, the mixed effects model of evolution, which allows for the detection of episodic positive selection (Murrell et al. 2012), and (2) FUBAR, the fast unconstrained Bayesian approximation model, which allows for the detection of sites under positive or purifying selection (Murrell et al. 2013).
To check the location of codons under selection at innate immunity genes, we identified and annotated domains within the studied proteins using SMART (Simple Modular Architecture Research Tool) which allows domains to be identified and annotated (Letunic and Bork 2018). Additionally, in TLRs using LRR finder (Offord et al. 2010) we identified leucine-rich repeats (LRRs) which form functionally important sites of these proteins, as they are involved in interactions with PAMPs or other components of the immune system.
Results
Phylogeny of cytochrome b
In cytB, we found 51 haplotypes in 68 individuals. The nucleotide evolution of cytB sequences was best described by the GTR + I model. The maximum likelihood tree (Fig. 2), despite relatively poor support of internal branches, well reflected the subdivision of European bank vole populations into four main lineages described by Kotlík et al. (2006). Haplotypes, whose assignment to mtDNA lineages was established in other studies, clearly grouped within clades detected in our analysis. This suggests that mitochondrial phylogeny from the current study follows the same pattern as reported previously.

Phylogeny of the bank vole cytochrome b, inferred by maximum likelihood method haplotypes from the current study are named by the sampling site; two haplotypes per each mitochondrial lineage described previously by Kotlík et al. (2006) are named by their Genbank accession numbers. Branch labels indicate the percent bootstrap frequencies greater than 50%. Colours correspond to lineages described by Kotlík et al. (2006): yellow—Western clade, green—Carpathian clade, blue—Balkan clade, red—Eastern clade. The putative nuclear pseudogenes are marked with purple. The number of sequences represented by each haplotype are given in brackets
Eleven haplotypes from individuals captured in sites Ard and Jur in France and Slo in Slovakia did not group within any clade, neither they formed a separate supported group (Fig. 2). Although they did not contain frame-shift mutations or premature stop codons characteristic for pseudogenes, they were considered nuclear paralogues, because they formed an early branching clade located closer to nuclear pseudogenes described by Filipi et al. (2015) than to any other known mitochondrial clades. The location of these putative pseudogenes within the tree suggests that nuclear insertion pre-dated the divergence between the mtDNA clades, and thus they were excluded from further analysis concerning cytB, as they are not reliable for mitochondrial phylogenetic inference. Notably, other samples from the three populations that included pseudogenes (i.e., Jur, Ard, and Slo) were clearly grouped within other clades what allowed us to assign them to the proper mtDNA lineages.
Disregarding the pseudogenes, six sites were assigned to a single mitochondrial lineage. Samples from Russia (Bak, Ily, Int) and Slovakia (Slo) were assigned to the Eastern lineage, all samples from Sweden (Swe) were assigned to Carpathian lineage, site Jur from France was assigned to the Western lineage (Figs. 1, 2). The three remaining sites included samples assigned to more than one lineage. In Poland (Pol), we found members of Carpathian and Eastern lineages, in France (Ard) there were individuals assigned to either Western or Carpathian lineages, in Germany (Ger)—from Western and Eastern lineages, and in Serbia (Ser)—Carpathian and Balkan. Although each lineage except Balkan included individuals from different sites and geographic areas, there were no haplotypes shared between sites except for one haplotype present in both, Ily and Int from Eastern lineage. One haplotype from Ard population was placed in the basal position to the Carpathian and Western lineages.
Population structure
To describe the population structure of the studied loci we used discriminant analysis of principal components (DAPC). First, we explored the structure with no prior assumption about the number of groups. The most probable number of clusters is identified as a point where increasing the number of clusters does not result in a considerable decrease in Bayesian Information Criterion (BIC), thus the classical BIC curve is expected to show an “elbow shape” (Jombart et al. 2010). In cytB, the structure was best described by 7–10 clusters. No structure was observed in TNFb, where BIC consistently decreased with increasing number of cluster. In the remaining loci the slope of the BIC curve levelled out after reaching 4–9 clusters, and this pattern was best visible in TLR2 (Figure S1). In cytB the resulting division resembled the geographic locations (Fig. 3), although DAPC identified three clusters within the samples assigned to the Eastern lineage (sites Bak, Ily, Int, Slo, indicated in Fig. 3 in blue, red and orange), and two clusters within the Western lineage (sites Jur and Ard, light green and light orange). In the immunity loci, the pattern was much less clear, and the membership probability of the samples was rather ambiguous in TLR2 and TNFa. In TLR2, some correspondence between geographic locations and clustering was seen, and the samples from Ural (sites Ily and Int) form a separate clade. Similarly, samples from Serbia grouped together in TLR5. Nonetheless, there was no consistent pattern across loci.

