
Abstract
Raine Syndrome (RS) is caused by biallelic loss-of-function mutations in FAM20C gene and characterized by hypophosphatemia, typical facial and skeletal features. Subperiosteal bone formation and generalized osteosclerosis are the most common radiological findings. Here we present a new case with RS. A 9-month-old male patient on a home-type ventilator was referred for hypophosphatemia. He was born with a weight of 3800 g to non-consanguineous parents. Prenatal ultrasound had demonstrated nasal bone agenesis. A large anterior fontanel, frontal bossing, exophthalmos, hypoplastic nose, high arched palate, low set ears, triangular mouth, and corneal opacification were detected on physical examination. Serial skeletal X-rays revealed diffuse osteosclerosis at birth which was gradually decreased by the age of 5 months with subperiosteal undermineralized bone formation and medullary space of long bone could be distinguishable with bone-within-a-bone appearance. At 9 months of age, hand X-ray revealed cupping of the ulna with loose radial bone margin with minimal fraying and osteopenia. Cranial computed tomography scan showed bilateral periventricular calcification and hydrocephalus in progress. The clinical, laboratory, and radiological examinations were consistent with RS. Molecular analyses revealed a compound heterozygous mutation in FAM20C gene (a known pathogenic mutation, c.1645C > T, p.Arg549Trp; and a novel c.863 + 5 G > C variant). The patient died due to respiratory failure at 17 months of age. This case allowed us to demonstrate natural progression of skeletal features in RS. Furthermore, we have described a novel FAM20C variant causing RS. Previous literature on RS is also reviewed.
Similar content being viewed by others


Introduction
Raine syndrome (RS) (OMIM #259775) is a rare osteosclerotic bone dysplasia which was first described in 1989 [1]. Generalized osteosclerosis and distinctive craniofacial anomalies including hypoplastic nose, midface hypoplasia, exophthalmos, and intracranial calcification are the characteristic features of the disease [2]. Sensorineural hearing loss, developmental delay, epilepsy, large fontanelle, and amelogenesis imperfecta were also reported in RS. Clinical heterogeneity from severe lethal phenotype to mild dysmorphic features with hypophosphatemia in older ages has been delineated [3,4,5]. In the lethal forms of the disease, majority of the patients die in the newborn/infancy period due to pulmonary hypoplasia or choanal atresia [3].
Biallelic loss-of-function mutations of FAM20C (Family with sequence similarity 20, member C) gene (OMIM#611061) were associated with the RS [2]. To date, around 50 individuals with RS have been reported with approximately 31 different mutations in FAM20C gene [HGMD-23 missense/nonsense, 5 splicing, 1 gross deletion, 1 small insertion, 1 complex rearrangement]. FAM20C (also known as DMP4) is located on chromosome 7p22.3 and encodes a protein kinase containing five functional regions [6]. FAM20C is localized within the Golgi apparatus and phosphorylates the S-x-E (Serine-x-Glutamic acid) motif which is found on a multiple secreted phosphoproteins, including small integrin-binding ligand N-linked glycoproteins (SIBLINGs) and fibroblast growth factor 23 (FGF23) [7, 8]. FAM20C has a crucial role in skeletal development and highly conserved among mammals and specifically expressed in the mineralized tissues [9,10,11]. SIBLINGs comprise dentin matrix protein 1 (DMP1), dentin sialophosphoprotein (DSPP), osteopontin (OPN), matrix extracellular phosphoglycoprotein (MEPE), and bone sialoprotein (BSP). The contribution of these phosphorylated secretory proteins to bone mineralization is complicated with both promoting and inhibiting roles to regulate proper hydroxyapatite crystal formation [12, 13]. Abnormal phosphorylation of these proteins leads to impairment of their function [7, 14]. Especially inactivation of OPN and MEPE is important, since knockout mouse models of these two genes are characterized by osteosclerosis [12, 15]. Therefore, impaired phosphorylation of OPN and MEPE could be the main reason of osteosclerosis in RS [16].
Recently, FAM20C knockout mice studies demonstrated the important role of FAM20C in the differentiation of osteoblasts/osteocytes and regulation of phosphate homeostasis via FGF23 [10]. FAM20C promotes FGF23 cleavage at the RXXR site by phosphorylating at Ser180 residue, which in turn reduces O-glycosylation at Thr178, and that mutant FAM20C with decreased kinase activity impairs FGF23 degradation and leads to elevated intact FGF23 leading to hypophosphatemia [17, 18].
