
Abstract
Background
Alanyl-tRNA synthetase 1 (AARS1) gene encodes a ubiquitously expressed class II enzyme that catalyzes the attachment of alanine to the cognate tRNA. AARS1 mutations are frequently responsible for autosomal dominant Charcot-Marie-Tooth disease type 2N (CMT2N).
Objective
To identify pathogenic mutation in the Korean patients with CMT and distal hereditary motor neuronopathy (dHMN).
Methods
We screened AARS1 mutations in 373 unrelated CMT families including 318 axonal CMT, 36 dHMN, and 19 intermediate CMT (Int-CMT) who were negative for 17p12 (PMP22) duplication or deletion using whole exome sequencing and targeted sequencing of CMT-related genes.
Results
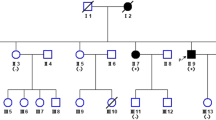
This study identified an early onset Int-CMT family harboring an AARS1 p.Arg329His mutation which was previously reported as pathogenic in French and Australian families. The mutation was located in the highly conserved tRNA binding domain and several in silico analyses suggested pathogenic prediction of the mutations. The patients harboring p.Arg329His showed clinically similar phenotypes of the early onset and electrophysiological intermediate type as those in Australian patients with same mutation. We also found a novel c.2564A>G (p.Gln855Arg) in a CMT2 patient, but its’ pathogenic role was uncertain (variant of uncertain significance).
Conclusion
This study suggests that the frequency of the AARS1 mutations appears to be quite low in Korean CMT. This is the first report of the AARS1 mutation in Korean CMT patients and will be helpful for the exact molecular diagnosis and treatment of Int-CMT patients.



Similar content being viewed by others

References
Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin S-Q, Jordanova A, Kremensky I, Christodoulou K, Middleton LT et al (2003) Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 72:1293–1299
Bansagi B, Antoniadi T, Burton-Jones S, Murphy SM, McHugh J, Alexander M, Wells R, Davies J, Hilton-Jones D, Lochmüller H et al (2015) Genotype/phenotype correlations in AARS-related neuropathy in a cohort of patients from the United Kingdom and Ireland. J Neurol 262:1899–1908
Beebe Mock K, Merriman ME, Schimmel P (2008) Distinct domains of tRNA synthetase recognize the same base pair. Nature 451:90–93
Birouk N, Gouider R, Le Guern E, Gugenheim M, Tardieu S, Maisonobe T, Le Forestier N, Agid Y, Brice A, Bouche P (1997) Charcot-Marie-Tooth disease type 1A with 17p11.2 duplication. Clinical and electrophysiological phenotype study and factors influencing disease severity in 119 cases. Brain 120:813–823
Chung KW, Suh BC, Shy ME, Cho SY, Yoo JH, Park SW, Moon H, Park KD, Choi KG, Kim S et al (2008) Different clinical and magnetic resonance imaging features between Charcot-Marie-Tooth disease type 1A and 2A. Neuromuscul Disord 18:610–618
Edvardson S, Cinnamon Y, Jalas C, Shaag A, Maayan C, Axelrod FB, Elpeleg O (2012) Hereditary sensory autonomic neuropathy caused by a mutation in dystonin. Ann Neurol 71:569–572
Gallardo E, Garcia A, Combarros O, Berciano J (2006) Charcot-Marie-Tooth disease type 1A duplication: spectrum of clinical and magnetic resonance imaging features in leg and foot muscles. Brain 129:426–437
Gonzalez M, McLaughlin H, Houlden H, Guo M, Yo-Tsen L, Hadjivassilious M, Speziani F, Yang X-L, Antonellis A, Reilly MM et al (2013) Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J Neurol Neurosurg Psychiat 84:1247–1249
Imaizumi N, Takeuchi Y, Hirano H, Torii T, Seki Y, Morimoto T, Miyamoto Y, Sakagami H, Yamauchi J (2019) Data on the effects of Charcot-Marie-Tooth disease type 2 N-associated AARS missense mutation (Arg329-to-His) on the cell biological properties. Data Brief 25:104029
Jordanova A, Irobi J, Thomas FP, Van Dijck P, Meerschaert K, Dewil M, Dierick I, Jacobs A, De Vriendt E, Guergueltcheva V et al (2006) Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet 38:197–202
Latour P, Thauvin-Robinet C, Baudelet-Mery C, Soichot P, Cusin V, Faivre L, Locatelli M-C, Mayencon M, Sarcey A, Broussolle E et al (2010) A major determinant for binding and aminoacylation of tRNA-Ala in cytoplasmic alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth Disease. Am J Hum Genet 86:77–82
Lin K-P, Soong B-W, Yang C-C, Huang L-W, Chang M-H, Lee I-H, Antonellis A, Lee Y-C (2011) The mutational spectrum in a cohort of Charcot-Marie-Tooth disease type 2 among the Han Chinese in Taiwan. PLoS One 6:e29393
Magy L, Mathis S, Le Masson G, Goizet C, Tazir M, Vallat JM (2018) Updating the classification of inherited neuropathies: results of an international survey. Neurology 90:e870–e876