Results of DAPC clustering algorithm applied to innate immunity loci with no prior assumption about group membership of the analysed individuals. Each bar represents an individual, and the populations of origin are given along x axis. Colour indicate different clusters, and the number of clusters is denoted by k
Next, we applied DAPC to characterize the variation in immune genes between actual populations (sampling sites). In cytB, PC1 differentiated samples from Balkan clade (Serbia) and from Sweden, which contained exclusively samples from Carpathian clade. Russian sites, assigned to the Eastern clade, were discriminated from the remaining clades along PC2, although this division was weaker that in the case of PC1. In immunity genes, we obtained no consistent pattern over loci, and likewise, the genetic differences between sites did not resemble either their geographical location or the assignment of the samples to mtDNA clades (Fig. 4). For instance, in TLR1, samples from Ard in France, assigned to Western mitochondrial lineage, were differentiated along the principal axis PC1, what repeated pattern revealed by the haplotype network (see below). In TLR2, three main clusters could be seen: one containing samples from Serbia (Ser) and Ural (Int, Ily), a second consisting of samples from Eastern Europe (Pol and Bak), and a third including the remaining populations. Again, the pattern differed from the one revealed by mitochondrial phylogenetic tree. No clear pattern was found in the remaining loci, although weak division along PC1 between samples from Eastern and Western Europe could be seen in TLR4.

Differences between sites (populations) summarized by DAPC. Only first and second principal component (PC1 and PC2) are shown. Individuals are represented as dots, and circles and colours indicate samples from respective sites
In cytB, the topology of the haplotype network generally followed the pattern expected from the geographic location of the samples and their division between mitochondrial lineages. In Figure S2, three main clusters are visible. In bottom left, a cluster groups samples from the Eastern lineage (sites Ger, Ily, Int, Pol), and in bottom right, samples from Western lineage (sites Ard and Jur) are clustered. The top of the network consist of a cluster including samples assigned to the Carpathian and Balkan lineages. Contrarily, networks showing genealogical relationships between the innate immunity haplotypes generally did not mirror the assignment of the samples to the mitochondrial lineages. In TLR loci, each network consisted of a few haplotypes present at several sites, and numerous rare alleles specific for particular sites (Figure S2). Even when rare haplotypes from geographically close sites grouped together, they usually adjoined haplotypes from a site located at a long distance. For instance, the TLR1 haplotypes from France (sites Ard and Jur) formed a distinct cluster within the network, yet this cluster was separated by one substitution from a haplotype present only in Russia, and two substitutions from haplotype from Poland. The genetic distance between haplotypes varied between loci: in TLR1, TNFa, TNFb most haplotypes were separated by few substitutions, while in TLR2 and TLR5 the network was sparse, and haplotypes were separated by several substitutions.
Selection patterns in innate immunity loci
Nucleotide diversity per site (π) and haplotype diversity (Hdiv) was higher at cytB than in immunity genes loci. The immune loci differed in terms of polymorphism: the highest number of segregating sites (S), and the highest average number of nucleotide differences between sequences (k), was found in TLR2, and the lowest in TNFb (Table 1). In TLR1, TLR2 the number of haplotypes exceeded 50, and in TNFb it was only 14. However, the haplotype diversity was high in all loci, exceeding 0.7 even in the least variable TNFb.
Species-wide Tajima’s D was significant and negative in TLR4 and TLR9, and marginally not significant (p = 0.053) in TLR5 (Table 2), which indicated an excess of rare alleles. Fay and Wu’s H test, which detects an excess of high/low frequency variants compared to intermediate frequency variants, was consistently negative and significant in all loci but TNFa. EW tests were significant in TLR2, 4, 5, and 9. The F statistics of EW test, an equivalent to expected homozygosity, was similar in TLR4, TLR5, and TLR9, varying from 0.12 to 0.15, and considerably lower in TLR2 (F = 0.04, Table 2).