Herein, we report a patient with RS having a novel variant in FAM20C gene which enabled us to observe changes in the phenotype and characteristics of the disease owing to a relatively longer term survival in spite of severe RS.
Case Report
A 9-month-old male was referred to our pediatric endocrinology clinic for evaluation of hypophosphatemia. He was the first born baby to unrelated Turkish parents. Family history was unremarkable with no effected family members. Mother was 22 years old and had an uncomplicated pregnancy; however, hypoplastic nasal bone and microcephaly of the baby had been detected on antenatal ultrasonography (USG). Neither amniocentesis nor further investigation for the dysmorphic findings were performed prenatally. The proband was born at term by normal vaginal delivery with a birth weight of 3.800 g (+ 1.5 SDS). Microcephaly, proptosis, and hypoplastic nasal bone with midface hypoplasia were noticed on examination. Intubation and transfer to neonatal intensive care unit (NICU) were required due to respiratory distress. He received mechanical ventilation and intensive medical support. Clavicular fractures including one healed fracture had been detected on X-ray taken at the first day of life. Hypocalcemia (5.4 mg/dl; N: 9–11) with elevated PTH (430 pg/m; N: 15–65) and low 25-OH Vit D (10.9 ug/L; N: 30–100) levels had been detected on 2nd day of life during routine screening for the fractures and treated with intravenous Ca and vitamin D supplementation. No other hypocalcemic event had been detected during the follow-up. Tracheostomy operation and gastrostomy tube insertion were performed at the postnatal 48th day and at 5.5 months of age, respectively. Echocardiography revealed mild insufficiency of mitral and tricuspid valves and pulmonary hypertension. Patient could be discharged to home with home ventilation at the age of 8 months. Although his cranial MRI was normal at birth, hydrocephaly was detected at 5 months of age (Suppl. Fig.). Ventriculoperitoneal shunt operation was performed at 9 months of age.
The patient was consulted to pediatric endocrinology at 9 months of age for hypophosphatemia. The patient’s height, weight, and head circumference were at − 1.1, − 2.1, and − 0.6 SDS, respectively. Midface hypoplasia, proptosis with corneal opacification, down slanting palpebral fissures, depression of nasal bridge, short hypoplastic nose, tented mouth, high arched palate, gum hypertrophy, protruding tongue, and micrognathia were detected. A cloverleaf skull with prominent forehead, bitemporal narrowing, open cranial sutures, and widely open anterior fontanelle (5 × 6 cm) with short neck were noticed. The patient had mild narrowing of the chest and was on home ventilation through the tracheostomy. Ophthalmological examination revealed optic atrophy and corneal opacification. Severe delay in developmental milestones was observed.
On biochemical evaluation, serum calcium was normal (9.6 mg/dl; N: 9–11); however, phosphorus was low (2.0 mg/dl; N: 4–6.5) and ALP was elevated (950 U/L; N: 116–450) with slightly increased PTH level (84.5 ng/L; N:15–65). 25-OH vitamin D was within normal range (28 ug/L; N: 30–100) and 1,25-dihydroxy vitamin D (1,25(OH)2D) concentration was mildly elevated (107 pg/ml; N: 24–86). Renal function tests and tubular reabsorption of phosphate (TRP) were normal (94.0% N: 85–100), but TmP/GFR was low (2.3 mg/dl, N: 4.8–8.0). Reviewing the previous hospital records showed that he had hypophosphatemia at 2nd day of life with hypocalcemia and persistent hypophosphatemia with normal calcium levels since 5 months of life (no phosphate level measured between 2nd day and 5 months of life), but these were not further investigated. Biochemical characteristics of the patient at admission and during follow-up are given in Suppl. Table 1.
Skeletal X-rays at 1st day of life and during the newborn period revealed diffuse osteosclerosis, fractures in clavicle, and pseudofractures at vertebro-costal junctions (Fig. 1). Osteosclerosis gradually reduced by 5 months of age and medullary space of long bones could be distinguishable with bone-within-a-bone appearance. First periosteal undermineralized bone formation leading to bone-within-a-bone appearance has been detectable at 4 months of age. Additionally, the patient had suffered bilateral scapula fractures at the age of 1 month (Fig. 1).