McLaughlin HM, Sakaguchi R, Liu C, Igarashi T, Pehlivan D, Chu K, Iyer R, Cruz P, Cherukuri PF, Hansen NF et al (2010) Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet 87:560–566
McLaughlin HM, Sakaguchi R, Giblin W, NISC Comparative Sequencing Program, Wilson TE, Biesecker L, Lupski JR, Talbot K, Vance JM, Zuchner S et al (2012) A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2 N (CMT2N). Hum Mutat 33:244–253
Motley WW, Griffin LB, Mademan I, Baets J, De Vriendt E, De Jonghe P, Antonellis A, Jordanova A, Scherer SS (2015) A novel AARS mutation in a family with dominant myeloneuropathy. Neurology 84:2040–2047
Murphy SM, Herrmann DN, McDermott MP, Scherer SS, Shy ME, Reilly MM, Pareyson D (2011) Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J Peripher Nerv Syst 16:191–198
Nam SH, Hong YB, Hyun YS, Nam DE, Kwak G, Hwang SH, Choi BO, Chung KW (2016) Identification of genetic causes of inherited peripheral neuropathies by targeted gene panel sequencing. Mol Cells 39:382–388
Nam DE, Yoo DH, Choi SS, Choi BO, Chung KW (2018) Wide phenotypic spectrum in axonal Charcot-Marie-Tooth neuropathy type 2 patients with KIF5A mutations. Genes Genomics 40:77–84
Rossor AM, Carr AS, Devine H, Chandrashekar H, Pelayo-Negro AL, Pareyson D, Shy ME, Scherer SS, Reilly MM (2017) Peripheral neuropathy in complex inherited diseases: an approach to diagnosis. J Neurol Neurosurg Psychiatry 88:846–863
Schimmel P, Ripmaster T (1995) Modular design of components of the operational RNA code for alanine in evolution. Trends Biochem Sci 20:333–334
Simons C, Griffin LB, Helman G, Golas G, Pizzino A, Bloom M, Murphy JL, Crawford J, Evans SH, Topper S et al (2015) Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am J Hum Genet 96:675–681
Swairjo MA, Otero FJ, Yang XL, Lovato MA, Skene RJ, McRee DE, Ribas de Pouplana L, Schimmel P (2004) Alanyl-tRNA synthetase crystal structure and design for acceptor-stem recognition. Mol Cell 13:829–841
Tsai P-C, Soong B-W, Mademan I, Huang Y-H, Liu C-R, Hsiao C-T, Wu H-T, Liu T-T, Liu Y-T, Tseng Y-T et al (2017) A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy. Brain 140:1252–1266
Vester A, Velez-Ruiz G, McLaughlin HM, NISC Comparative Sequencing Program, Lupski JR, Talbot K, Vance JM, Zuchner S, Roda RH, Fischbeck KH et al (2013) A loss-of-function variant in the human histidyl-tRNA synthetase (HARS) gene is neurotoxic in vivo. Hum Mutat 34:191–199
Weedon MN, Hastings R, Caswell R, Xie W, Paszkiewicz K, Antoniadi T, Williams M, King C, Greenhalgh L, Newbury-Ecob R et al (2011) sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 89:308–312
Weterman MAJ, Kuo M, Kenter SB, Gordillo S, Karjosukarso DW, Takase R, Bronk M, Oprescu S, van Ruissen F, Witteveen RJW et al (2018) Hypermorphic and hypomorphic AARS alleles in patients with CMT2N expand clinical and molecular heterogeneities. Hum Mol Genet 27:4036–4050
Zhao Z, Hashiguchi A, Hu J, Sakiyama Y, Okamoto Y, Tokunaga S, Zhu L, Shen H, Takashima H (2012) Alanyl-tRNA synthetase mutation in a family with dominant distal hereditary motor neuropathy. Neurology 78:1644–1649
Acknowledgements
We thank the patients and their families for consenting to participation and sample donation for this study. This work was supported by Grants from the National Research Foundation (2016R1A5A2007009, 2018R1A4A1024506 and 2019R1A2C1087547) and the Korean Health Technology R&D Project, Ministry of Health and Welfare (HI14C3484 and HI16C0426), Republic of Korea.
Author information
Authors and Affiliations
Contributions
BOC and KWC were major contributors to the experimental design. AJL and DEN performed molecular genetic works. BOC and SHN collected patient samples. BOC and KWC were involved in writing the manuscript and data analysis and interpretation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
AJL, DEN, YJC, SHN, BOC, and KWC declare that they have no conflict of interest.
Ethics approval and consent to participate
All participants provided written informed consent, and the study protocol was approved by the Institutional Review Boards for Sungkyunkwan University School of Medicine, Samsung Medical Center (SMC, 2014-08-057-002), and Kongju National University (KNU-IRB-2018-06).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lee, A.J., Nam, D.E., Choi, Y.J. et al. Alanyl-tRNA synthetase 1 (AARS1) gene mutation in a family with intermediate Charcot-Marie-Tooth neuropathy. Genes Genom 42, 663–672 (2020). https://doi.org/10.1007/s13258-020-00933-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-020-00933-9