Since the outcome of the classical neutrality tests described above may be biased due to demographic events that affected studied populations, we computed compound tests (Zeng et al. 2006, 2007) what allowed us to test for selection regardless of other processes that might affect the site-frequency spectrum, such as population expansion, bottleneck or strong structure. All compound tests (DH, HEW, DHEW) gave consistently significant results for TLR4, TLR5, and TLR9, indicating that these loci are under positive selection (Tables 2, 3).
Using site-specific, phylogenetically controlled dN/dS selection tests, we identified codons under selection in each studied locus (Table 4). The FUBAR model identified codons under purifying selection in all studied loci, and codons under pervasive diversifying selection in all loci but TNFa. The MEME model indicated codons under episodic positive selection in all loci except for TNFa and TLR9. In TNFa only signatures of purifying selection were found, and in TNFb each model indicated just one codon under selection. Overall, the highest number of positively and negatively selected codons was found in TLR2, and majority of codons under selection were located within leucine-rich repeats. In TLR1 and TLR4 only codons affected by pervasive purifying selection were located within LRRs. Moreover, in TLR2, TLR4, and TLR9 several codons under pervasive purifying selection were located in highly conserved, intracellular signalling TIR domain.
Discussion
The impact of post-glacial demography on genetic variation in Palaearctic has been comprehensively studied using neutral markers, such as mitochondrial DNA, but still not much is known how these events shaped variation in the coding parts of the genome, particularly in genes that are expected to experience selective pressure associated with adaptations to local environmental conditions. In the present paper, we studied species-wide variation at seven innate immunity loci in ten populations assigned to different mtDNA lineages corresponding to different glacial refugia. We have found clear differences between population structure at selectively neutral cytB and innate immunity genes what suggest the major role of selection over demography in shaping the diversity and population structure of these genes.
Population structure based on cytochrome b
The large-scale population structure of the bank vole based on mtDNA generally confirmed the pattern described previously (e.g., Filipi et al. 2015; Deffontaine et al. 2005). Populations located in the central part of the species range (Poland, Germany, and Serbia) included voles assigned to more than one mtDNA lineage, while sites located in the eastern and northern parts of the species range were assigned to single lineages (Eastern or Carpathian, respectively). Contrarily to the previous studies, where samples from France were all assigned to the Western lineage (Deffontaine et al. 2005), in French site Ard we found an individual assigned to the Carpathian lineage, and another one occupying a basal position in the tree. As suggested by Filipi et al. (2015), such a position may be an artefact caused by a low number of informative positions in short cytB sequences (Filipi et al. 2015), yet the cytB fragment analysed in the current study was 1074 bp.
Recent development of whole-genome sequencing techniques allows for inferring the population structure using large sets of SNP (single-nucleotide polymorphisms) scattered through the genome. Although they provide much detailed insight into the genetic variation, recent studies have shown that genome-wide random SNP markers give similarly good estimates of population structure as microsatellites (Liu et al. 2005). On the other hand, in the case of bank voles, spatial pattern derived from microsatellite analysis was concordant to that based on mtDNA (Tarnowska et al. 2019) what indicates that mtDNA variation reliably reflects the large-scale neutral variation in this species.
Our phylogenetic analysis revealed a group haplotypes that fell outside the main clades. Similar pattern was found in a study of bank vole mitogenomes (Filipi et al. 2015) and the group of haplotypes forming an early branching clade was interpreted as resulting from a transposition of mtDNA into nuclear genome. According to Filipi et al. (2015), this event occurred before the diversification of the contemporary mtDNA haplotypes. For this reason, these samples could not be used to infer post-glacial recolonization, yet in each of ours sampling sites that included putative pseudogenes, we also identified haplotypes specific for each proper mtDNA lineage. The presence of pseudogenes did not affect our results, as in the current paper the division between mtDNA lineages was used as a background for an analysis focused on the spatial variation in the innate immunity loci.
Spatial variation in immunity loci
As expected, in the innate immunity loci we found haplotypes shared between sites assigned to different mtDNA lineages what suggest that they have originated before the split of the population between glacial refugia (Morger et al. 2015). Alleles shared between lineages were more frequent in cytokines (TNFa and TNFb) than in toll-like receptors (TLRs) which are more likely to evolve under parasite-driven selection (Kloch et al. 2018). This pattern suggests long-time retention of multiple lineages what may be contributed to the role of balancing selection in shaping immune genetic diversity (Quéméré et al. 2015; Morger et al. 2015).