X-rays of the patient showing diffuse osteosclerosis and clavicular fractures at first day of life (a), bilateral scapular fracture at 4th week of life (b), and undermineralization and periosteal new bone formation first started to appear at 4th month of life (c), become obvious at 5th month of life, leading diffuse bone-within-a-bone appearance (d, e)
At the age of 9 months, cupping of ulna with loose radial bone margin with minimal fraying and osteopenia, and hypoplastic distal phalanges were observed in the hand X-ray (Fig. 2). Lateral radiograph of vertebrate showed sclerotic central core with relatively undermineralized periphery of vertebral bodies. The patient was started on phosphate (30 mg/kg/day) and calcitriol (25 µg/day) treatment at 9 months of age due to hypophosphatemia, low TmP/GFR, and rickets findings on radiography. After six months of phosphate and calcitriol treatment, ulnar cupping improved, but bone-within-a-bone appearance and undermineralized periosteal bone did not improve significantly.

X-rays showing cupping of ulna with loose radial bone margin with minimal fraying and osteopenia at the age of 9 months (a1) and improvement after 6 months of treatment with oral phosphate and calcitriol (a2). Lateral radiograph of vertebrate showed sclerotic central core with relatively undermineralized periphery of vertebral body before (b1) and after treatment (b2). The last radiographs of the patient at 15 months of age showing sclerotic broad ribs and undermineralized periosteal bones on sclerotic primary silhouette (arrows) creating bone-within-a-bone appearance (c and d)
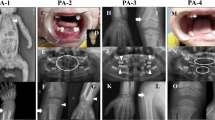
Cranial MRI and CT showed thickening of cranial bones, pyriform aperture stenosis/agenesis, and small posterior fossa with deformed bone (Suppl. Fig.). Bilateral periventricular calcification with cerebral atrophy, hypoplasia of corpus callosum, and triventricular hydrocephaly before the shunt operation were also observed (Suppl. Fig.). Three-dimensional computed tomography (CT) scan showed enlarged anterior fontanel and evidence of craniosynostosis (Fig. 3). Orbital magnetic resonance imaging (MRI) and CT revealed optic nerve and chiasma atrophy, microphthalmia of the left eye with hyperdensity in vitreous body related to intraocular hemorrhage, and shallow orbital walls.

Skull X-rays showed cloverleaf skull with sclerotic cranial bones, large bulging anterior fontanelle, hypoplastic nasal bone (a). The three-dimensional (3D) CT of the patient (b) in comparison with the normal 3D CT in a similar aged child (c) showed enlarged bulging anterior fontanel, absent cranial sutures indicating craniosynostosis, shallow orbital fossa, prominent frontal bone, underdeveloped midface with agenetic nasal bones, and hypoplastic nasal aperture
Chest X-rays and high resolution CT findings were consistent with interstitial lung diseases with normal large airways and basal atelectasis, areas of ground-glass attenuation and consolidation, suggesting aspiration. Additionally, lateral thickening of the ribs and the chest asymmetry had been detected.
Renal USG revealed nephrocalcinosis at 1st and 30th day of life; however, at 11 month of age, it was resolved.
During endocrinological follow-up, bone markers were evaluated before treatment. C-telopeptide (CTX) and Procollagen I, N-terminal peptide (P1NP) levels were normal for his age [1.27 µg/l (N: 0.54–1.71) and 820 µg/l (N: 400–1500), respectively]. Deoxypyridinoline (DPD) in urine was > 42 nM DPD/mM creatinine (N: 13.4–40)). Although the compliance to treatment was good, biochemical findings were not significantly improved during the follow-up. Oral phosphate (up to 45 mg/kg/day) and calcitriol (up to 1 µg/day) were increased but PTH elevation persisted. Nevertheless, radiological findings of rickets were improved with some decrease in ALP (Fig. 2, Suppl. Table 1).
The clinical, laboratory, and radiological examinations were consistent with RS in our patient, who died at 17 months of age due to respiratory failure.
Molecular Analyses
Patient’s DNA isolated from peripheral blood using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) in accordance with manufacturer’s instructions. After DNA isolation, all exons and exon–intron boundaries of FAM20C gene were amplified with PCR and sequenced at IlluminaMiseq platform. PCR conditions and primers will be provided upon request. Data were analyzed with Sophia-DDM-v4 software. Heterozygous c.1645C > T (p.Arg549Trp) missense mutation and novel heterozygous c.863 + 5G > C intronic variation were detected (Fig. 4). First mutation is a known to be pathogenic according to ClinVar and Human Genome Mutation Database (HGMD). Second mutation is not present at these databases and GnomAD, 1000 Genome, and ESP5400 databases. According to in silico analysis with Mutation Taster and Human Splicing Finder; c.863 + 5G > C variation is predicted to affect splicing. Since there were no patient derived cells or total RNA material, we could not further evaluate the effect of this novel mutation on expression of FAM20C. After considering of all this bioinformatic processes and our patient’s phenotype, this variation accepted as a pathogenic mutation. Segregation analysis revealed that his father had heterozygous c.1645C > T (p.Arg549Trp) and his mother had heterozygous c.863 + 5G > C mutation and the patient was only child of the family.

Scheme of FAM20C gene showing described mutations and the current patient’s heterozygous c.1645C > T (p.Arg549Trp) missense and novel heterozygous c.863 + 5G > C intronic mutations. Mutations given with underlined letters show lethal genotypes
Discussion
Raine Syndrome is a rare skeletal disorder with variable clinical heterogeneity from lethal to non-lethal and isolated teeth/bone phenotypes [1,2,3,4,5, 19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40]. It is characterized by typical facial features and osteosclerosis. Molecular testing of FAM20C gene is confirmatory for the diagnosis when accompanied by typical phenotypic and radiological findings. Here we report a RS case with hypophosphatemia having compound heterozygous mutation in FAM20C gene including one novel variant. Although our case had severe/lethal phenotype of the disease, he had survived for 17 months. Therefore, we could have chance to evaluate radiological and clinical evolution of the disease, which will contribute to our understanding of the functional loss of FAM20C protein in humans.
Previously reported genetically documented RS cases with their clinico-radiological findings and biochemical parameters of bone metabolism are presented in supplementary table 2 [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40]. So far, nearly 20 cases with non-lethal RS have been described. The majority of the patients who have lethal form of the disease die in the newborn/infancy period due to respiratory failure [3]. Lung size is one of the important factor that may predict survival; however, choanal atresia/stenosis also contributes to mortality.
Genotype phenotype correlation had been suggested for lethality in RS, since, mutations in lethal and non-lethal phenotypes affect different domains of the protein. FAM20C mutations located in the signal sequence (SS) domain can affect the secretion of protein because of improper protein stabilization/folding, while the mutations effecting C-terminal domain (CCD) may cause loss of function of the kinase activity [3, 4, 14, 16]. The majority of the lethal mutations are located in the conserved CCD [2, 3]. The c.1645C > T (p.Arg549Trp) mutation detected in our patients was localized in the CCD of the protein and reported in a lethal RS case in homozygous state [2, 29]. The maternally inherited novel c.863 + 5G > C variant is located in the intronic region of the gene, which might have a deleterious effect on splicing for the proper formation of FAM20C protein as analyzed with in silico tools. Our patient had severe/lethal genotype; however, he had survived up to the 17 months of age due to prenatal detection of hypoplastic nasal bone and early intervention with intensive care settings, which enabled him to survive through most critical neonatal period. However, he had severe neurological delay and never gained any developmental milestones and stayed in a vegetative state. Genotypes and clinical features of previous cases with RS have been summarized including the present case in Fig. 4 and supplementary Table 2.
Typical facial features, clinical, laboratory, and radiological findings were typical for the RS in the present case. Midface cranial deformities lead to major dysmorphic features of the disease and tightening supraorbital rim decreases the orbital volume causing exophthalmos, as in our case. Furthermore, our patient had optic atrophy with hypoplastic optic nerve, periventricular calcifications, corpus callosum hypoplasia, and hydrocephalus requiring shunt operation at nine months of age. Intracranial calcifications are commonly reported in RS [3]; however, hydrocephalus [27, 28, 33], optic atrophy [19], and corpus callosum dysgenesis [37] are reported rarely. Here we showed that hydrocephalus was not congenital and triventricular, suggesting that underdeveloped posterior fossa led to obstruction of CSF circulation with age. Similar to our patient, previously reported cases with hydrocephalus required shunt operation at 6 to 8 months of age [27, 28, 33]. These cases were also most severely affected non-lethal RS cases with severe mental and motor delay [27, 28] or lethal cases live long enough to develop hydrocephalus [33]. Thus, it must be kept in mind that patients with RS can develop hydrocephalus with time and cranial imaging is required in case of neurological deterioration.