In each TLR locus we found a large number of private alleles unique for each site which generally did not form apparent clusters in the haplotype networks. Such an inconsistency over haplotype networks constructed for several loci suggests that the variation at these loci was shaped differently by the selection (Avise and Ball 1990). Moreover, within-site variance indicated by DAPC analysis was much higher than between-site variance, and the clusters grouped samples from various location. Similar results were reported in a phylogeographic study of bank vole MHC DQA gene, where over 90% of observed variation was distributed among individuals rather than populations, and grouping of MHC alleles did not correspond to the mtDNA clades (Malé et al. 2012). Also, a previous study of TLR2 in bank voles revealed strong differentiation between sites, a large number of private haplotypes, and no associations between TLR2 and mtDNA clades (Morger et al. 2015).
In numerous studies, a major role of parasite-driven selection in shaping the diversity of immunity genes was shown (Tollenaere et al. 2008; Kloch et al. 2010; Tschirren et al. 2012). The bank vole parasites display high genetic variability across various geographic scales (e.g., Paziewska et al. 2011; Sakka et al. 2015), what may explain the structure of haplotype networks observed in bank vole TLRs: common haplotypes may be associated with old pathogen lineages, while numerous private alleles suggests younger adaptations to local, parasite-driven selection. The phylogeographic variation at innate immunity loci studied in the current paper suggest that majority of the observed variation resulted from local, parasite-driven selection that occurred independently in each location, regardless on the large-scale demographic processes, including post-glacial colonization patterns.
A global excess of rare alleles, detected in the current study in all studied genes, may be explained by a large number of private alleles in each site. Again, this result suggests that local selection, likely due to evolutionary pressure from parasites, plays a crucial role in shaping variation at innate immunity loci over a large geographic scale. A considerable number of private alleles may indicate selection even if the population-specific test do not produce significant results (Sjöstrand et al. 2014). An excess of rare variants, usually interpreted as an evidence of a negative selection removing deleterious mutations, may also indicate positive selection and demographic events such as change in population size or population subdivision (Biswas and Akey 2006; Charlesworth et al. 1993). Although this explanation seem plausible for our study, we are aware that the high number of rare variants may be a result of low sample sizes for some populations.
Differences in selection between two families of innate immunity genes
The two families of innate immunity proteins studied in the current paper differ in function, and thus they are expected to experience different strength and direction of selection. TLRs acting as pattern-recognition proteins are considered an afferent arm of the response, and thus are expected to evolve under parasite-driven selection promoting high variability, while cytokines contributing to the signal transduction are considered an efferent arm, and are expected to be more evolutionary constrained (Beutler 2004; Chapman et al. 2016). In the components of the efferent arm, low diversity and signatures of purifying selection were previously reported in beta-defensins in mallards (Chapman et al. 2016) but interleukins Il1b, Il2 in field voles displayed signatures of balancing selection (Turner et al. 2012), and variation at these loci was associated with a resistance against parasites (Turner et al. 2011). Relatively high variation in TNFa found in our study suggests it evolves under parasite-driven selection, and indeed, associations between SNPs located in the promoter of TNFa gene and susceptibility to Puumala virus has been reported in bank voles (Guivier et al. 2010).
Recent studies have shown that innate immunity loci more often show signatures of balancing selection in a small geographic scale (Ferrer-Admetlla et al. 2008; Turner et al. 2012; Quéméré et al. 2015; Kloch et al. 2018), and on a larger geographical the positive and/or negative selection was usually reported (Nakajima et al. 2008; Wlasiuk and Nachman 2010; Babik et al. 2015). This may be explained by the fact that, contrary to positive selection, it is difficult to detect balancing selection over a long evolutionary scale (Charlesworth 2006). The neutrality tests widely used tool to infer selection are sensitive to demographic assumptions. For instance, negative values of Tajima’s D, as detected in TLR4, TLR5, and TLR9 in the current study, are commonly interpreted as indicative of negative (purifying) selection, which eliminates rare, deleterious alleles but such a result may be also a sign of population expansion but it can be also a result of the selective sweep. However, we find this explanation not likely. In mice, the genomic organization of regions adjacent to the studied loci differs considerably between TLR4, which is flanked by ~ 0.5 Mb non-coding sequences, and the remaining two neighbouring many genes. Thus, if sweep was the reason of the negative values of Tajima’s D in the studied loci, one would likely observe differences between TLR4 and TLR5 and 9. Using compound tests (Zeng et al. 2006, 2007) we circumvent rather rigorous assumptions of the classic neutrality tests. The compound tests (DH, HEW, DHEW) gave consistently significant results for TLR4, TLR5, and TLR9, indicating that these loci are under positive selection. In TNFb only HEW was significant. The HEW test is powerful in detecting positive selection but also vulnerable to population structure, so this supports our finding that TNFb gene diversity is shaped rather by demography than positive selection.