Generalized osteosclerosis is the most common radiological finding of RS. In our case, serial skeletal X-rays revealed diffuse osteosclerosis at birth, which was gradually reduced by the age of 5 months. Medullary space of long bones became distinguishable with “bone-within-a bone” appearance (Figs. 2, 3). This type of bone changes has not been reported previously in RS. Although we did not have any histopathological tissue analysis, we hypothesize that this appearance might develop as a result of hypomineralized periosteal bone formation on a sclerotic background bone during the course of the disease. Reducing osteosclerosis and visible medullary space occurred at the same period. We think that undermineralized periosteal bone formation or decreasing osteosclerosis could be related to the hypophosphatemia and related osteomalacia. In case of inherited hypophosphatemia, hypophosphatemia and rickets develop after birth following cessation of transplacental phosphate transport, which is also evident in a few RS cases studied shortly after birth [31]. In line with that, undermineralization, but not osteosclerosis, had been detected in conditional Fam20c knockout mice in older ages [10, 41]. However, all reported cases with generalized osteosclerosis in RS were detected at birth or shortly after birth. Only a few cases could be studied for long-term radiological features in milder form (non-lethal) of the disease. Fradin et al. [20] reported generalized osteosclerosis at birth which decreased with age, and the patient had normal appearing bones with distinct corticomedullary differentiation and normal cortical thickness at 3 years of age. Additionally, RS cases presenting in older ages had preserved corticomedullary junction or only focal sclerosis and mildly increased bone mineral density [5, 20, 24, 28, 37, 39]. Therefore, it seems that osteosclerosis and especially periosteal bone formation is related to the severity of the RS. Most severe form with early postnatal lethality showed extensive sclerotic periosteal bone formation which could be detected prenatally [2, 29, 31]. However, cases with non-lethal RS have osteosclerosis at birth which decreased with age and remained as focal sclerosis or mildly elevated bone mineral density. No case similar to that presented here had been reported with evolution of radiological findings with severe form of the disease in the literature.
Biallelic loss-of-function mutations in FAM20C gene cause FGF23-related hypophosphatemia mainly by reduction of the phosphorylation of FGF23 protein and probably partially by reduced transcription of DMP1 [10,11,12, 26]. Overexpression of DMP1 fails to rescue bone phenotype and hypophosphatemia in Fam20c knockout mice suggesting that DMP1 is not the major causative factor [18]. Although this mechanism is clearly known today, biochemical data of previous RS cases have not always been reported (Suppl. Table 2) [2, 30, 31, 36,37,38]. FGF23 levels could be studied in a few cases and shown to be elevated [4, 5, 28, 39], but elevated urinary phosphate excretion had been shown in almost all cases including the case herein [24, 27, 28, 34, 39]. However, serum phosphate levels were found to be normal in some of the patients [4, 20, 32,33,34,35]. Additionally, the development of hypophosphatemia in cases with normal phosphate levels is also detected in later ages, in puberty and in adulthood [34]. Therefore, in case of normal phosphate levels, follow-up for the development of hypophosphatemia is required. There are only a few data about the effect of treatment with alfacalcidol/calcitriol and oral phosphorus in RS. In this report, we could not normalize PTH and phosphate levels in our patient in spite of dose increments, but phosphate doses could not be increased further due to elevated PTH levels. However, some improvement was seen in ALP and rickets findings. Mild hyperparathyroidism can be seen at initial presentation due to inhibitory effect of FGF23 on 11,25(OH)2D, leading to poor calcium absorption from gastrointestinal tract in hypophosphatemic rickets. After the initiation of phosphorus, hyperparathyroidism is mostly due to higher phosphate doses relative to calcitriol doses. However, in our patient with RS we could not normalize PTH in spite of calcitriol dose increment. Poor treatment response similar to our case was also detected in two non-lethal but severe RS [27, 28]. These observations suggest that in severe RS cases, much higher than regular doses of calcitriol could be required. Alternatively, novel treatment options such as anti-FGF23 monoclonal antibody could be tried. However, in milder form of RS, spontaneous improvement of hypophosphatemia and/or no rachitic changes have been observed in older patients [4].