In the present paper, we identified codons under purifying selection in all innate immunity loci, which agrees with previous reports on selection acting on TLRs (Mukherjee et al. 2014; Alcaide and Edwards 2011). The codons under negative selection were evenly distributed in the extracellular domain and in the conserved TIR domain while positively selected codons, both under episodic and pervasive selection, were mostly located in the extracellular domain and within LLRs. The extracellular domain, where leucine-rich repeats responsible for PAMP binding are located, are expected to evolve under positive diversifying selection as a result of host–pathogen interactions (Fornůsková et al. 2013). Similarly, positive selection at specific codons was found in TLR1, TLR3 and TLR4 in pipits (Gonzalez-Quevedo et al. 2015). In rodents, positively selected codons were located close to sites involved in pathogen binding or heterodimerization with TLR1 (Tschirren et al. 2011). Episodic positive selection was confirmed in birds (Grueber et al. 2013). The highest number of both positively and negatively selected sites was found in TLR2, which was previously shown to evolve under parasite-driven selection (Tschirren et al. 2013; Quéméré et al. 2015), and it is maintained over evolutionary time in European bank vole populations (Morger et al. 2015).
The fact that episodic and pervasive selection were detected in the same codons in most of the studied loci suggests that either pervasive selection is responsible for shaping the diversity of the studied loci, or the episodic effects are masked by continuously acting selection (Grueber et al. 2013). This result may also be attributed to the large geographic scale of our study, and episodic selection caused by local host–pathogen interactions should be easier to detect on a smaller geographical scale.
Conclusions
Our results showed that the variation of the innate immunity loci in bank voles in a considerable part of its range does not mirror the genetic structure in mtDNA. No congruence suggest that variation at innate immunity loci was shaped by local, parasite-driven selection rather than demographic effects. Although we identified haplotypes shared between sites, most variation was observed within sites, and in most loci we detected a large number of private alleles. We found no uniform pattern of evolution among innate immunity loci, neither between nor within gene families, what suggests that selection operates differently at each locus, and the results from a single locus should not be generalized. To gain comprehensive insight into the evolution of innate immunity components, a multi-locus approach seems crucial.
Data archiving
Sequences analysed in the current paper have been submitted to GenBank. The datasets used during the current study are available from the corresponding author upon request.
References
Alcaide M, Edwards SV (2011) Molecular evolution of the toll-like receptor multigene family in birds. Mol Biol Evol 28:1703–1715
Avise JC, Ball RM (1990) Principles of genealogical concordance in species concepts and biological taxonomy. Oxf Surv Evol Biol 7:45–67
Babik W, Dudek K, Fijarczyk A, Pabijan M, Stuglik M, Szkotak R, Zieliński P (2015) Constraint and adaptation in newt toll-like receptor genes. Genom Biol Evol 7:81–95
Baca M, Nadachowski A, Lipecki G, Mackiewicz P, Marciszak A, Popović D, Socha P, Stefaniak K, Wojtal P (2017) Impact of climatic changes in the Late Pleistocene on migrations and extinctions of mammals in Europe: four case studies. Geol Q 61(2):291–304
Bernatchez L, Landry C (2003) MHC studies in nonmodel vertebrates, what have we learned about natural selection in 15 years. J Evol Biol 16:363–377
Beutler B (2004) Innate immunity, an overview. Mol Immunol 40:845–859
Biswas S, Akey JM (2006) Genomic insights into positive selection. Trends Genet 22:437–446
Bonneaud C, Perez-Tris J, Federici P, Chastel O, Sorci G (2006) Major histocompatibility alleles associated with local resistance to malaria in a passerine. Evolution 60:383–389
Chapman JR, Hellgren O, Helinm AS, Kraus RH, Cromie RL, Waldenström J (2016) The evolution of innate immune genes, purifying and balancing selection on β-defensins in waterfowl. Mol Biol Evol 33:3075–3087
Charlesworth D (2006) Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet 2:e64
Charlesworth B, Morgan MT, Charlesworth D (1993) The effect of deleterious mutations on neutral molecular variation. Genetics 134:1289–1303
Czarnomska SD, Niedziałkowska M, Borowik T, Jędrzejewska B (2019) Winter temperature correlates with mtDNA genetic structure of yellownecked mouse population in NE Poland. PLoS ONE 14:e0216361
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 30:772
Deffontaine V, Libois R, Kotlik P, Sommer R, Nieberding C, Paradis E, Searle JB, Michaux JR (2005) Beyond the Mediterranean peninsulas: evidence of central European glacial refugia for a temperate forest mammal species, the bank vole (Clethrionomys glareolus). Mol Ecol 14:1727–1739
Delport W, Poon AFY, Frost SDW, Kosakovsky Pond SL (2010) Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457
Ewens WJ (1972) The sampling theory of selectively neutral alleles. Theor Popul Biol 3:87–112
Fay JC, Wu CI (2000) Hitchhiking under positive Darwinian selection. Genetics 155:1405–1413
Ferrer-Admetlla A, Bosch E, Sikora M, Marquès-Bonet T, Ramírez-Soriano A, Muntasell A, Navarro A, Lazarus R, Calafell F, Bertranpetit J, Casals F (2008) Balancing selection is the main force shaping the evolution of innate immunity genes. J Immunol 181:1315–1322
Filipi K, Marková S, Searle JB, Kotlík P (2015) Mitogenomic phylogenetics of the bank vole Clethrionomys glareolus, a model system for studying end-glacial colonization of Europe. Mol Phylogenet Evol 82:245–257
Fornůsková A, Vinkler M, Pagès M, Galan M, Jousselin E, Cerqueira F, Morand S, Charbonnel N, Bryja J, Cosson J-F (2013) Contrasted evolutionary histories of two Toll-like receptors Tlr4 and Tlr7 in wild rodents (MURINAE). BMC Evol Biol 13:194
Fumagalli M, Pozzoli U, Cagliani R, Comi GP, Bresolin N et al (2010) Genome-wide identification of susceptibility alleles for viral infections through a population genetics approach. PLoS Genet 6:e1000849
Gonzalez-Quevedo C, Spurgin LG, Illera JC, Richardson DS (2015) Drift, not selection, shapes toll-like receptor variation among oceanic island populations. Mol Ecol 24:5852–5863
Grueber C, Wallis GP, Jamieson I (2013) Genetic drift outweighs natural selection at toll-like receptor TLR immunity loci in a re-introduced population of a threatened species. Mol Ecol 22:4470–4482
Guivier E, Galan M, Ribas A, Xuéreb A, Chaval Y, Olsson G, Essbauer S, Henttonen H, Voutilainen L, Cosson JF, Charbonnel N (2010) Tnf-α expression and promoter sequences reflect the balance of tolerance/resistance to Puumala hantavirus infection in European bank vole populations. Infect Genet Evol 10:1208–1217
Haukisalmi V, Henttonen H (2000) Variability of helminth assemblages and populations in the bank vole Clethrionomys glareolus. Pol J Ecol 48:219–231
Hewitt G (2000) The genetic legacy of the quaternary ice ages. Nature 405:907–913
Jepson A, Banya W, Sisay-Joof F, Hassan-King M, Nunes C, Bennett S, Whittle H (1997) Quantification of the relative contribution of major histocompatibility complex (MHC) and non-MHC genes to human immune responses to foreign antigens. Infect Immunol 65:872–876
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components, a new method for the analysis of genetically structured populations. BMC Genet 11:94
Kloch A, Babik W, Bajer A, Sinski E, Radwan J (2010) Effects of an MHC-DRB genotype and allele number on the load of gut parasites in the bank vole Myodes glareolus. Mol Ecol 19:255–265
Kloch A, Wenzel MA, Laetsch DR, Michalski O, Bajer A, Behnke J, Welc-Falęciak R, Piertney SB (2018) Signatures of balancing selection in toll-like receptor (TLR) genes—novel insights from a free-living rodent. Sci Rep 8:8361
Klütsch CFC, Manseau M, Wilson PJ (2012) Phylogeographical analysis of mtDNA data indicates postglacial expansion from multiple glacial refugia in woodland caribou (Rangifer tarandus caribou). PLoS One 7:e52661