Hypocalcemia has also been reported in lethal cases shortly after birth in RS [31, 40]. Our patient had hypocalcemia at the second day of life with elevated PTH and low 25-OH Vit D levels. Similarly, hypocalcemia with elevated PTH and low 25-OH Vit D has been detected in one non-lethal but severe RS case at 2 months of age and treated with vitamin D and calcium, but continued to have hypophosphatemia and rickets [28]. Thus, it seems that patients with RS are prone to hypocalcemia in early neonatal period most probably related to the abrupt cessation of transplacental calcium transport, especially in vitamin D deficiency settings. On the other hand, either increasing Ca influx to the bone or decreased bone resorption might be operative in osteosclerotic conditions and contribute to the development of hypocalcemia. Hypocalcemia should be checked at early postnatal hours in these cases even in the absence of symptoms as in our case.
Perivascular, white matter, and basal ganglia calcifications are common and reported in 95% of RS cases (Suppl. Table 2). Nephrocalcinosis is not examined in most of the cases; however, 6 cases with nephrocalcinosis including the case reported herein were identified. Ectopic soft tissue calcifications and calcification in different organ systems such as ovaries, liver, and spleen have been also reported in RS [20, 21, 25]. It seems that intracranial calcification is a consistent feature of the disease and independent from serum calcium and phosphate, as it has been described at birth and/or antenatal USG [2, 31, 38]. Furthermore, nephrocalcinosis also appears to develop independently from calcium and phosphate status since it was present at the first day of life in our case and has also been described in a case with normal serum calcium and phosphate levels [20]. Although mechanism of nephrocalcinosis, intracranial and soft tissue calcification in RS is not known, we hypothesize that FAM20C deficiency leading to impaired phosphorylation of OPN and BSP might have a role in calcifications (15), since OPN and BSP are localized to matrix vesicle and regulate extracellular matrix calcification. Additionally, it has been shown that SIBLING derived protease-resistant phosphorylated peptide (ASARM peptide) is also phosphorylated by FAM20C and non-phosphorylated ASARM peptides enhance mineralization while phosphorylated ones inhibit mineralization [42, 43]. Thus, phosphorylation defects related to the FAM20C deficiency of SIBLING or its degradation product (ASARM peptide) might cause dysregulated mineralization in bone and soft tissues in RS.
In conclusion, we described a novel variant in FAM20C gene and evolution of skeletal features in a new case with lethal RS. RS should be recognized among the rare causes of FGF23-related hypophosphatemia and sclerotic bone dysplasia. Changing morbidities associated with progression of biochemical and radiologic characteristics in RS require close monitoring.
References
Raine J, Winter RM, Davey A, Tucker SM (1989) Unknown syndrome: microcephaly, hypoplastic nose, exophthalmos, gum hyperplasia, cleft palate, low set ears, and osteosclerosis. J Med Genet 26:786–788
Simpson MA, Hsu R, Keir LS, Hao J, Sivapalan G, Ernst LM et al (2007) Mutations in FAM20C are associated with lethal osteosclerotic bone dysplasia (Raine syndrome), highlighting a crucial molecule in bone development. Am J Hum Genet 81:906–912
Faundes V, Castillo-Taucher S, Gonzalez-Hormazabal P, Chandler K, Crosby A, Chioza B (2014) Raine syndrome: an overview. Eur J Med Genet 57:536–542
Rafaelsen SH, Raeder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S et al (2013) Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res 28:1378–1385
Takeyari S, Yamamoto T, Kinoshita Y, Fukumoto S, Glorieux FH, Michigami T et al (2014) Hypophosphatemic osteomalacia and bone sclerosis caused by a novel homozygous mutation of the FAM20C gene in an elderly man with a mild variant of Raine syndrome. Bone 67:56–62