Kosakovsky Pond SL, Frost SD (2005) Not so different after all, a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222
Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD (2006) GARD, a genetic algorithm for recombination detection. Bioinformatics 22:3096–3098
Kotlík P, Deffontaine V, Mascheretti S, Zima J, Michaux JR, Searle JB (2006) A northern glacial refugium for bank voles (Clethrionomys glareolus). Proc Natl Acad Sci USA 103:14860–14864
Kotlík P, Marková S, Vojtek L, Stratil A, Šlechta V, Hyršl P, Searle JB (2014) Adaptive phylogeography: functional divergence between haemoglobins derived from different glacial refugia in the bank vole. Proc R Soc Biol Sci 281:20140021
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Leigh JW, Bryant D (2015) PopART: Full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Letunic I, Bork P (2018) 20 years of the SMART protein domain annotation resource. Nucleic Acids Res 46:D493–D496
Liu N, Chen L, Wang S, Oh C, Zhao H (2005) Comparison of single-nucleotide polymorphisms and microsatellites in inference of population structure. BMC Genet 6:S26
Mäkinen HS, Merilä J (2008) Mitochondrial DNA phylogeography of the three-spined stickleback (Gasterosteus aculeatus) in Europe—evidence for multiple glacial refugia. Mol Phylogenet Evol 46:167–182
Malé PJG, Martin JF, Galan M, Deffontaine V, Bryja J, Cosson JF, Michaux J, Charbonnel N (2012) Discongruence of Mhc and cytochrome b phylogeographical patterns in Myodes glareolus (Rodentia: Cricetidae). Biol J Linn Soc 105:881–899
Moeller DA, Tenaillon MI, Tiffin P (2007) Population structure and its effects on patterns of nucleotide polymorphism in teosinte (Zea mays ssp. parviglumis). Genetics 176:1799–1809
Morger J, Råberg L, Hille SM, Helsen S, Štefka J, Al-Sabi MM, Kapel ChMO, Mappes T, Essbauer S, Ulrich RG, Bartolommei P, Mortelliti A, Balčiauskas L, van den Brink NW, Rémy A, Bajer A, Cheprakov M, Korva M, García-Pérez AL, Biek R, Withenshaw S, Tschirren B (2015) Distinct haplotype structure at the innate immune receptor Toll-like receptor 2 across bank vole populations and lineages in Europe. Biol J Linn Soc 116:124–133
Mukherjee S, Ganguli D, Majumder PP (2014) Global footprints of purifying selection on toll-like receptor genes primarily associated with response to bacterial infections in humans. Genom Biol Evol 6:551–558
Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL (2012) Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8:e1002764
Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K (2013) FUBAR: a fast, unconstrained Bayesian approximation for inferring selection. Mol Biol Evol 30:1196–1205
Nakajima T, Ohtani H, Satta Y, Uno Y, Akari H, Ishida T, Kimura A (2008) Natural selection in the TLR-related genes in the course of primate evolution. Immunogenetics 60:727–735
Nielsen R (2001) Statistical tests of selective neutrality in the age of genomics. Heredity 86:641–647
Offord V, Coffey TJ, Werling D (2010) A web application for the identification of leucine-rich repeats and an integrative Toll-like receptor database. Dev Comp Immunol 34:1035–1041
Paziewska A, Harris PD, Zwolińska L, Bajer A, Siński E (2011) Recombination within and between species of the alpha proteobacterium Bartonella infecting rodents. Microb Ecol 61:134–145
Pluzhnikov A, Donnelly P (1996) Optimal sequencing strategies for surveying molecular genetic diversity. Genetics 144:1247–1262
Quéméré E, Galan M, Cosson J, Klein F, Aulagnier S, Gilot-Fromont E, Merlet J, Bonhomme M, Hewison AJ, Charbonnel N (2015) Immunogenetic heterogeneity in a widespread ungulate, the European roe deer (Capreolus capreolus). Mol Ecol 24:3873–3887
R Core Team (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol Biol Evol 34:3299–3302
Sakka H, Henttonen H, Baraket G, Amel S-H, Michaux J (2015) Phylogeography analysis and molecular evolution patterns of the nematode parasite Heligmosomum mixtum based on mitochondrial DNA sequences. Acta Parasitol 60:85–98