Zhang H, Zhu Q, Cui J, Wang Y, Chen MJ, Guo X et al (2018) Structure and evolution of the Fam20 kinases. Nat Commun 9(1):1218
Ishikawa HO, Xu A, Ogura E, Manning G, Irvine KD (2012) The Raine syndrome protein FAM20C is a golgi kinase that phosphorylates bio-mineralization proteins. PLoS ONE 7:e42988
Tagliabracci VS, Wiley SE, Guo X, Kinch LN, Durrant E, Wen J et al (2015) A single kinase generates the majority of the secreted phosphoproteome. Cell 161:1619–1632
Wang X, Hao J, Xie Y, Sun Y, Hernandez B, Yamoah AK et al (2010) Expression of FAM20C in the osteogenesis and odontogenesis of mouse. J Histochem Cytochem 58:957–967
Wang X, Wang S, Li C, Gao T, Liu Y, Rangiani A et al (2012) Inactivation of a novel FGF23 regulator, FAM20C, leads to hypophosphatemic rickets in mice. PLoS Genet 8:e1002708
Kawasaki K, Weiss KM (2003) Mineralized tissue and vertebrate evolution: the secretory calcium-binding phosphoprotein gene cluster. Proc Natl Acad Sci USA 100:4060–4065
Steitz SA, Speer MY, McKee MD, Liaw L, Almeida M, Yang H et al (2002) Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am J Pathol 161:2035–2046
Narayanan K, Srinivas R, Ramachandran A, Hao J, Quinn B, George A (2001) Differentiation of embryonic mesenchymal cells to odontoblast-like cells by overexpression of dentin matrix protein 1. Proc Natl Acad Sci USA 98:4516–4521
Tagliabracci VS, Engle JL, Wen J, Wiley SE, Worby CA, Kinch LN et al (2012) Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science 336:1150–1153
Gowen LC, Petersen DN, Mansolf AL, Qi H, Stock JL, Tkalcevic GT et al (2003) Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J Biol Chem 278:1998–2007
Kinoshita Y, Hori M, Taguchi M, Fukumoto S (2014) Functional analysis of mutant FAM20C in Raine syndrome with FGF23-related hypophosphatemia. Bone 67:145–151
Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H et al (2014) Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci USA 111:5520–5525
Wang X, Wang J, Yuan B, Lu Y, Feng JQ, Qin C (2014) Overexpression of Dmp1 fails to rescue the bone and dentin defects, in Fam20C knockout mice. Connect Tissue Res 55:1–16
Shalev SA, Shalev E, Reich D, Borochowitz ZU (1999) Osteosclerosis, hypoplastic nose, and proptosis (Raine syndrome): further delineation. Am J Med Genet. 86(3):274–277
Fradin M, Stoetzel C, Muller J, Koob M, Christmann D, Debry C et al (2011) Osteosclerotic bone dysplasia in siblings with a Fam20C mutation. Clin Genet 80:177–183
Al Mane KA, Coates RK, McDonald P (1996) Intracranial calcification in Raine syndrome. Pediatr Radiol 26:55–58
Vogel P, Hansen GM, Read RW, Vance RB, Thiel M, Liu J et al (2012) Amelogenesis imperfecta and other biomineralization defects in Fam20a and Fam20c null mice. Vet Pathol 49:998–1017
Kan AE, Kozlowski K (1992) New distinct lethal osteosclerotic bone dysplasia (Raine syndrome). Am J Med Genet 43(5):860–864
Al-Mane K, Al-Dayel F, McDonald P (1998) Intracranial calcification in Raine syndrome: radiological pathological correlation. Pediatr Radiol 28(11):820–823
Mamedova E, Dimitrova D, Przhiyalkovskaya E, Buryakina S, Vasilyev E, Tiulpakov A et al (2019) Non-lethal Raine syndrome in a middle-aged woman caused by a novel FAM20C mutation. Calcif Tissue Int 105(5):567–572
Oya K, Ishida K, Nishida T, Sato S, Kishino M, Hirose K et al (2017) Immunohistochemical analysis of dentin matrix protein 1 (Dmp1) phosphorylation by Fam20C in bone: implications for the induction of biomineralization. Histochem Cell Biol 147:341–351
Simpson M, Scheuerle A, Hurst J, Patton M, Stewart H, Crosby A (2009) Mutations in FAM20C also identified in non-lethal osteosclerotic bone dysplasia. Clin Genet 75:271–276
Mahmood N, Donne A, Weber A, Dharmaraj P (2014) Raine syndrome: a review and a report of metabolic bone disease as a new link. Research. 10.13070/rs.en.1.890.
Kingston HM, Freeman JS, Hall CM (1991) A new lethal sclerosing bone dysplasia. Skelet Radiol 20:117–119
Al-Gazali LI, Jehier K, Nazih B, Abtin F, Haas D, Sadagahatian R (2003) Further delineation of Raine syndrome. Clin Dysmorphol 12:89–93
Hulskamp G, Wieczorek D, Rieder H, Louwen F, Hörnig-Franz I, Rickert CH et al (2003) Raine syndrome: report of a family with three affected sibs and further delineation of the syndrome. Clin Dysmorphol 12:153–160
Kochar GS, Choudhary A, Gadodia A, Gupta N, Simpson MA, Crosby AH et al (2010) Raine syndrome: a clinical, radiographic and genetic investigation of a case from the Indian subcontinent. Clin Dysmorphol 19:153–156
Ababneh FK, Alswaid A, Youssef T, Al Azzawi M, Crosby A, Albalwi MA (2013) Hereditary deletion of the entire FAM20C gene in a patient with Raine syndrome. Am J Med Genet A. 161(12):3155–3160
Acevedo AC, Poulter JA, Alves PG, de Lima CL, Castro LC, Yamaguti PM et al (2015) Variability of systemic and oro-dental phenotype in two families with non-lethal Raine syndrome with FAM20C mutations. BMC Med Genet 16:8
Seidahmed MZ, Alazami AM, Abdelbasit OB, Al Hussein K, Miqdad AM, Abu-Sa’da O et al (2015) Report of a case of Raine syndrome and literature review. Am J Med Genet A. 167A(10):2394–2398
Elalaoui SC, Al-Sheqaih N, Ratbi I, Urquhart JE, O'Sullivan J, Bhaskar S et al (2016) Non-lethal Raine syndrome and differential diagnosis. Eur J Med Genet 59:577–583
Sheth J, Bhavsar R, Gandhi A, Sheth F, Pancholi D (2018) A case of Raine syndrome presenting with facial dysmorphy and review of literature. BMC Med Genet 19(1):76
Tamai K, Tada K, Takeuchi A, Nakamura M, Marunaka H, Washio Y et al (2018) Fetal ultrasonographic findings including cerebral hyperechogenicity in a patient with non-lethal form of Raine syndrome. Am J Med Genet A 176(3):682–686
Rolvien T, Kornak U, Schinke T, Amling M, Oheim R (2019) A novel FAM20C mutation causing hypophosphatemic osteomalacia with osteosclerosis (mild Raine syndrome) in an elderly man with spontaneous osteonecrosis of the knee. Osteoporos Int 30(3):685–689
Hung CY, Rodriguez M, Roberts A, Bauer M, Mihalek I, Bodamer O (2019) A novel FAM20C mutation causes a rare form of neonatal lethal Raine syndrome. Am J Med Genet A 179(9):1866–1871
Liu P, Ma S, Zhang H, Liu C, Lu Y, Chen L et al (2017) Specific ablation of mouse Fam20C in cells expressing type I collagen leads to skeletal defects and hypophosphatemia. Sci Rep 7(1):3590
Martin A, David V, Laurence JS, Schwarz PM, Lafer EM, Hedge AM, Rowe PS (2008) Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP. Endocrinology 149(4):1757–1772
Staines KA, Mackenzie NC, Clarkin CE, Zelenchuk L, Rowe PS, MacRae VE, Farquharson C (2012) MEPE is a novel regulator of growth plate cartilage mineralization. Bone 51(3):418–430
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Human and Animal Rights and Informed Consent
Written informed consent forms were obtained according to 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
223_2020_694_MOESM1_ESM.pptx
Supplementary file1 (PPTX 3524 kb)—Supplementary Figure. A-B. Two axial cranial MRI slices of the patient taken in five months of interval reveal development of skull shape deformity and enlargement of ventricles. A. Early T1 axial MRI with GD at postnatal 1st day. B: Later T2 images within same level. C-D. Cranial CT showed thickening of cranial bones, pyriform aperture stenosis/agenesis, moderate hydrocephalus and periventricular calcifications.
Rights and permissions
About this article

Cite this article
Eltan, M., Alavanda, C., Yavas Abali, Z. et al. A Rare Cause of Hypophosphatemia: Raine Syndrome Changing Clinical Features with Age. Calcif Tissue Int 107, 96–103 (2020). https://doi.org/10.1007/s00223-020-00694-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-020-00694-3