Schmitt T (2007) Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Front Zool 4:11
Sjöstrand AE, Sjödin P, Jakobsson M (2014) Private haplotypes can reveal local adaptation. BMC Genet 15:61
Stewart JR, Lister AM, Barnes I, Dalén L (2010) Refugia revisited: individualistic responses of species in space and time. Proc Biol Sci 277:661–671
Tajima F (1989) The effect of change in population size on DNA polymorphism. Genetics 123:597–601
Tarnowska E, Niedziałkowska M, Jędrzejewska B (2019) Genetic structure of bank vole populations in the contact zone of two lineages in north-eastern Poland. Mamm Biol 96:93–101
Tollenaere C, Bryja J, Galan M, Cadet P, Deter J, Chaval Y, Berthier K, Ribas Salvador A, Voutilainen L, Laakkonen J, Henttonen H, Cosson JF, Charbonnel N (2008) Multiple parasites mediate balancing selection at two MHC class II genes in the fossorial water vole: insights from multivariate analyses and population genetics. J Evol Biol 21:1307–1320
Tschirren B, Råberg L, Westerdahl H (2011) Signatures of selection acting on the innate immunity gene Toll-like receptor 2 TLR2 during the evolutionary history of rodents. J Evol Biol 24:1232–1240
Tschirren B, Andersson M, Scherman K, Westerdahl H, Råberg L (2012) Contrasting patterns of diversity and population differentiation at the innate immunity gene toll-like receptor 2 (TLR2) in two sympatric rodent species. Evolution 66:720–731
Tschirren B, Andersson M, Scherman K, Westerdahl H, Mittl PR, Raberg L (2013) Polymorphisms at the innate immune receptor TLR2 are associated with Borrelia infection in a wild rodent population. Proc R Soc Biol Sci 280:20130364
Turner AK, Begon M, Jackson JA, Bradley JE, Paterson S (2011) Genetic diversity in cytokines associated with immune variation and resistance to multiple pathogens in a natural rodent population. PLoS Genet 7:e1002343
Turner AK, Begon M, Jackson JA, Paterson S (2012) Evidence for selection at cytokine loci in a natural population of field voles Microtus agrestis. Mol Ecol 21:1632–1646
Watterson G (1978) The homozygosity test of neutrality. Genetics 88:405–417
Wielstra B, Crnobrnja-Isailović J, Litvinchuk SN, Reijnen BT, Skidmore AK, Sotiropoulos K, Toxopeus AG, Tzankov N, Vukov T, Arntzen JW (2013) Tracing glacial refugia of Triturus newts based on mitochondrial DNA phylogeography and species distribution modeling. Front Zool 10:13
Wlasiuk G, Nachman NW (2010) Adaptation and constraint at Toll-like receptors in primates. Mol Biol Evol 2:2172–2186
Wójcik JM, Kawałko A, Marková S, Searle JB, Kotlík P (2010) Phylogeographic signatures of northward post-glacial colonization from high-latitude refugia: a case study of bank voles using museum specimens. J Zool 281:249–262
Zeng K, Fu Y-X, Shi S, Wu CI (2006) Statistical tests for detecting positive selection by utilizing high-frequency variants. Genetics 174:1431–1439
Zeng K, Shi S, Wu C-I (2007) Compound test for the detection of hitchhiking under positive selection. Mol Biol Evol 24:1898–1908
Acknowledgements
We wish to thank A. Bajer, N. Charbonnel, D. A. Dawson, V. Jovanovic, N. Shchipanov, A. Sandor, S. Sommer, and L. Raberg who kindly provided bank vole samples. We also thank W. Babik who provided access to an Illumina MiSeq platform, to K. Dudek who prepared the Nextera library, and A. Karnkowska who granted access to computational cluster.
Funding
The work was in part supported by Grant no. DEC-2012/07/B/NZ8/00058 from the Polish National Science Centre to A. K. The funding body did not interfere with the design of the study, data collection, analysis, or interpretation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declare that they have no competing interests, neither financial nor non-financial.
Additional information
Handling editor: Allan McDevitt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kloch, A., Biedrzycka, A. Post-glacial phylogeography and variation in innate immunity loci in a sylvatic rodent, bank vole Myodes glareolus. Mamm Biol 100, 141–154 (2020). https://doi.org/10.1007/s42991-020-00016-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42991-020-00016-y